
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometallated phosphonium ylide-based iridium(III) photocatalysts†
Oussama
Fayafrou
,
Juliette
Zanzi
,
Carine
Duhayon
 ,
Jean-Baptiste
Sortais
,
Olivier
Baslé
* and
Yves
Canac
*
,
Jean-Baptiste
Sortais
,
Olivier
Baslé
* and
Yves
Canac
*
LCC-CNRS, Université de Toulouse, CNRS, UPS, Toulouse, France. E-mail: olivier.basle@lcc-toulouse.fr; yves.canac@lcc-toulouse.fr
First published on 26th May 2025
Abstract
A novel class of cyclometallated photoactive iridium(III) complexes containing phosphonium ylide ligands has been synthesized and thoroughly characterized. Their photophysical and redox properties have been systematically investigated and employed in photocatalytic applications. Notably, these complexes exhibit high activity in visible-light-mediated EnT and SET processes.
In recent decades, photoluminescent transition-metal complexes have emerged as highly versatile compounds, attracting considerable interest for a multitude of applications.1 The two main families of luminescent metal complexes are based on octahedral ruthenium and iridium centers, typically coordinated with bipyridine and phenylpyridine ligands, respectively.2 In particular, cyclometallated iridium(III) complexes possess well-defined photophysical and redox properties,3 making them highly efficient and widely applicable for photocatalytic applications.4 These properties can be precisely tuned by carefully selecting the coordinating ligands and modifying their neighbouring environment.5 A wide variety of ligands, particularly those featuring a coordinating carbon atom, have been extensively studied to improve the efficiency and selectivity of iridium(III) photocatalysts.6 However, phosphonium ylides (P-ylides) behaving as C-sp3 carbon ligands have been unexplored in this context, thus providing an opportunity to evaluate their properties and potential in photocatalysis. Phosphonium ylides are globally neutral η1-carbon ligands,7 characterized by strong σ-donor and weak π-acceptor properties, exhibiting superior donor strength compared to N-heterocyclic carbenes.8 Initially renowned as key intermediates in Wittig-type reactions, they have also demonstrated remarkable coordination chemistry with both main group elements9 and transition metals.10 Their extreme electronic features result in exalted reactivity, which influence the selectivity of catalytic transformations.11 Recently, we reported a photoluminescent ruthenium(II) bipyridyl complex featuring a phosphonium ylide ligand as well as its photocatalytic activity,12 which constitutes a rare case of luminescent P-ylide metal complex13 and the first photocatalyzed reaction involving P-ylides. As mentioned, the absence to date of photoluminescent iridium complexes involving P-ylides prompted us to develop a new family of cyclometallated iridium(III) complexes incorporating such C-sp3 carbon ligands (Fig. 1). These complexes should make it possible to reach higher triplet energy states and different redox potentials than the Ru(II) P-ylide analogue,12 thus broadening the scope of catalytic reactions, both for energy transfer (EnT) and single-electron transfer (SET) processes.
 | ||
| Fig. 1 Representation of targeted cyclometallated P-ylide Ir(III) complexes. | ||
The phosphonium pre-ligand [1](I) used in this study was prepared in high yield via a one-step procedure following a recently reported method.12 The two targeted bis-cyclometallated iridium complexes [2a](PF6) and [2b](PF6) were then synthesized through a sequential process. First, the KHMDS base was added to deprotonate the pre-ligand [1](I), generating the free P-ylide at −78 °C in THF (Scheme 1). Subsequently, chloro-bridged Ir(III) dimeric precursors [Ir(ppy)2-μ-Cl]2 or [Ir(dF(CF3)ppy)2-μ-Cl]2 were introduced and after anion exchange, the formation of cyclometallated P-ylide Ir(III) complexes [2a](PF6) and [2b](PF6) was observed. The latter were isolated as air and moisture stable yellow crystalline solids, in 82% and 75% yields, respectively. The coordination of P-ylide ligand in [2a](PF6) and [2b](PF6) was confirmed by 31P NMR spectroscopy, showing a deshielded single resonance at δP = 50.2 ppm and 51.3 ppm, respectively. These values compared to the precursor [1](I) (δP = 17.1 ppm), indicate successful C-ylide coordination. 13C NMR spectroscopy further supports this assignment with the ylide resonance appearing with an upfield shift (δCH2 = −2.6 ppm for [2a](PF6) and −1.6 ppm for [2b](PF6)), and both exhibiting the expected doublet multiplicity (1JCP = 20.8 Hz and 23.2 Hz, respectively).
