
Synthesis, structure, and properties of twisted π-conjugated molecules featuring three-dimensional π–π interactions in solid states†
Mai
Nagase
 ab,
Ryu
Yoshida
ab,
Sachiko
Nakano
a,
Takashi
Hirose
c and
Yasutomo
Segawa
*ab
ab,
Ryu
Yoshida
ab,
Sachiko
Nakano
a,
Takashi
Hirose
c and
Yasutomo
Segawa
*ab
aInstitute for Molecular Science, Myodaiji, Okazaki 444-8787, Japan. E-mail: segawa@ims.ac.jp
bThe Graduate University for Advanced Studies, SOKENDAI, Myodaiji, Okazaki 444-8787, Japan
cInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan
First published on 19th June 2025
Abstract
Thiophene-containing twisted π-conjugated molecules were synthesized by introducing methyl groups to sterically hindered positions. Three-dimensional π–π stacking was observed in the solid state, and the hole-transporting ability up to 1.85 × 10−4 cm2 V−1 s−1 was confirmed by constructing field effect transistor devices.
Nonplanar π-conjugated molecules are receiving increased attention because the multiple and intricate intermolecular interactions in the solid state are beneficial for organic functional materials.1–3 Unlike planar π-conjugated molecules, which typically adopt conventional packing structures, nonplanar molecules exhibit alternative packing arrangements with unique electronic properties.4 For instance, twisted two-blade molecules and spiro-linked compounds have been reported to form unusual two-dimensional (2D) π-stacking configurations. In addition, 3D network assemblies have been observed for twisted two- and three-blade molecules. While two-blade molecules resulted in shifted stacking, in three-blade molecules, π–π interactions between the blades and neighbouring molecules contribute to forming three-dimensional networks.5–13
Thiophene rings, owing to their unique properties, play a pivotal role in the field of structural organic chemistry and organic electronics. Thiophene-containing π-conjugated molecules give rise to characteristic electronic and photophysical properties.14–17 Synthesis of thiophene-containing nonplanar π-conjugated molecules can be achieved using several strategies such as the introduction of non-hexagonal rings, inducing steric repulsions of bulky substituents or helically fused structures, and incorporation of spiro linkages.18–22 Among these methods, the introduction of sterically demanding substituents provides a direct means of inducing molecular distortion in a concise synthetic step.
Herein, we present the synthesis of twisted thiophene-containing π-conjugated molecules 1 and 2 (Fig. 1) by tetramethylation reactions of corresponding quasiplanar precursors. The twisted structures were confirmed by X-ray crystallography, and characteristic 2D and 3D π–π stacking modes were observed in the crystal of 1 and 2, respectively. The isomerization barriers of the twisted molecules were studied by theoretical calculations. Photophysical measurements revealed that the frontier molecular orbital energies of 1 and 2 were affected by the twisted structures. The transfer integrals of 1 in the crystalline state were calculated, and the p-type organic field-effect transistor (OFET) performance of 1 was confirmed experimentally.
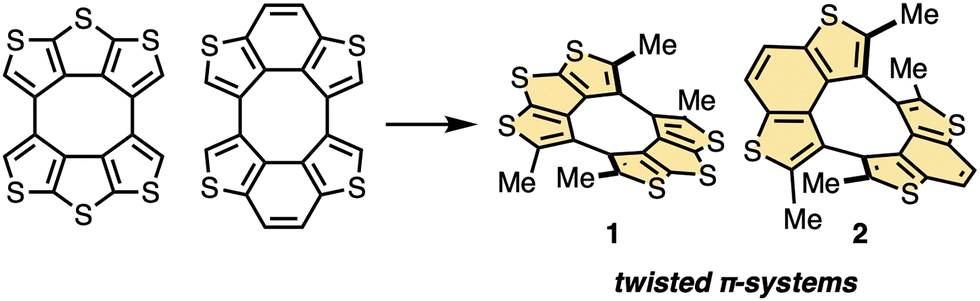 | ||
| Fig. 1 Structures of twisted thiophene-containing π-conjugated molecules 1 and 2. | ||
The tetramethylated dithieno[2,3-b:3′,2′-d]thiophene dimer 1 and tetramethylated benzo[1,2-b:4,3-b′]dithiophene dimer 2 were synthesized by methylation of corresponding precursors 423 and 7 (Fig. 2). Compound 7 was prepared from cyclic thiophene tetramer 3via three steps including base-mediated formylation, Wittig reaction, and alkene metathesis reactions according to reported procedures.24 Tetramethylation of 4 and 7 took place by using an excess amount of lithium diisopropylamide (LDA) and dimethyl sulfate in THF. All products were identified by 1H and 13C NMR spectroscopy as well as high-resolution mass spectrometry.
