
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–nitrogen–carbon catalysts for electrochemical CO2 reduction: from design to industrial applications
Shengyao
Wang
,
Ahmed
Badreldin
and
Ying
Li
 *
*
J. Mike Walker ’66 Department of Mechanical Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: yingli@tamu.edu
First published on 12th June 2025
Abstract
The electrochemical CO2 reduction reaction (eCO2RR) offers a promising route for converting CO2 into value-added chemicals and fuels using renewable electricity. Developing efficient, stable, and scalable catalysts is key to advancing this technology for commercialization. As non-precious metal catalysts, transition metal–nitrogen–doped carbon (M–N–C) materials have demonstrated excellent catalytic performance due to their tunable electronic structure, high activity, and structural stability. Herein, we provide a comprehensive overview of our group's work in designing and optimizing M–N–C catalysts for the eCO2RR, focusing on metal site engineering, carbon substrate modification, and heteroatom doping strategies to enhance electrocatalytic efficiency and selectivity. We have also discussed the challenges and progress in scaling up the synthesis of M–N–C catalysts, integrating M–N–C materials into membrane electrode assembly (MEA) electrolyzers, and employing tandem electrocatalytic systems to achieve multi-carbon products. Comparisons between tandem catalysts and tandem electrolyzers based on M–N–C materials are presented. The potential of coupling the eCO2RR with thermocatalysis for producing other high-value products is also briefly discussed in this review. We envision that the M–N–C catalyst-based eCO2RR will offer a viable pathway for cost-effective CO2 utilization, while future research may focus on demonstrating long-term stability in large-scale electrolyzers and the development of efficient tandem reactor systems to further validate the commercialization potential.
 Shengyao Wang | Shengyao Wang is currently a second-year PhD student in the J. Mike Walker ’66 Department of Mechanical Engineering at Texas A&M University (TAMU), under the supervision of Prof. Ying Li. Her research focuses on the design and optimization of MNC-type and copper-based catalysts for electrochemical CO2 reduction reactions. |
 Ahmed Badreldin | Dr Ahmed Badreldin obtained his PhD degree in Chemical Engineering from Texas A&M University (TAMU) and is currently a postdoctoral researcher with the J. Mike Walker ’66 Department of Mechanical Engineering at TAMU under the supervision of Prof. Ying Li. His primary research focus has been on theoretical and experimental approaches towards electrocatalysts, electrolytes, and electrolyzer designs particularly focused on integrated and direct CO2 reduction routes for C1 and C2+ products. He has also been actively working on near-neutral pH water electrolysis, thermochemical CO2 reduction to solid carbon nanomaterials, advanced oxidation and reduction processes, and engineered osmosis for water desalination. |
 Ying Li | Dr Ying Li is a Professor and Pioneer National Resources Faculty Fellow in the J. Mike Walker ’66 Department of Mechanical Engineering and Artie McFerrin Department of Chemical Engineering at Texas A&M University. His research interests include nanomaterials synthesis, carbon capture and utilization, catalysis for energy conversion, energy storage, water treatment and desalination. His recent research focuses on catalytic conversion of CO2 into fuels and value-added products through photo-, thermo-, and electro-chemical approaches. He is a Fellow of the Royal Society of Chemistry and he is the recipient of a U.S. National Science Foundation CAREER Award. |
1. Introduction
As global energy consumption continues to rise, fossil fuels are still the main energy source for modern society.1,2 However, the increasing emissions of carbon dioxide (CO2) into the atmosphere due to fossil fuel consumption significantly contribute to climate change and serious environmental consequences.3 In recent years, to reduce CO2 emissions and for better carbon management, researchers have developed various carbon capture, utilization, and storage (CCUS) strategies.4,5 Based on the form of energy input, CO2 reduction is generally classified into thermocatalytic, electrocatalytic, and photocatalytic processes.6–11 Among them, the electrochemical CO2 reduction reaction (eCO2RR) has attracted rapidly increasing attention because it can convert CO2 into value-added chemicals and fuels, such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), ethylene (C2H4), and ethanol (C2H5OH), under mild operating conditions. In particular, when powered by renewable electricity, the eCO2RR has the potential to achieve carbon neutrality, or even negative carbon emissions. Thus, it is considered one of the key technologies to achieve a net-zero society in the future.12Despite being a promising technology, many scientific and technical challenges should be addressed before the eCO2RR can be applied at the industrial scale. First, CO2 has a highly stable molecular structure, with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond dissociation energies as high as 750–806 kJ mol−1, requiring high activation energy for reduction.13 Second, the eCO2RR involves a complex multi-electron and multi-proton transfer pathway, and competition between different reaction routes often leads to low target product selectivity.14 For example, the reduction of CO2 to CH4 requires eight electron transfers, while the reduction to HCOOH requires only two electron transfers, but the equilibrium potentials of common eCO2RR products are all around 1.1 V, which only differ by about ±0.1 V, making them thermodynamically non-selective.15 Additionally, the hydrogen evolution reaction (HER) is a major side reaction of the eCO2RR, especially at low overpotentials, and the HER can compete with CO2 reduction and significantly lower the faradaic efficiency (FE), or selectivity, of the target products. Therefore, developing highly efficient and selective electrocatalysts that can reduce the required overpotential (i.e., voltage needed to sustain a certain production rate or current density) and suppress side reactions is key to advancing this technology.
O bond dissociation energies as high as 750–806 kJ mol−1, requiring high activation energy for reduction.13 Second, the eCO2RR involves a complex multi-electron and multi-proton transfer pathway, and competition between different reaction routes often leads to low target product selectivity.14 For example, the reduction of CO2 to CH4 requires eight electron transfers, while the reduction to HCOOH requires only two electron transfers, but the equilibrium potentials of common eCO2RR products are all around 1.1 V, which only differ by about ±0.1 V, making them thermodynamically non-selective.15 Additionally, the hydrogen evolution reaction (HER) is a major side reaction of the eCO2RR, especially at low overpotentials, and the HER can compete with CO2 reduction and significantly lower the faradaic efficiency (FE), or selectivity, of the target products. Therefore, developing highly efficient and selective electrocatalysts that can reduce the required overpotential (i.e., voltage needed to sustain a certain production rate or current density) and suppress side reactions is key to advancing this technology.