 | ||
| Scheme 1 Synthesis of P-ylide Ir(III) bis(phenylpyridyl) [2a](PF6) and [2b](PF6) complexes from the phosphonium pre-ligand [1](I). | ||
Additionally, 1H NMR spectra reveal the presence of two diastereotopic H-atoms of the [–CH2–PPh2Pyr+] sequence as a doublet of doublets in the high-field zone (1.1–1.8 ppm), further confirming the formation of the targeted P-ylide complexes. The X-ray crystal structures of the Ir(III) complexes [2a](PF6) and [2b](PF6) are illustrated in Fig. 2. Both complexes display a slightly distorted octahedral coordination geometry around the Ir(III) center with two phenylpyridine ligands in combination with the cis-chelating pyridyl-phosphonium ylide moiety.
 | ||
| Fig. 2 Perspective views of the cationic part of [2a](PF6) (left) and [2b](PF6) (right) with thermal ellipsoids drawn at the 30% probability level. Selected bond lengths [Å], and angles (°). [2a](PF6): C1–P1 = 1.751(4); C1–Ir1 = 2.209(4); C7–Ir1 = 2.047(4); C19–Ir1 = 2.007(4); N1–Ir1 = 2.166(3); N2–Ir1 = 2.085(4); N3–Ir1 = 2.050(4); P1–C1–Ir1 = 107.5(2); C1–Ir1–C7 = 174.99(16); C1–Ir1–N1 = 85.51(14); N1–Ir1–C19 = 175.64(14); [2b](PF6): C1–P1 = 1.752(3); C1–Ir1 = 2.193(3); C7–Ir1 = 2.047(3); C19–Ir1 = 1.994(3); N1–Ir1 = 2.156(3); N2–Ir1 = 2.072(3); N3–Ir1 = 2.054(2); P1–C1–Ir1 = 106.75(15); C1–Ir1–C7 = 176.81(12); C1–Ir1–N1 = 85.33(11); N1–Ir1–C19 = 172.43(11). | ||
The C–Ir ylide bond distances are comparable in both complexes ([2a](PF6): 2.209(4) Å; [2b](PF6): 2.193(3) Å) and with the rare example of P-ylide Ir(III) complexes.14 These C–Ir bonds are significantly longer than the C7–Ir1 bond ([2a](PF6): 2.047(4) Å; [2b](PF6): 2.047(3) Å), where C7 represents the coordinated carbon trans to the ylide, and the C19–Ir1 bond ([2a](PF6): 2.007(4) Å; [2b](PF6): 1.994(3) Å) where C19 is the coordinated carbon trans to the pyridine. This difference is consistent with the respective hybridization states of the carbon atoms (C-sp3vs. C-sp2). However, the C7–Ir1 bonds exhibit an elongation compared to the C19–Ir1 bonds due to the trans influence of the strongly donating ylide ligand. In the 5-membered metallacycle, the CH2 ylide is out of the plane defined by the other four atoms. Compared to other Ir–N bonds, the elongation of the Ir1–N1 bonds ([2a](PF6): 2.166(3) Å; [2b](PF6): 2.156(3) Å) can be rationalized by the trans influence of the cyclometallated C19 carbon atom. Indeed, the Ir–N2 ([2a](PF6): 2.085(4) Å; [2b](PF6): 2.072(3) Å) and Ir1–N3 ([2a](PF6): 2.050(4) Å; [2b](PF6): 2.054(2) Å) bond distances trans to each other in both complexes remain shorter.