 | ||
| Fig. 2 Synthesis of twisted π-molecules 1 and 2. Reaction conditions: (i) LDA; Me2SO4, THF, 0 °C to rt; (ii) n-BuLi; DMF, THF, −78 °C to rt; (iii) Ph3PCH2Br, n-BuLi, THF, −10 °C to rt; (iv) Grubbs 2nd catalyst, THF, reflux. LDA = lithium diisopropylamide. DMF = N,N-dimethylformamide. THF = tetrahydrofuran. | ||
Compounds 1 and 2 were subjected to X-ray crystallographic analysis to elucidate the structural characteristics in the solid state (Fig. 3). Suitable single crystals were formed by the diffusion of pentane vapor into benzene solutions of 1 or dichloroethane solutions of 2. It was confirmed that compounds 1 and 2 adopted twisted structures due to steric hindrance of the methyl groups. The twist angles between the planes of the rings highlighted in blue in Fig. 3a and b are 40° (1) and 54° (2), which are larger than those of corresponding precursors 4 (0°)23 and 7 (46°). A slipped stacking structure is observed by two π-planes of the dithienothiophene moieties in 1 (Fig. 3c), which differs from the dimeric pitched π-stacking of 4.19,20 In contrast, compound 2 forms a sheet-like structure along its ab plane (Fig. 3d). The packing structures were altered significantly by methylation.
 | ||
| Fig. 3 (a) and (b) X-ray crystal structures of 1 (a) and 2 (b) with thermal ellipsoids set at 50% probability; gray = carbon; yellow = sulfur. Hydrogen atoms were omitted for clarity. (c) and (d) Packing mode of 1 (c) and 2 (d). | ||
Density functional theory (DFT) calculations were performed to obtain structural insights, with the optimized geometries determined at the B3LYP-D3/6-31G(d) level. The calculated twist angles between the two planes of 1 and 2 are 40° and 55°, respectively, which are quite similar to those observed in the X-ray crystal structures. Compounds 1 and 2 have two stable conformations, i.e. twisted D2 symmetric structures and stair-like C2v symmetric ones owing to the steric repulsion between methyl groups. The twisted structures of compounds 1 and 2 (1-D2 and 2-D2 in Fig. 4) are 11.0 and 26.2 kcal mol−1 more stable than the corresponding stair-like structures 1-C2v and 2-C2v, respectively. The isomerization barriers were calculated to be 28.1 (1-TS) and 51.0 (2-TS) kcal mol−1, indicating that interconversion between 1-D2 and 1-C2v takes place smoothly in solution under ambient temperature whereas isomerization of 2 is quite difficult to occur. Compared with non-methylated derivatives 4 and 7, twisted angles and isomerization barriers are increased due to the steric effect of methyl groups (Fig. 4 and ESI†).
 | ||
| Fig. 4 (a) and (b) Local minima and transition states of 1 (a) and 2 (b) with relative Gibbs free energies (kcal mol−1) calculated at B3LYP-D3/6-31G(d) level of theory. | ||
The UV-vis absorption and fluorescence spectra of 1, 2, 4, and 7 were recorded in dichloromethane (Fig. 5a). Broadened absorption spectra were obtained with molar absorption coefficients (ε) being 1.84 × 104 M−1 cm−1 at 275 nm for 1, 1.85 × 104 M−1 cm−1 at 339 nm for 2, and 1.24 × 104 M−1 cm−1 at 334 nm for 7, where ε could not be determined for 4 because of low solubility. The blue-shift of the longest wavelength absorption bands of 1 and 2 compared with those of 4 and 7 were observed, which are consistent with the slight increase in the HOMO–LUMO gap upon methylation (4.26 eV (4), 4.35 eV (1), 4.05 eV (7), and 4.08 eV (2) as shown in Fig. 5b). The S0–S1 transition wavelengths calculated by time-dependent (TD) DFT also consistent with the experimental results (323 nm (4), 318 nm (1), 339 nm (7), and 346 nm (2)). No fluorescence was observed from the dichloromethane solution of 1 and 4, whereas weak fluorescence 2 and 7 was detected (Fig. 5a) with the quantum yields (ΦF) of 3.3% (2) and 1.3% (7) and the lifetime (τ) of 4.6 ns (2) and 2.5 ns (7), respectively (see ESI† for detail). Using the equations ΦF = kr (kr + knr)−1 and τ = (kr + knr)−1, the radiative (kr) and nonradiative (knr) decay rate constants were calculated for two compounds 2 and 7: for 2, kr = ΦFτ−1 = 7.2 × 106 s−1 and knr = (1 − ΦF) τ−1 = 2.1 × 108 s−1; and for 7, kr = 5.2 × 106 s−1 and knr = 3.9 × 108 s−1.