Among the various products of the eCO2RR, CO stands out because of its demonstrated superior selectivity and industrial viability. The FE of CO2 reduction to CO has reached nearly 100% in recent studies, whereas C2+ products require multiple CO2 molecules to co-adsorb and react, leading to significantly lower selectivity.16 For instance, the highest FE reported for ethylene, ethanol, and n-propanol is about 83%, 81%, and 30%, respectively.17–19 Since CO formation follows a two-electron transfer process, it avoids the complex multi-step pathways required for C2+ products, resulting in faster reaction kinetics, lower overpotential, and reduced electricity costs.20 Beyond its high selectivity, CO also offers significant advantages in product separation. For liquid products like formic acid it requires energy-intensive separation processes (∼$60 per ton), whereas CO, a gaseous product, can be efficiently purified using pressure swing adsorption (PSA) technology, with a separation cost of approximately $10 per ton, making it highly suitable for large-scale implementation.21 Techno-economic analysis (TEA) has shown that the levelized production cost of CO via the eCO2RR is approximately $0.45 per kg, slightly lower than that of formic acid ($0.47 per kg), and both are below their respective 2023 market prices ($0.6 per kg and $0.68 per kg).22 Additionally, CO has a much larger industrial market compared to formic acid.23 CO is a key intermediate in syngas (H2/CO) production, which is widely utilized in the Fischer–Tropsch (F–T) process to synthesize gasoline, diesel, and other high-value hydrocarbons, as well as in metal refining and chemical synthesis.24,25 Therefore, electrocatalytic CO2 reduction to CO represents the most economical and technically feasible approach for large-scale CO2 electrochemical conversion.
As early as the 1980s, Hori et al. discovered that gold (Au) electrocatalysts could efficiently reduce CO2 to CO at low potentials, achieving faradaic efficiencies of up to 91% and partial current densities of 3.7 mA cm−2.26 In pursuit of a more cost-effective alternative, Lu et al. first developed a nanoporous silver (Ag) catalyst for the eCO2RR, which exhibited a CO faradaic efficiency (FECO) of up to 92%.27 In recent years, researchers have further optimized the nanostructure of Ag to enhance CO selectivity and reaction activity. For example, Liu's group synthesized five-fold twinned Ag nanowires (NWs) using a bromide-mediated polyol method, achieving a maximum FECO of 99.3% and an energy efficiency (EE) of 61.3%.28 However, pure Ag nanoparticles have a high surface energy, making it challenging to maintain their structure and size during long-term operation.29 The electrochemical reduction of CO2 to CO is strongly influenced by the size, crystal structure, and morphology of Ag nanoparticles. Moreover, as a noble metal, silver is relatively expensive and not earth-abundant, posing inherent cost and scalability limitations. While some Ag-based catalysts can reach FECO above 90%, achieving a higher FE close to 100% often requires precise size control and surface structure modifications, which not only increase synthetic complexity but also further elevate catalyst costs.30 Due to these challenges, researchers have begun exploring more economically viable non-precious metal catalysts.
Transition metal–nitrogen–carbon (M–N–C) catalysts have emerged as a key research focus in the field of CO2 reduction due to their high catalytic activity, excellent selectivity, and structural stability.31 The single-atom metal sites coordinate with various nitrogen functional groups in the carbon framework, such as pyridinic nitrogen, pyrrolic nitrogen, and graphitic nitrogen, forming well-defined M–Nx active centers, or single-atom catalysts (SACs).32 These coordination environments not only modulate the electronic structure of the active sites, enhancing CO2 adsorption and activation, but also contribute to catalyst stability by preventing metal atom aggregation or leaching during operation.33 The catalytic activity of M–N–C is primarily attributed to atomically dispersed metal centers, instead of being dictated by specific nanoparticle sizes, which enables high CO selectivity across a broad range of operating conditions.34
Unlike metal nanoparticles, where multiple adjacent metal sites can facilitate C–C coupling via a Langmuir–Hinshelwood (LH) mechanism, M–N–C catalysts feature isolated atomic sites that hinder the co-adsorption and interaction of CO intermediates, thereby limiting the formation of C2+ products.35 This inherent site isolation makes M–N–C catalysts particularly selective toward CO production. Additionally, under working potentials, the metal sites in M–N–C SACs experience charge accumulation effects, which promote the chemisorption of CO2. While this enhances CO2 activation, the adsorbed *CO2 preferentially undergoes protonation to form *COOH rather than *OCHO, ultimately favoring CO selectivity.36
Many recent studies have shown that M–N–C catalysts can inherently achieve CO faradaic efficiencies exceeding 95% across a wide range of formulations.37–39 Compared to noble metal catalysts, M–N–C systems are composed of earth-abundant transition metals (e.g., Fe, Co, Ni) and nitrogen-doped carbon materials, offering advantages such as lower cost, greater scalability, and tunable catalytic properties. As a subclass of SACs, M–N–C catalysts exhibit near-maximum atomic utilization, tunable coordination environments, strong metal–support interactions, and excellent electrical conductivity, making them highly promising electrocatalysts for the eCO2RR.40,41
In the exploration of M–N–C catalysts, research has primarily focused on three key aspects: (1) optimizing metal centers by selecting suitable metal species and tuning their coordination environments to enhance CO2 reduction activity and selectivity; (2) improving catalyst stability and electron modulation through nitrogen coordination and other heteroatom doping; and (3) engineering the base carbon substrates, such as graphene, CNTs, and graphitic carbon nitride (g-C3N4), to optimize electron transport and enhance catalytic stability.