The UV-visible absorption spectra of complexes [2a](PF6) and [2b](PF6), recorded in CH2Cl2 solution at room temperature (RT) at a concentration of 5.0 × 10−5 mol L−1, are presented in Fig. 3 with the corresponding photophysical data summarized in Table 1. Both Ir(III) complexes exhibit intense absorption bands below 300 nm ([2a](PF6), λmax = 261 nm, ε = 60572 M−1 cm−1; [2b](PF6), λmax = 255 nm, ε = 54936 M−1 cm−1), attributed to spin-allowed π → π* ligand-centered (LC) transitions. Weak broad absorption beyond 380 nm and up to 500 nm ([2a](PF6), λmax = 393 nm, ε = 6126 M−1 cm−1; [2b](PF6), λmax = 395 nm, ε = 5736 M−1 cm−1), is assigned to mixed metal-to-ligand-charge-transfer/intraligand (MLCT/IL) transitions.15 The solution photoluminescence (PL) spectra of complexes [2a](PF6) and [2b](PF6) were measured in degassed CH2Cl2 at RT. Upon photoexcitation at 430 nm, both Ir(III) complexes exhibited intense visible-region luminescence, with PL peaks observed at λmax = 506 nm for [2a](PF6) and λmax = 486 nm for [2b](PF6). The PL spectra of both complexes displayed structured profiles, with [2a](PF6) showing a clear shoulder at longer wavelengths. The halogenated complex [2b](PF6) is characterized by a significant blue shift of 20 nm, which reflects its increased 3MLCT energy. This spectral shift can be rationalized by considering the electronic resonance and inductive effects of the halogen substituents on aryl groups. Next, the solution photoluminescence quantum yields and excited-state lifetimes of [2a](PF6) and [2b](PF6) were measured in degassed CH2Cl2 (Table 1). As expected, these values are superior to those of related Ru(II) P-ylide bipyridyl complex recently reported (τ = 62 ns and ϕlum < 0.01).12
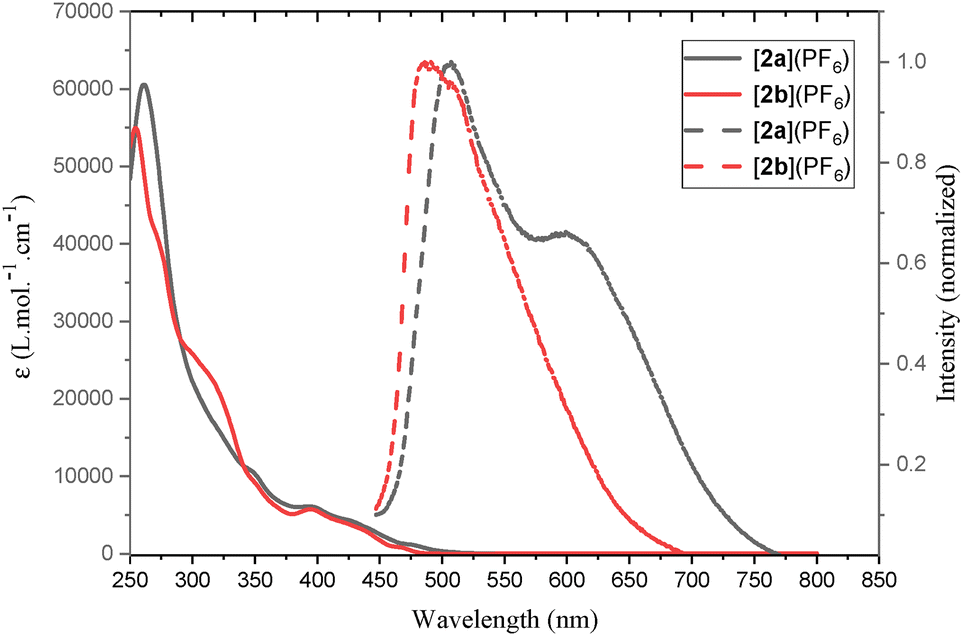 | ||
| Fig. 3 UV-Vis absorbance (full lines) and emission (dashed lines, λex = 430 nm) in CH2Cl2 of [2a](PF6) and [2b](PF6). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
A photostability study was then conducted for both Ir(III) complexes. Their stability under irradiation was monitored by recording 1H NMR spectra at different time intervals. After 24 hours of exposure to 460 nm light, the spectroscopic analysis revealed excellent resistance to photodegradation, with conservation of about 90% and 60% of the initial 1H NMR integration for [2a](PF6) and [2b](PF6), respectively (ESI).