 | ||
| Fig. 5 (a) UV-vis absorption spectra of 1, 2, 4, and 7 and fluorescence spectra of 2 and 7 excited upon 330 nm in CH2Cl2. (b) The frontier molecular orbitals and their energies of 1, 2, 4, and 7 calculated using B3LYP-D3/6-31G(d) level of theory (isovalue: 0.02). | ||
Since the packing structure of 1 appeared to extend over three dimensions through π–π stacking, electronic properties were investigated by calculation using the obtained X-ray crystal structure. Hole transfer integrals, the indicators that describe the strength of the interaction between occupied molecular orbitals, were calculated using the Amsterdam Density Functional (ADF) program with PW91 level of theory and DZP basis sets.25–27 The transfer integrals between two molecules were 3.0, 7,4, 13.7, 25.5, and 26.8 meV, and the largest transfer integral was found between the molecules where the short contact (Å) was within the sum of van der Waals radii (Fig. 6a and b).
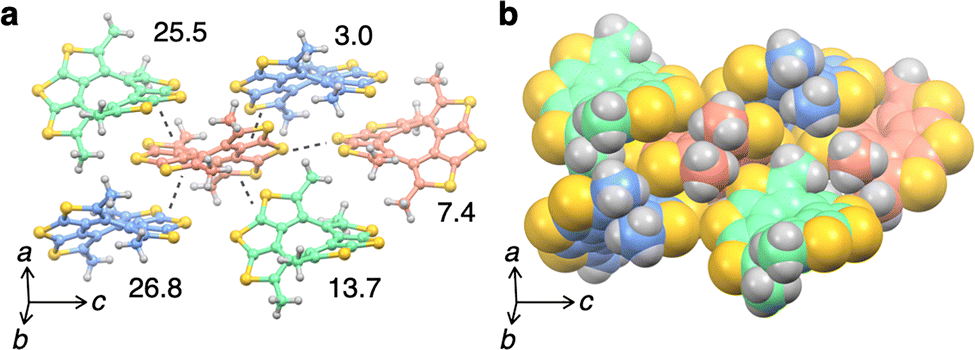 | ||
| Fig. 6 (a) Hole transfer integrals (meV) in the crystal of 1 calculated using ADF with PW91/DZP. Each crystallographically independent molecule was colored differently. (b) Packing model of 1 with space-filling models. | ||
Motivated by the multidirectional hole transfer ability of 1 indicated by ADF calculations, p-type OFET devices were fabricated and its hole mobility was measured. The p-type OFET devices were fabricated in a bottom-contact/bottom-gate configuration by forming a thin film of 1 by vapor deposition on SiO2 substrates modified with hexamethyldisilazane (HMDS) and pentafluorothiophenol (PFBT) (see ESI† for details). The thin layer of 1 exhibited semiconducting behavior, and the average hole mobility of eight independent devices was estimated to be 1.85 × 10−4 ± 1.6 × 10−5 cm2 V−1 s−1 with a threshold voltage of −4.5 V and an on/off current ratio of 7.0 × 103. The transfer and output characteristics of the device are shown in Fig. 7. These results suggested that 1 could be a useful nonplanar π-conjugated molecule for semiconductor applications.
 | ||
| Fig. 7 Transfer (a) and output (b) characteristics of the OFET device for the thin-film layer (40 nm) of compound 1 prepared by vapor deposition on HMDS/PFBT modified SiO2 surface. | ||
In summary, we have investigated the structural, optical, and electronic properties of novel twisted thiophene-containing π-conjugated molecules 1 and 2. The synthesis of 1 and 2 was performed by tetramethylation reactions of corresponding quasiplanar precursors 4 and 7, respectively, owing to the high reactivity of the α-positions of thiophene moieties. The twisted structures and characteristic π–π stacking modes of 1 and 2 in the solid state were confirmed by X-ray crystallography. The isomerization barriers of the twisted molecules 1 and 2 were calculated to be 28.1 (1-TS) and 51.0 (2-TS) kcal mol−1, which are much higher than those of corresponding precursors 4 and 7, respectively, because of the steric effect of methyl groups. Photophysical measurements revealed that the frontier molecular orbital energies of 1 and 2 were affected by the twisted structures. The transfer integrals of 1 in crystalline state were calculated by ADF indicating the existence of multidirectional hole transporting ability. The performance of 1 as a p-type organic field effect transistor (mobility: 1.85 × 10−4 cm2 V−1 s−1) was confirmed experimentally. The non-planar twisted molecule 1, synthesized via a simple methylation reaction, demonstrated semiconducting properties, providing useful guidance for the design of future semiconducting material molecules.