Our research group has made seminal contributions to the aforementioned key areas in advancing M–N–C catalysts for the eCO2RR, and this feature article summarizes our past accomplishments focusing on metal site engineering, nitrogen and other heteroatom doping strategies, variations in carbon substrates, and discussion of metal-free nitrogen-doped carbon (NC) systems. Additionally, a review of the progress from the larger M–N–C research community is incorporated to provide an in-depth analysis of how the recent material innovations influence catalytic performance. Furthermore, a perspective is presented to give insights on the future development of M–N–C catalysts, highlighting key advancements in scalable synthesis, industrial implementation in electrolyzers, application of M–N–C catalysts in tandem reactions, and the integration of electrocatalysis with thermocatalysis for production beyond CO. This article aims to provide a comprehensive understanding of the key design principles of M–N–C catalysts for the eCO2RR and the critical requirements for large-scale industrial applications.
2. Rational design of M–N–C catalysts
Significant progress has been made in the development of M–N–C catalysts for the eCO2RR. In this section, we review key optimization strategies by discussing how the modulation of metal centers, nitrogen coordination, and carbon substrates affects electrocatalytic performance. The discussion is based on our representative studies, providing insights into the structure–performance relationship of M–N–C catalysts.2.1 Metal sites
Notwithstanding, these early-explored electrocatalysts still face challenges such as low activity, large overpotentials, and limited CO selectivity. Furthermore, differences in carbon and nitrogen precursors, synthesis methods, and heat treatment conditions lead to variations in carbon structure, metal site distribution, and nitrogen doping levels. These inconsistencies make it difficult to directly compare results across studies. As a result, the relationship between catalytic activity and metal center type remained unclear at the early-stage, and the catalytic mechanisms of different metals in the CO2RR were not fully understood during earlier explorations. A systematic screening of metal centers was needed to guide the rational design of high-performance M–N–C catalysts.
In an effort to address these limitations, our group synthesized five M–N–C catalysts (Cr, Mn, Fe, Co, and Ni) using a unified solid-phase pyrolysis strategy and systematically compared their eCO2RR performance and possible reduction mechanisms.45 All five synthesized catalysts exhibited M–N coordination environments with similar nitrogen configurations, including pyridinic N (∼398.5 eV), graphitic N (∼400.7 eV), and oxidized N (∼402.8 eV). Additionally, they maintained very close metal loading levels of 2.49 ± 0.30 wt%, ensuring valid activity comparisons. Electrochemical tests revealed that Fe–N–C and Ni–N–C significantly enhanced CO2-to-CO selectivity compared to metal-free N–C catalysts. Fe–N–C demonstrated the lowest overpotential, achieving a maximum FECO of 87% at 0.38 V, while Ni–N–C exhibited the highest CO selectivity (96% FECO at 0.64 V) and a TOF of 1060 h−1 (Fig. 1a). In contrast, Mn–N–C and Cr–N–C favored the competing HER. Although Co–N–C exhibited some CO2 reduction activity at low overpotentials, its CO selectivity decreases substantially at higher applied voltages compared to Ni–N–C. These results highlight Fe and Ni as the most effective metal centers for the eCO2RR, whereas Mn, Co, and Cr display weaker activity due to electronic and coordination limitations. To further investigate the CO2RR mechanism, Tafel slope analysis was carried out (Fig. 1b). Ni–N–C and Co–N–C showed relatively low slopes of 118 and 147 mV dec−1, respectively, which suggest that the rate-determining step involves the initial one-electron transfer to CO2, forming a surface-adsorbed *CO2− intermediate. This intermediate then reacts with another electron and two protons to generate *CO and H2O, followed by CO desorption (Fig. 1c). In contrast, Fe–N–C, Mn–N–C, and Cr–N–C exhibited higher slopes of around 200 mV dec−1. The rate-determining step in these cases may involve a proton-coupled electron transfer process, where CO2 is first converted to a *COOH intermediate. This intermediate is further reduced by one proton and one electron to form *CO and H2O, followed by desorption (Fig. 1c). These differences likely originate from variations in the electronic structure of M–N sites, which affect the binding strength of CO2 and related intermediates. The analysis provides a basis for tuning metal centers in M–N–C catalysts to optimize eCO2RR activity and reaction pathways.
 | ||
| Fig. 1 (a) Summarization of maximum FECO and CO TOF at the corresponding overpotentials of M–N–C at different metal sites. (b) Tafel slopes for M–N–Cs with different metal sites. (c) Proposed CO2RR mechanisms on M–N–C.45 Reprinted with Permission. Copyright 2018 Elsevier. (d) Illustration of M–N4 coordination environments: M–N4–C10vs. M–N2+2–C8.46 Reprinted with permission. Copyright 2018 American Chemical Society. (e) Schematic of CoN5/HNPCSs.49 Reprinted with permission. Copyright 2018 American Chemical Society. (f) FECO at different applied potentials over NiSA–Nx–C.50 Reprinted with permission. Copyright 2020 Wiley. | ||
In one of our studies, two distinct N4 coordination configurations were identified (Fig. 1c): (1) M–N4–C10 (bulk site), where metal atoms are embedded within the graphitic layer and fully surrounded by carbon atoms; and (2) M–N2+2–C8 (edge site), where metal atoms are anchored at the edges of two adjacent N-doped graphitic layers, forming coordination bonds with two nitrogen atoms from each respective layer.46 This structural arrangement induces localized distortion in the carbon skeleton, deviating the metal site from a planar structure.47 A MOF-derived synthesis method was successfully applied to prepare atomically dispersed Fe–N–C and Co–N–C catalysts without metallic phases. We systematically studied two typical N4-coordinated configurations in M–N–C structures through combined experimental and theoretical investigations. Density functional theory (DFT) calculations revealed that for Fe and Co metal centers, the M–N2+2–C8 structure serves as the primary active site for the CO2RR. As shown in Fig. 1d, the reaction enthalpy change (ΔH) between the rate-limiting COOH* to CO* + OH* step for M–N2+2–C8 is approximately −1.18 eV, whereas that of M–N4–C10 exceeds +1.11 eV. This suggests that the M–N2+2–C8 structure has a thermodynamically downhill COOH* dissociation step. In contrast, M–N4–C10, with its metal active site deeply embedded in the carbon framework, exhibits weak *COOH binding (positive ΔH). This increases the required energy barrier for COOH* dissociation, effectively slowing down the eCO2RR rate and enhancing competition from the HER, as HER is thermodynamically more favorable than eCO2RR at these sites.