The influence of the P-ylide ligand on the electrochemical properties of complexes [2a](PF6) and [2b](PF6) was investigated using cyclic voltammetry in CH2Cl2 solution with TBAPF6 as the supporting electrolyte (Table 1). Upon anodic scanning, both complexes exhibited a reversible one-electron oxidation process, occurring at E1/2 = 1.04 V vs. SCE for [2a](PF6) and E1/2 = 1.55 V vs. SCE for [2b](PF6). This oxidation process is formally assigned to the Ir(III)/Ir(IV) redox couple. The presence of fluorine atoms on the aromatic rings induces a significant positive anodic shift (+0.49 V). In the cathodic region, complexes [2a](PF6) and [2b](PF6) exhibited a reversible one-electron reduction wave with a smaller positive shift (+0.16 V) observed from [2a](PF6) (E1/2 = −1.59 V vs. SCE) to [2b](PF6) (E1/2 = −1.43 V vs. SCE). According to previous studies of similar compounds, these reduction events are likely attributed to the reduction of the phenylpyridine ligands.16
Based on their triplet energy states estimated from their emission spectra, complexes [2a](PF6) (ET ∼ 56.5 kcal mol−1) and [2b](PF6) (ET ∼ 58.8 kcal mol−1) were first selected for evaluating the photocatalytic activity of P-ylide ligands in Ir(III)-based visible-light photocatalysis driven by energy transfer.17 The first transformation involves the EnT [2 + 2] cycloaddition of (E)-1-(1-methyl-1H-imidazol-2-yl)-3-phenylprop-2-en-1-one (ET ∼ 49.7 kcal mol−1)18 with styrene, using [2a](PF6) and [2b](PF6) as triplet sensitizers under blue LED irradiation (460 nm) (Table 2, left). With 0.5 mol% (0.5 M) of [2a](PF6), the photocycloaddition proceeded with 72% yield in CH2Cl2 (entry 1). Under identical conditions, the cycloaddition product was obtained with a similar yield with [2b](PF6) (70%, entry 3). However, further optimization by reducing the catalyst loading highlighted the remarkable performance of catalyst [2b](PF6). Indeed, while [2a](PF6) (0.05 mol%, 0.5 M in CH2Cl2) led to the product in only 28% yield, [2b](PF6) appeared to be much more efficient affording in the same conditions the cycloaddition adduct in 70% yield (entries 2 and 5). The catalyst loading of [2b](PF6) can even be lowered to 0.01 mol% (0.5 M in CH2Cl2) while maintaining satisfactory yield (67%, entry 6). In MeCN, the product was formed in 53% yield with 0.01 mol% of [2b](PF6) (entry 7). Under the following conditions (0.5 mol%, 0.2 M in CH2Cl2), a kinetic study was performed to evaluate the efficiency of [2b](PF6). These results revealed that the reaction proceeds rapidly, achieving full conversion in just 2.5 hours and thus evidencing the potential of complex [2b](PF6) for this transformation (ESI), which is not accessible by the Ru(II) P-ylide analogue (ET ∼ 43.0 kcal mol−1).12 Based on this, in a second application, we investigated the more energetically demanding intramolecular EnT [2 + 2] cycloaddition of 4-(but-3-enyloxy)quinolone under visible-light irradiation (Table 2, right).19 The 3MLCT energy of [2a](PF6) (ET ∼ 56.5 kcal mol−1) suggests that the cycloaddition of 4-(but-3-enyloxy)quinolone (ET ∼ 57.8 kcal mol−1)20 is slightly endothermic. This was experimentally confirmed, as only 5% yield was obtained using [2a](PF6) (0.5 mol%, 0.5 M in DCM) (entry 8). In contrast, [2b](PF6), which exhibits a higher 3MLCT energy (ET ∼ 58.8 kcal mol−1), effectively promoted the reaction. In our experiments, using 0.5 mol% of [2b](PF6) at 0.5 M concentration under 460 nm LED irradiation resulted in complete conversion of the quinolone substrate (entry 9). Decreasing the catalyst loading of [2b](PF6) to 0.1 mol% led to the product in moderate yield (58%, entry 10). With 0.05 mol% of [2b](PF6), the cycloadduct was formed in 24% yield (entry 11). It should be noted that the reaction can also be carried out in MeCN with generally similar yields (ESI†). A kinetic study of the cycloaddition was conducted using photocatalyst [2b](PF6) (0.5 mol%, 0.1 M in MeCN) indicating a conversion of about 40% after 4 hours, and of 86% after 10 hours of reaction which corresponds to full completion (ESI†). These results therefore underscore the excellent catalytic performance of [2b](PF6), including at low catalyst loadings, even surpassing those of the reference Ir(ppy)3 (entries 12–14) and some of the reported Ir(III) catalysts.21