This work was supported by FOREST program (JPMJFR211R to Y. S.) from JST, JSPS KAKENHI (JP22K19038 and JP25K01758 to Y. S.), Murata Science and Education Foundation, Iketani Science and Technology Foundation, and Foundation of public interest of Tatematsu. We thank Prof. Hiroshi Yamamoto (Institute for Molecular Science), Prof. Akinori Saeki, and Prof. Fumitaka Ishiwari (Osaka University) for the support of experiments. Fabrication of OFET devices and those evaluation were conducted using Custom Evaluation Service of OFET performance by Tokyo Chemical Industry, Co. Ltd. M. N. is a recipient of JSPS Research Fellowship for Young Scientists (DC2) and IMS SRA fellowship. This work was conducted in IMS supported by ARIM (JPMXP1224MS5001). Calculations were partially performed using the resources of the Research Center for Computational Science, Okazaki, Japan (24-IMS-C232). Computation time for ADF calculations was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University.
Conflicts of interest
There are no conflicts to declare.Data availability
Crystallographic data have been deposited at the CCDC under 2417361 (7), 2417362 (1), and 2417363 (2). The data supporting this article have been included as the ESI† (pdf) and CartesianCoordinates.xyz (xyz) files.Notes and references
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- M. Ball, Y. Zhong, Y. Wu, C. Schenck, F. Ng, M. Steigerwald, S. Xiao and C. Nuckolls, Acc. Chem. Res., 2015, 48, 267–276 CrossRef CAS PubMed.
- M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef CAS PubMed.
- L. Yang, X. Zhu, Q. Zhou, C. Qi, Q. Wang, F. Shi, M. Zhu, G. Chen, D. Wang, X. Liu, L. Wang, D. Zhang, H. Li and S. Xiao, J. Mater. Chem. A, 2024, 12, 7005–7014 RSC.
- S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390–7394 CrossRef CAS PubMed.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A. M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740–743 CrossRef PubMed.
- K. Kato, K. Takaba, S. Maki-Yonekura, N. Mitoma, Y. Nakanishi, T. Nishihara, T. Hatakeyama, T. Kawada, Y. Hijikata, J. Pirillo, L. T. Scott, K. Yonekura, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2021, 143, 5465–5469 CrossRef CAS PubMed.
- L. Shan, D. Liu, H. Li, X. Xu, B. Shan, J. Bin Xu and Q. Miao, Adv. Mater., 2015, 27, 3418–3423 CrossRef CAS PubMed.
- Y. Yang, L. Yuan, B. Shan, Z. Liu and Q. Miao, Chem. – Eur. J., 2016, 22, 18620–18627 CrossRef CAS PubMed.
- D. Xia, X. Guo, M. Wagner, M. Baumgarten, D. Schollmeyer and K. Müllen, Cryst. Growth Des., 2017, 17, 2816–2821 CrossRef CAS.
- T. Fujikawa, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 7763–7768 CrossRef CAS PubMed.
- T. Fujikawa, N. Mitoma, A. Wakamiya, A. Saeki, Y. Segawa and K. Itami, Org. Biomol. Chem., 2017, 15, 4697–4703 RSC.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184–10190 CrossRef CAS PubMed.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed.
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- M. Miyasaka, A. Rajca, M. Pink and S. Rajca, Chem. – Eur. J., 2004, 10, 6531–6539 CrossRef CAS PubMed.
- C. Zhao, L. Xu, Y. Wang, C. Li and H. Wang, Chin. J. Chem., 2015, 33, 71–78 CrossRef CAS.
- K. Fukuzumi, Y. Nishii and M. Miura, Angew. Chem., Int. Ed., 2017, 56, 12746–12750 CrossRef CAS PubMed.
- D. Waghray, C. de Vet, K. Karypidou and W. Dehaen, J. Org. Chem., 2013, 78, 11147–11154 CrossRef CAS PubMed.
- A. Rajca, M. Miyasaka, M. Pink, H. Wang and S. Rajca, J. Am. Chem. Soc., 2004, 126, 15211–15222 CrossRef CAS PubMed.
- L. Li, S. Zhao, B. Li, L. Xu, C. Li, J. Shi and H. Wang, Org. Lett., 2018, 20, 2181–2185 CrossRef CAS PubMed.
- G. R. Stephenson, C. Silvia and D. Julien, Synlett, 2014, 701–707 CrossRef CAS.
- V. I. Minkin, Pure Appl. Chem., 1999, 71, 1919–1981 CrossRef CAS.
- ADF 2022.101, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com.
- C. Fonseca Guerra, J. G. Snijders, G. Te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details (PDF). CCDC 2417361 (7), 2417362 (1) and 2417363 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02387d |
| This journal is © The Royal Society of Chemistry 2025 |