Besides Fe- and Co-based M–N–Cs, another study of our group further explored the structure–performance relationship on single-atom Ni–N–C catalysts, which were synthesized through calcination of chemically doped ZIF-8.48 Electrochemical tests in H-cell demonstrated a high FECO of 96% at an overpotential of −0.57 V. Theoretical calculations further supported that Ni–N2+2 sites exhibited enhanced CO2 reduction activity compared to bulk-hosted Ni–N4 configurations, as they facilitate the breaking of the C–O bond in the limiting COOH* intermediate. These findings collectively highlight the crucial role of coordination environment engineering in optimizing M–N–C catalysts for the CO2RR.
Furthermore, besides the well-accepted M–N4 structure, other configurations (M–Nx; x = 5, 3, 2) have been reported in M–N–C catalysts, whereas the x value is determined through direct or indirect evidence from characterization or computation.51–53 Pan et al. designed a catalyst based on a single-atom Co–N5 coordination structure (Fig. 1e) by anchoring atomically dispersed Co onto nitrogen-doped porous carbon spheres (HNPCSs) via a polymer-derived strategy.49 X-ray absorption near-edge structure (XANES) results indicated that the oxidation state of Co ranged between +2 and +3, while extended X-ray absorption fine structure (EXAFS) fitting further verified a coordination number of approximately 5, with no detectable Co–Co coordination peak, confirming the atomically dispersed nature of Co. The Co–N5 catalyst, with a loading of 3.54 wt%, achieved a FECO of 99.4% at −0.79 V vs. RHE and maintained an FECO above 90% over a wide potential range from −0.57 to −0.88 V vs. RHE. Combined experimental and DFT studies confirmed that the Co–N5 site serves as the active center for CO2 activation and COOH* intermediate formation, while also promoting efficient CO desorption.
Gong et al. developed a strategy to regulate the coordination environment of single-atom Ni catalysts using a bimetallic MgNi-MOF-74 precursor.50 By varying the pyrolysis temperature, they successfully synthesized a series of Ni single-atom catalysts (NiSA–Nx–C) with different nitrogen coordination numbers. XANES analysis revealed that the oxidation state of Ni in all NiSA–Nx–C samples ranged between Ni0 and Ni2+, while EXAFS fitting confirmed the atomic dispersion of Ni and the presence of different Ni–N coordination states (2, 3, 4). Electrochemical CO2 reduction tests demonstrated that NiSA–N2–C exhibited a high FECO of 98% at −0.8 V vs. RHE, with a TOF of 1622 h−1, significantly outperforming NiSA–N3–C and NiSA–N4–C (Fig. 1f). DFT calculations supported the experimental findings, indicating that the Ni–N2 configuration facilitates CO2 activation by stabilizing the COOH* intermediate and reducing the reaction energy barrier for CO2 reduction.
Overall, the coordination number and type of nitrogen in M–Nx sites endow them with unique catalytic properties. However, the optimal M–Nx coordination structure for the CO2RR remains a subject of debate. For example, some studies suggest that Co–N2 sites exhibit superior CO2 reduction activity compared to Co–N4 and Co–N3 sites while others report that Co–N4 is the most active site.54,55 Additionally, research has shown that synthesis conditions, such as pyrolysis temperature, play a crucial role in tuning the coordination environment. For instance, M–N4 sites may transform into M–N2 sites at elevated temperatures.56 Sa et al. prepared Ni–N3 sites through thermal transformation of planar Ni–N4 structures, which exhibited a 4.7-fold increase in TOF compared to their NiPc/CNT with Ni–N4 structure. In situ XANES further revealed a Ni2+ to Ni+ reduction in Ni–N3.57 Liu et al. employed operando XAS and near-ambient XPS to investigate the catalytic evolution of a Ni SAC during the eCO2RR, revealing a reversible Ni2+/Ni+ redox transition and suggesting that the Ni–N4 structure in the Ni+ state serves as the active site for CO2 activation.58 This work highlighted the dynamic redox behavior of Ni–N4 sites under operating conditions and underscored the importance of real-time characterization for identifying true active species. Nevertheless, in most studies, the identification of M–Nx coordination structures relies on ex situ XAS fitting, which only captures static information before or after the CO2RR. This approach cannot reveal potential structural evolution under dynamic electrochemical environments, potentially overlooking the true active state during catalysis. Therefore, future research should integrate advanced characterization techniques (e.g., in situ XAS, X-ray photoelectron spectroscopy (XPS)) with theoretical calculations to achieve a deeper understanding of the structure–performance relationship in M–N–C catalysts and further optimize their catalytic activity.59
2.2 Nitrogen source and other heteroatom doping
To that end, we developed a novel synthesis strategy for M–N–C catalysts using sulfamethoxazole (SMX), a pharmaceutical waste found in wastewater, as the nitrogen source by first adsorbing SMX onto a carbon substrate before pyrolysis.37 Unlike conventional one-step pyrolysis of solid precursors, this approach enables higher nitrogen utilization efficiency by ensuring intimate contact between the nitrogen source and carbon substrate prior to pyrolysis. However, the choice of nitrogen precursor is crucial, as not all nitrogen-containing compounds exhibit strong affinity for CNTs. SMX demonstrates strong π–π interactions with carbon materials in aqueous solution. This enables efficient adsorption onto commercial CNTs before pyrolysis, ensuring more uniform nitrogen distribution across the carbon surface. By adsorbing SMX onto commercial carbon nanotubes (CNTs) from an aqueous solution under stirring, followed by drying at 60 °C overnight and subsequent pyrolysis at 650 °C for 3 hours under an argon atmosphere, we successfully synthesized Fe–N–C catalysts with 0.5 at% Fe loading. The Fe content in the catalyst originates from intrinsic metal impurities in commercial CNTs, thus no addition of extra metal is necessary. Structural characterization confirmed the single-atom Fe–N4 site in CNT-SMX. Brunauer–Emmett–Teller (BET) analysis showed a surface area of 170.6 m2 g−1, significantly higher than untreated CNT (81.1 m2 g−1), indicating that SMX promotes microporous structure formation, thereby improving CO2 mass transfer during the eCO2RR.