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| PC | Cat (mol%) | [C] (M) | Yield (%) | PC | Cat (mol%) | [C] (M) | Yield (%) | ||
| a In MeCN. [Ir] = Ir(ppy)3. Yield in % was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. PC = photocatalyst. | |||||||||
| 1 | 2a | 0.5 | 0.5 | 72 | 8 | 2a | 0.5 | 0.5 | 5 |
| 2 | 2a | 0.05 | 0.5 | 28 | 9 | 2b | 0.5 | 0.5 | 100 |
| 3 | 2b | 0.5 | 0.5 | 70 | 10 | 2b | 0.1 | 0.5 | 58 |
| 4 | 2b | 0.1 | 0.5 | 74 | 11 | 2b | 0.05 | 0.5 | 24 |
| 5 | 2b | 0.05 | 0.5 | 70 | 12 | [Ir] | 0.5 | 0.5 | 80 |
| 6 | 2b | 0.01 | 0.5 | 67 | 13 | [Ir] | 0.1 | 0.5 | 43 |
| 7a | 2b | 0.01 | 0.5 | 53 | 14 | [Ir] | 0.05 | 0.5 | 20 |

To take advantage of the excited-state redox potentials of complexes [2a](PF6) and [2b](PF6) (ESI†), the third application involves a reductive cross-coupling reaction between an imine and an olefin using a visible-light photoredox catalysis approach.22 The reaction was carried out by irradiating N-benzylideneaniline in the presence of 3 equiv. of methyl acrylate, 1.5 equiv. of tributylamine, and [2a](PF6) or [2b](PF6) under 460 nm blue LED light for 24 hours in ethanol (Table 3). Complex [2a](PF6) (2 mol%) exhibited very low efficiency in this transformation, yielding only 8% of the coupling product (entry 1). In contrast, [2b](PF6) in the same loading demonstrated excellent catalytic performance, achieving 87% yield (entry 2). Upon reducing the catalyst charge of [2b](PF6) to 1 mol%, a decrease in efficiency was observed, with the yield dropping to 60% (entry 3). According to the generally accepted mechanism,22 the difference in activity between the catalysts can be rationalized by the excited state potential values E(Ir3+*/2+) (+0.86 V for [2b](PF6), +1.12 V for [2b](PF6)) favouring the reductive quenching step with the amine in the latter case. Additionally, various solvents were evaluated to determine their impact on catalytic performance showing with [2b](PF6) the formation of the product in 81 and 71% yields in MeCN and DMF, respectively (entries 5 and 6). Complex [2b](PF6) was also demonstrated to surpass complexes Ir[F(CF3)ppy]2(bpy)PF6 and Ir(ppy)2(dtbbpy)PF6 (entries 7 and 8),22 thus acting as an efficient catalyst for the reductive coupling of imines with olefins.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| PC | Cat (mol%) | [C] (M) | Yield (%) | PC | Cat (mol%) | [C] (M) | Yield (%) | ||
| a MeCN, 24 h. b DMF, 24 h. c From ref. 22, [IrF] = Ir[F(CF3)ppy]2(bpy)PF6, [IrtBu] = Ir(ppy)2(dtbbpy)PF6. Yield in % was determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. | |||||||||
| 1 | 2a | 2 | 0.1 | 8 | 5a | 2b | 2 | 0.1 | 81 |
| 2 | 2b | 2 | 0.1 | 87 | 6b | 2b | 2 | 0.1 | 71 |
| 3 | 2b | 1 | 0.1 | 60 | 7c | [IrF] | 2 | 0.1 | 84 |
| 4 | 2b | 1 | 0.5 | 47 | 8c | [IrtBu] | 2 | 0.1 | 57 |

A new class of C-sp3 carbon ligands, namely phosphonium ylides has been incorporated into cyclometallated iridium complexes, leading to the development of air-stable photoluminescent Ir(III) complexes. These complexes represent the first reported examples of luminescent P-ylide-based Ir(III) compounds. Full characterization revealed that they exhibit well-defined optical and redox properties. Furthermore, these complexes demonstrate excellent performance, even at low catalyst loadings, effectively participating in three distinct reactions under visible-light excitation driven by EnT or SET mechanisms.