eCO2RR tests demonstrated the superior performance of CNT-SMX. In an H-cell at −0.76 V vs. RHE, CNT-SMX achieved a FECO of 91.5% (Fig. 2a) with a CO generation current of 14 mA cm−2, whereas CNT-melamine (prepared using the same aqueous adsorption approach but using melamine) only reached 60.8% FECO and 1 mA cm−2. Further flow-cell tests confirmed the high performance of CNT-SMX at industrially relevant conditions, maintaining 97.5% CO selectivity at 300 mA cm−2 and above 98% FE towards CO for over 24 hours of operation at a constant current density of 100 mA cm−2. Compared to previous reports on M–N–C catalysts, CNT-SMX required 2 to 4 orders of magnitude less nitrogen precursor while maintaining high CO selectivity and current density.
 | ||
| Fig. 2 (a) CO faradaic efficiency of CNT catalysts using SMX and melamine as nitrogen sources.37 Reproduced with permission. Copyright 2023 Elsevier. (b) and (c) Schematic illustration of the fabrication process for Fe–NS–C and CO FEs on Fe–S–C, Fe–N–C, FeS@NC, and Fe–NS–C.67 Reproduced with permission. Copyright 2020 Elsevier. (d) and (e) Schematic of CO production via eCO2RR over a Ni–SNC electrocatalyst and FECO on different catalysts (NC, SNC, Ni–NC and Ni–SNC).68 Reproduced with permission. Copyright 2022 Elsevier. (f) and (g) TOF for CO of Ni–N2Cl/C, Ni0.2–N2Cl/C, and Ni–N2/C; FEs of CO and H2 for Ni–N2Cl/C.69 Reproduced with permission. Copyright 2023 Wiley. | ||
XPS analysis provided insight into the nitrogen doping efficiency of CNT-SMX. CNT-SMX synthesized via the solution method achieved a nitrogen atomic concentration of 0.80 at%, comparable to CNT-melamine prepared by the traditional dry process (0.77 at%). Remarkably, this was achieved using only 1/100 the amount of nitrogen precursor compared to melamine usage, demonstrating a significantly higher nitrogen utilization efficiency. This suggests that the lower performance of CNT–melamine is likely due to its weaker adsorption on CNT surfaces, leading to lower nitrogen incorporation efficiency. This multi-layer adsorption likely increases the probability of nitrogen-containing intermediates encountering intrinsic metal impurities in CNTs during pyrolysis, thereby promoting the formation of M–N active sites. Additionally, our method eliminates the need for post-pyrolysis acid washing, simplifying the synthesis process and reducing environmental concerns associated with metal-containing effluents. These findings highlight a sustainable approach of converting wastes (SMX) and unwanted products (metal impurities in CNTs) into useful products (CNT-SMX catalysts).
Ever since, solution-based adsorption strategies have been applied to improve nitrogen utilization efficiency and tailor M–N coordination environments. Zhao et al. synthesized nitrogen-doped carbon materials using a solution-based method to investigate the curvature effect of carbon materials on the eCO2RR.66 In their synthesis process, oxidized carbon nanotubes (OCNTs) were mixed with dicyandiamide (DICY) in ultrapure water, followed by ultrasonic treatment and overnight stirring. The mixture was then freeze-dried and annealed under a nitrogen atmosphere to achieve nitrogen doping. Electrochemical tests demonstrated that the resulting nitrogen-doped CNTs (N-CNTs) achieved nearly 100% FECO at −0.6 V vs. RHE and maintained 91% FECO over a 12 hours stability test, showcasing excellent selectivity and stability.
Using 5,5′-ethylenedithio-2,2′-bipyridine (EDTO) as a sulfur-containing ligand, we synthesized Fe–NS–C catalysts via a polymerization–pyrolysis–etching–annealing process, as shown in Fig. 2b. BET analysis revealed that Fe–NS–C exhibits a surface area of 1353 m2 g−1, significantly larger than Fe–N–C (639 m2 g−1) and Fe–S–C (440 m2 g−1). Fe–NS–C achieved a FECO of 98% at −0.8 V vs. RHE, outperforming Fe–N–C (89%) and Fe–S–C (64%) at the same applied potential (Fig. 2c). Furthermore, Fe–NS–C maintained stable CO selectivity for over 30 hours of continuous operation with minimal current density decay. DFT calculations further demonstrated that S-doping shifts the Fermi level of Fe–N4 upward, increasing electron density at the active site. This modulation reduces the free energy barrier for COOH* intermediate formation, thereby enhancing CO2RR kinetics and improving catalytic efficiency. These findings highlight S-doping as a key strategy for improving Fe–N–C catalysts, offering enhanced activity, selectivity, and stability.
Notably, the effectiveness of sulfur doping is not limited to Fe-based catalysts; recent studies have demonstrated its ability to enhance CO2RR performance in other M–N–C systems as well. Yang et al. developed a sulfur-doped single-atom catalyst Ni–SNC (Fig. 2d) by calcining sulfate-doped Zn/Ni ZIF at 1000 °C under an argon atmosphere.68 Structural analysis confirmed the formation of Ni–N3–S active sites, which were shown to effectively lower the energy barrier for CO2 reduction. DFT results showed that compared to Ni–NC, the Ni–SNC catalyst exhibited an energy barrier of only 0.69 eV for CO2-to-COOH* conversion, significantly lower than the 2.02 eV barrier for Ni–NC. eCO2RR tests demonstrated that Ni–SNC achieved an FECO above 90% over a potential range of −0.6 to −0.9 V vs. RHE, with a peak FECO of 95% at −0.8 V (Fig. 2e). The introduction of sulfur was found to enhance local charge density, facilitating COOH* formation and improving CO desorption, thereby enhancing CO2RR performance.
Beyond sulfur doping, researchers have also explored other heteroatom dopants, such as chlorine, phosphorus, and fluorine, to further tailor the electronic structure of M–N–C catalysts for the CO2RR.70–72 For instance, Song et al. developed a Ni–N2Cl/C catalyst using a chlorine-functionalized carbon support, achieving a TOF of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 808 h−1—significantly higher than that of Ni–N2/C (1476 h−1), as shown in Fig. 2f.69 TOF represents the number of catalytic cycles per active site per hour, making it a key indicator of the intrinsic activity of a catalyst. The significantly higher TOF of Ni–N2Cl/C suggests that chlorine functionalization enhances the catalytic efficiency of individual active sites. This catalyst exhibited an FECO of 97.6% at −0.8 V vs. RHE (Fig. 2g) and maintained a CO faradaic efficiency above 80% over a potential range from −0.6 to −1.2 V vs. RHE. Yang et al. developed a phosphorus-doped Ni–N–C catalyst (Ni-NPC) to enhance CO2RR performance.73 eCO2RR tests demonstrated that Ni-NPC achieved a high FECO of 92% and a current density of 22.6 mA cm−2 at −0.8 V vs. RHE. DFT calculations revealed that P-doping modulated the charge distribution, significantly reducing the free energy barrier for COOH* formation, thereby enhancing CO2 reduction kinetics.
808 h−1—significantly higher than that of Ni–N2/C (1476 h−1), as shown in Fig. 2f.69 TOF represents the number of catalytic cycles per active site per hour, making it a key indicator of the intrinsic activity of a catalyst. The significantly higher TOF of Ni–N2Cl/C suggests that chlorine functionalization enhances the catalytic efficiency of individual active sites. This catalyst exhibited an FECO of 97.6% at −0.8 V vs. RHE (Fig. 2g) and maintained a CO faradaic efficiency above 80% over a potential range from −0.6 to −1.2 V vs. RHE. Yang et al. developed a phosphorus-doped Ni–N–C catalyst (Ni-NPC) to enhance CO2RR performance.73 eCO2RR tests demonstrated that Ni-NPC achieved a high FECO of 92% and a current density of 22.6 mA cm−2 at −0.8 V vs. RHE. DFT calculations revealed that P-doping modulated the charge distribution, significantly reducing the free energy barrier for COOH* formation, thereby enhancing CO2 reduction kinetics.
2.3 Carbon substrate
In our early efforts to optimize carbon substrates, our group developed a SiO2-confined pyrolysis strategy to construct Fe–N doped on a CNT/CNS hierarchical hybrid carbon structure (NFe–CNT/CNS).77 SiO2 acts as a confinement template, preventing Fe particle aggregation while inducing the transformation of CNTs into sheet-like carbon nanosheets (CNS) at high temperatures. Compared to NFe@CNT (3.86 at% N, 0.52 at% Fe), NFe-CNT/CNS exhibits higher nitrogen (4.64 at%) and iron (0.66 at%) contents. The hybrid structure synergistically enhances both electron transport and active site exposure by combining the high conductivity of CNTs with the large surface area and abundant defects of CNS. The interconnected CNT/CNS network further facilitates mass transport during the CO2RR. Electrochemical tests show the catalyst achieved a FECO of 69% at −0.19 V vs. RHE, significantly better than that of polycrystalline silver (−1.04 V) and cobalt porphyrin MOF (−0.58 V), which are known CO2RR catalysts. Moreover, a low Tafel slope of 89 mV dec−1 indicates that CO2RR on NFe-CNT/CNS has a faster initial electron transfer to a CO2 molecule compared to that on NFe@CNT, indicative of a higher kinetic rate. This study highlights the importance of rationally designing engineered carbon structures to enhance CO2RR performance. The synergistic effect of the CNT/CNS composite provides a promising strategy for optimizing future M–N–C catalysts.
Inspired by the advantages of the early-discovered hierarchical carbon structure, our group further optimized the design of Fe–N–C catalysts by oxidatively (partially) unzipping CNTs to form graphene nanoribbons (GNRs), leading to the construction of a hierarchical CNT@GNR structure with dispersed Fe–N4 sites (Fig. 3a).78 A mild oxidation treatment using KMnO4 partially unzipped the CNTs, generating GNRs while retaining the CNT backbone, forming an interconnected hierarchical network. During this process, residual Fe nanoparticles (originating from the CNT growth catalyst) were directly converted into single-atom Fe–N4 active sites, which were uniformly dispersed on the CNT@GNR structure. This hybrid structure significantly improved CO2 reduction performance by increasing Fe–N4 site exposure and enhancing electron transport and gas diffusion. As shown in Fig. 3a, oxidation degree played a crucial role in determining the final carbon structure and catalytic performance. When the KMnO4-to-CNT mass ratio was below 3, oxidation was insufficient, limiting GNR formation and Fe–N4 site exposure. BET analysis (Fig. 3b) showed that Fe–N/CNT@GNR-2 had a specific surface area of 224 m2 g−1, more than double that of Fe–N/CNT (104 m2 g−1). As a result, Fe–N/CNT@GNR achieved a CO partial current density of 22.6 mA cm−2 with a FECO of 98% at −0.76 V vs. RHE, outperforming Fe–N/CNT (82% FE) and Fe–N/GNR (92% FE) (Fig. 3c). Conversely, excessive oxidation led to GNR stacking, which hindered mass transport. The best performance was achieved with Fe–N/CNT@GNR-2, which balanced the high conductivity of CNTs with the large surface area of GNRs, maximizing Fe–N4 site exposure and optimizing the CO2 reduction pathway. DFT calculations indicate that the Fe–N/CNT@GNR-2 hybrid structure exhibits excellent CO2 reduction catalytic activity by enhancing CO2 adsorption, lowering the COOH formation energy barrier, facilitating CO desorption, and suppressing the HER, which aligns with the superior catalytic performance observed experimentally.