This work was supported by the CNRS and the Agence Nationale de la Recherche (ANR-21-CE07-0026 “LYMACATO” grant to OF, JBS and YC; ANR-20-CE07-0021 “SMASH” grant to JZ, YC and OB). The authors thank Gabor Molnar from LCC-Toulouse for excited state lifetime measurements and INSA-Toulouse for luminescence quantum yield measurements.
Data availability
The data supporting this article were included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Li, Y. Chen, J. Wang and C. Yang, Coord. Chem. Rev., 2021, 433, 213755 CrossRef CAS; (b) C. Wegeberg and O. S. Wenger, JACS Au, 2021, 1, 1860 CrossRef CAS PubMed; (c) L. Cho-Cheung Lee and K. Kam-Wim Lo, Chem. Rev., 2024, 124, 8825 CrossRef PubMed.
- (a) Y. Chi, T. K. Chang, P. Ganesan and P. Rajakannu, Coord. Chem. Rev., 2017, 346, 91 CrossRef CAS; (b) J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540 RSC; (c) J. Shen, T. W. Rees, L. Ji and H. Chao, Coord. Chem. Rev., 2021, 443, 214016 CrossRef CAS.
- (a) C. Caporale and M. Massi, Coord. Chem. Rev., 2018, 363, 71 CrossRef CAS; (b) F. Monti, A. Baschieri, L. Sambri and N. Armaroli, Acc. Chem. Res., 2021, 54, 1492 CrossRef CAS PubMed; (c) R. C. Silva, F. S. M. Canisares, L. F. Saraiva, A. M. Pires and A. M. Lima, Dalton Trans., 2024, 53, 5466 RSC.
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 52 CrossRef CAS; (b) D. N. Tritton, F. K. Tang, G. B. Bodedla, F. W. Lee, C. S. Kwan, K. C. F. Leung, X. Zhu and W. Y. Wong, Coord. Chem. Rev., 2022, 459, 214390 CrossRef CAS; (c) Y. L. Li, A. J. Li, S. L. Huang, J. J. Vittal and G. Y. Yang, Chem. Soc. Rev., 2023, 52, 4725 RSC.
- (a) Y. M. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438 CrossRef CAS PubMed; (b) G. Zhou, C. L. Ho, W. Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499 CrossRef CAS; (c) X. Yang, N. Sun, J. Dang, Z. Huang, C. Yao, X. Xu, C. L. Ho, G. J. Zhou, D. Ma, X. Zhao and W. Y. Wong, J. Mater. Chem. C, 2013, 1, 3317 RSC; (d) A. F. Henwood and E. Zysman-Colman, Chem. Commun., 2017, 53, 807 RSC.
- (a) R. Visbal and M. Concepción Gimeno, Chem. Soc. Rev., 2014, 43, 3551 RSC; (b) M. A. Kinzhalov, E. V. Grachova and K. V. Luzyanin, Inorg. Chem. Front., 2022, 9, 417 RSC; (c) H. Amouri, Chem. Rev., 2023, 123, 230 CrossRef CAS PubMed.