 | ||
| Fig. 3 (a) Schematic illustration of transforming multiwalled CNTs into Fe–N/CNT@GNR and structural evolution from CNTs to CNT@GNR to GNR by adjusting the KMnO4:CNT mass ratios. (b) BET surface area for Fe–N/CNT@GNR with different KMnO4:CNT mass ratios. (c) Faradaic efficiency of CO production for Fe–N/CNT, Fe–N/CNT@GNR-1/2/3, and Fe–N/GNR.78 Reprinted with permission. Copyright 2020 American Chemical Society. (d) Schematic diagram of the process of synthesizing Ni–N-CNS/CNT using the CVD method.81 Reprinted with permission. Copyright 2021 American Chemical Society. (e) Schematic diagram of the 3D mesoporous carbon skeleton synthesized from self-grown templates.83 Reprinted with permission. Copyright 2017 Royal Society of Chemistry. (f) and (g) CO desorption and TOF comparison of edge-hosted vs. bulk-hosted Fe–N4 sites; comparison of CO faradaic efficiency for N–G and Fe–N–G catalysts with and without pores.84 Reprinted with permission. Copyright 2020 American Chemical Society. (h) Comparison of CO faradaic efficiency for graphite and petroleum coke as the carbon substrate.39 Reprinted with permission. Copyright 2020 American Chemical Society. (i) Comparison of CO faradaic efficiency for corn starch and graphene oxide as the carbon substrate.85 | ||
Two-dimensional graphene and one-dimensional carbon nanotubes each exhibit excellent properties but also have certain limitations. Graphene tends to undergo interlayer stacking, while CNTs have a relatively low density of active sites.79,80 Although the CNT@GNR structure effectively enhances CO2RR activity, its synthesis process still requires oxidation and exfoliation steps, which may limit scalability. To address this challenge and improve material scalability, we developed a one-step chemical vapor deposition (CVD) method to synthesize a Ni- and N-co-doped carbon nanosheet/carbon nanotube (CNS/CNT) composite structure.81 In this approach, pre-synthesized Ni-containing nitrogen-doped carbon nanosheets (Ni–N–CNS) serve as the growth substrate, while C2H2 is introduced as the carbon source to induce in situ CNT growth, forming an interconnected 3D hierarchical network (Fig. 3d). Compared with conventional CVD methods, this strategy eliminates multiple processing steps, avoiding strong acid or oxidant pretreatment and post-synthesis washing, thereby significantly reducing synthesis costs and improving scalability. Electrochemical tests showed that after 20 minutes of CVD growth, the Ni–N–CNS/CNT catalyst achieved a FECO of 90% at −0.66 V vs. RHE in H-cell tests, with a CO partial current density of 18.9 mA cm−2. In flow-cell tests, the catalyst maintained FECO at 85% under a high current density of 600 mA cm−2, outperforming many previously reported M–N–C catalysts.
Many recent studies have also explored hybrid carbon structures for the eCO2RR. Zhang et al. developed a MOF-derived 1D/3D N-doped porous carbon (NPC) material and investigated the spatial confinement effect on the eCO2RR.82 Using Zn-MOF-74 as a precursor, high-temperature carbonization produced 1D carbon nanorods and 3D N-doped carbon framework hybrid structures. This design significantly enhanced CO2 adsorption and charge transfer, leading to a CO/H2 ratio of 5.06 at −0.55 V vs. RHE, with a CO production rate of 31 mmol g−1 h−1, which is 13.7 times and 21.4 times higher than MOF-74-derived 1D porous carbon and N-doped carbon from molecular sieve carbonization, respectively. This enhancement was attributed to the electron transfer capability from the 3D N-doped carbon framework to the 1D carbon structure, along with improved CO2 adsorption and mass transport properties. These findings reinforce the crucial role of interconnected hybrid carbon frameworks in achieving high stability, selectivity, and catalytic activity for the CO2RR.
To overcome the limitations of traditional template-assisted methods, we developed a self-growth-templating strategy using a vitamin B12 (VB12) and NaCl co-assembly system to synthesize a 3D mesoporous N/P/Co co-doped carbon (MPC) framework via one-step high-temperature pyrolysis.83 This method simplifies the conventional hard templating approach while enhancing both the porous structure and electronic properties of the catalyst. In this strategy, VB12 acts as both the C and N precursor and the Co pre-coordination agent, ensuring uniform metal dispersion and forming stable Co–N structures during high-temperature processing. Meanwhile, NaCl serves as a sacrificial template, promoting the formation of a mesoporous carbon framework during pyrolysis (Fig. 3e) and being easily removed via water washing, thereby eliminating the need for additional chemical etching or activation steps.
The resulting 3D mesoporous carbon framework exhibits highly interconnected pore channels (∼30 nm in diameter) and a large specific surface area (1036 m2 g−1), with uniformly distributed N, P, and Co dopants. Structural analysis revealed that pyridinic and pyrrolic N were bonded with the Co forming Co−N coordination, while P exists as P–O/P–C species, modulating the electronic environment of carbon. In CO2RR tests, the catalyst achieved a maximum FECO of 62% at −0.19 V vs. RHE, with a low Tafel slope of 129 mV dec−1. Additionally, in long-term stability tests, the FECO exhibited only minor degradation, demonstrating good structural stability and anti-deactivation capabilities. This 3D N/P/Co co-doped mesoporous carbon framework, synthesized via a one-step method without additional hard templates, simplifies the conventional catalyst preparation process and offers a more efficient and scalable design strategy for optimizing carbon substrates in M–N–C systems.
Beyond mesoporous carbon, graphene-based materials have also been widely used as a carbon support in M–N–C catalysts. Conventional bulk Fe–N4 sites on graphene tend to strongly adsorb CO, hindering desorption and limiting catalytic performance. To overcome this issue, we developed a pore-edge tailoring strategy, introducing nanoscale pores into the graphene framework to optimize Fe–N4 electronic environments and enhance CO formation efficiency.84 Using H2O2 chemical etching of graphene oxide followed by high-temperature reduction and nitridation, we created a porous Fe–N–G-p catalyst with Fe–N4 sites anchored at the pore edges of the reduced graphene structure. Structural analysis confirmed the presence of edge-located Fe–N4 sites, while electrochemical tests showed that Fe–N–G-p achieved a FECO of 94% at −0.58 V vs. RHE (Fig. 3f), a 13% increase over non-porous Fe–N–G, with a threefold enhancement in TOF for CO generation. DFT calculations revealed that Fe–N4 sites at pore edges exhibited a downward shift in the d-band center, weakening Fe–C interactions and facilitating CO desorption, thereby accelerating CO2RR kinetics. As shown in Fig. 3g, the calculated CO adsorption energy for edge-hosted Fe–N4 was −0.72 eV, significantly weaker than that of bulk-hosted Fe–N4 (−1.06 eV), indicating that CO desorption is much easier at the pore edges. The DFT results showcased good consistency with experimental observations. Moreover, compared to non-porous Fe–N–G, Fe–N–G-p exhibited a higher electrochemical active surface area (ECSA) and lower charge transfer resistance (RCT), further supporting the role of pore-edge Fe–N4 sites in facilitating the CO2RR.