- (a) E. P. Urriolabeitia, Top. Organomet. Chem., 2010, 30, 15 CrossRef CAS PubMed; (b) D. A. Valyaev and Y. Canac, Dalton Trans., 2021, 50, 16434 RSC; (c) Y. Canac, Chem. Record., 2023, 23, e202300187 CrossRef CAS PubMed.
- (a) Y. Canac, C. Lepetit, M. Abdalilah, C. Duhayon and R. Chauvin, J. Am. Chem. Soc., 2008, 130, 8406 CrossRef CAS PubMed; (b) Y. Canac and C. Lepetit, Inorg. Chem., 2017, 56, 667 CrossRef CAS PubMed.
- A. Sarbajna, V. S. V. S. N. Swamy and V. H. Gessner, Chem. Sci., 2021, 12, 2016 RSC.
- (a) H. Schmidbaur, Angew. Chem., Int. Ed. Engl., 1983, 22, 907 CrossRef; (b) W. C. Kaska and K. A. Ostoja Starzewski, in Ylides and Imines of Phosphorus, ed. A. W. Johnson, John Wiley & Sons, New York, 1993 Search PubMed.
- (a) R. Taakili, C. Barthes, A. Goëffon, C. Lepetit, C. Duhayon, D. A. Valyaev and Y. Canac, Inorg. Chem., 2020, 59, 7082 CrossRef CAS PubMed; (b) Y. Shi, B. W. Pan, J. S. Yu, Y. Zhou and J. Zhou, Chem. Cat. Chem., 2021, 13, 129 CrossRef CAS.
- O. Fayafrou, E. Lognon, C. Duhayon, J.-B. Sortais, A. Monari, O. Baslé and Y. Canac, Chem. Commun., 2024, 60, 13602 RSC.
- (a) A. Johnson and M. C. Gimeno, Chem. – Eur. J., 2020, 26, 11256 CrossRef CAS PubMed; (b) R. Visbal, N. Rosado, J. Zapata-Rivera and M. C. Gimeno, Inorg. Chem., 2024, 63, 6589 CrossRef CAS PubMed.
- Y. Li, L. Maser, L. Alig, Z. Ke and R. Langer, Dalton Trans., 2021, 50, 954 RSC.
- L. Schmid, F. Glaser, R. Schaer and O. S. Wenger, J. Am. Chem. Soc., 2022, 144, 963 CrossRef CAS PubMed.
- T. Y. Li, X. Liang, L. Zhou, C. Wu, S. Zhang, X. Liu, G. Z. Lu, L. S. Xue, Y. X. Zheng and J. L. Zuo, Inorg. Chem., 2015, 54, 161 CrossRef CAS PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- (a) E. M. Sherbrook, H. Jung, D. Cho, M. H. Baik and T. P. Yoon, Chem. Sci., 2020, 11, 856 RSC; (b) J. Zanzi, Z. Pastorel, C. Duhayon, E. Lognon, C. Coudret, A. Monari, I. M. Dixon, Y. Canac, M. Smietana and O. Baslé, JACS Au, 2024, 4, 3049 CrossRef CAS PubMed.
- K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186 CrossRef CAS PubMed.
- L. Schlosser, D. Rana, P. Pflüger, F. Katzenburg and F. Glorius, J. Am. Chem. Soc., 2024, 146, 13266 CrossRef CAS PubMed.
- (a) J. Zheng, W. B. Swords, H. Jung, K. L. Skubi, J. B. Kidd, G. J. Meyer, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 13625 CrossRef CAS PubMed; (b) R. Takagi and T. Tanimoto, Org. Biomol. Chem., 2022, 20, 3940 RSC.
- Q. Lefebvre, R. Porta, A. Millet, J. Jia and M. Rueping, Chem. – Eur. J., 2020, 26, 1363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopic, crystallographic and catalytic details. CCDC 2443033 and 2443034. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02320c |
| This journal is © The Royal Society of Chemistry 2025 |