The above studies show that the pore structure and edge structure can effectively optimize the active site distribution and electronic environment of M–N–C catalysts, and thus enhance the CO2 reduction activity. In addition to the direct effect on the catalytic sites, the pore structure may also indirectly regulate the reaction microenvironment, thus affecting the kinetics and selectivity of CO2 reduction.89,90
Recently, Zhao et al. investigated the influence of mesoporous structures on the eCO2RR, revealing how pore architectures affect both CO2 transport and local reaction conditions.91 By tuning precursor carbonization, they synthesized Fe–N–C catalysts with dominant 9 nm mesopores (p-Fe–N–C) and 3 nm micropores (s–Fe–N–C). While p-Fe–N–C exhibited a higher CO partial current density (24.6 mA cm−2 at −0.65 V vs. RHE) than s-Fe–N–C (15.3 mA cm−2), its CO selectivity was lower due to an enhancement in the parasitic HER. The study demonstrates that although mesopores facilitate CO2 diffusion, they also buffer the local pH, which prevents the accumulation of hydroxide ions (OH−). This reduces the suppression of the HER, thereby increasing H2 generation and slightly decreasing CO selectivity in mesopore-rich Fe–N–C catalysts. These findings highlight the need for a delicate balance in pore structure optimization, where enhancing CO2 mass transport must be carefully controlled to avoid side reactions. This further reinforces the importance of integrating electronic and structural design strategies in M–N–C catalysts to optimize CO2 electroreduction performance.
Beyond utilizing industrial waste, we also explored biomass-derived materials as sustainable carbon sources. Specifically, we investigated corn starch (CS) as a bio-based carbon source and developed an environmentally friendly catalyst synthesis method.85 In this approach, corn starch was used to effectively adsorb Ni from simulated wastewater, followed by nitrogen-doping pyrolysis to synthesize single-atom Ni–N–C catalysts (CS–N–Ni). This process eliminates the need for post-treatment to remove excess metals via acid washing. As shown in Fig. 3i, the CS–N–Ni catalyst achieved a FECO of 92% at −0.8 V (vs. RHE), comparable to that of graphene oxide (GO)-derived Ni–N–C catalysts, which exhibited an FE of CO of 81%. Both catalysts displayed similar current densities (∼11.5 mA cm−2), confirming that CS can serve as an effective alternative to GO without compromising electrocatalytic performance. More importantly, CS is an abundant and renewable resource, whereas GO synthesis remains costly and requires extensive chemical processing. The potential of converting two wastes (Ni from wastewater and CS from agriculture wastes) into valuable catalyst products makes our method truly sustainable.
Overall, our research group developed two strategies for synthesizing CO2 reduction catalysts based on industrial byproducts and renewable biomass as carbon precursors. These strategies provide complementary advantages—industrial waste-derived catalysts offer economic feasibility while biomass-derived catalysts enable environmental sustainability. It is important to note that sustainability and large-scale production of carbon substrates are not limited to specific precursors but rather represent a scalable research direction. The core contribution of this study lies in validating the feasibility of sustainable carbon substrates and proposing a research framework for sustainable CO2 reduction catalyst development. Looking ahead, as more industrial byproducts and biomass resources become available, the optimal choice of carbon precursors will depend on material availability, economic considerations, and application scenarios.
2.4 Metal-free nitrogen-doped carbon catalysts
In 2009, Dai et al. first reported a metal-free nitrogen-doped carbon nanotube (N-CNT) catalyst for the oxygen reduction reaction (ORR) under alkaline conditions.93 Vertically aligned nitrogen-doped carbon nanotubes (VA-NCNTs) were synthesized, demonstrating superior catalytic activity and durability compared to commercial platinum–carbon (Pt/C) catalysts. This groundbreaking work spurred interest in metal-free carbon-based electrocatalysts across various electrochemical applications. Notwithstanding, N–C catalysts exhibit low catalytic activity and limited suppression of the HER during the eCO2RR, resulting in poor selectivity and efficiency.94 Some studies have shown that the concentration and electronic structure of N-doped sites play a crucial role in catalytic performance, particularly pyridinic nitrogen (Pyri-N), which interacts directly with CO2 molecules and is considered the primary active site for the eCO2RR.95,96To enhance activity and selectivity, we developed a fluorine and nitrogen co-doped mesoporous carbon (NF-C) catalyst using a sacrificial soft-templating approach (Fig. 4a).97 The synthesis employed dicyandiamide, sucrose, and ammonium fluoride, where g-C3N4 acted as a hard/soft template and fluorine/nitrogen sources were released upon thermal decomposition. Compared to N–C, NF-C exhibited a higher nitrogen content (14.1 at% vs. 10.8 at%) and a significantly increased pyridinic-N fraction (72% vs. 40%), confirming that fluorine doping enhances Pyri-N site formation. BET analysis further revealed that NF-C-950 possessed nearly double the surface area of N–C-950 (197 m2 g−1vs. 105 m2 g−1), suggesting improved CO2 mass transport properties. At −0.6 V vs. RHE, the FECO of NF-C-950 reached 90%, significantly higher than that of N–C-950 (64%), while the CO partial current density increased to 1.9 mA cm−2, nearly 15 times higher than that of N–C-950 (0.13 mA cm−2) (Fig. 4b). DFT calculations further revealed that fluorine doping reduced the *COOH adsorption energy (from 1.54 to 0.42 eV) and increased the thermodynamic preference for the CO2RR over the HER, explaining the enhanced activity and selectivity.
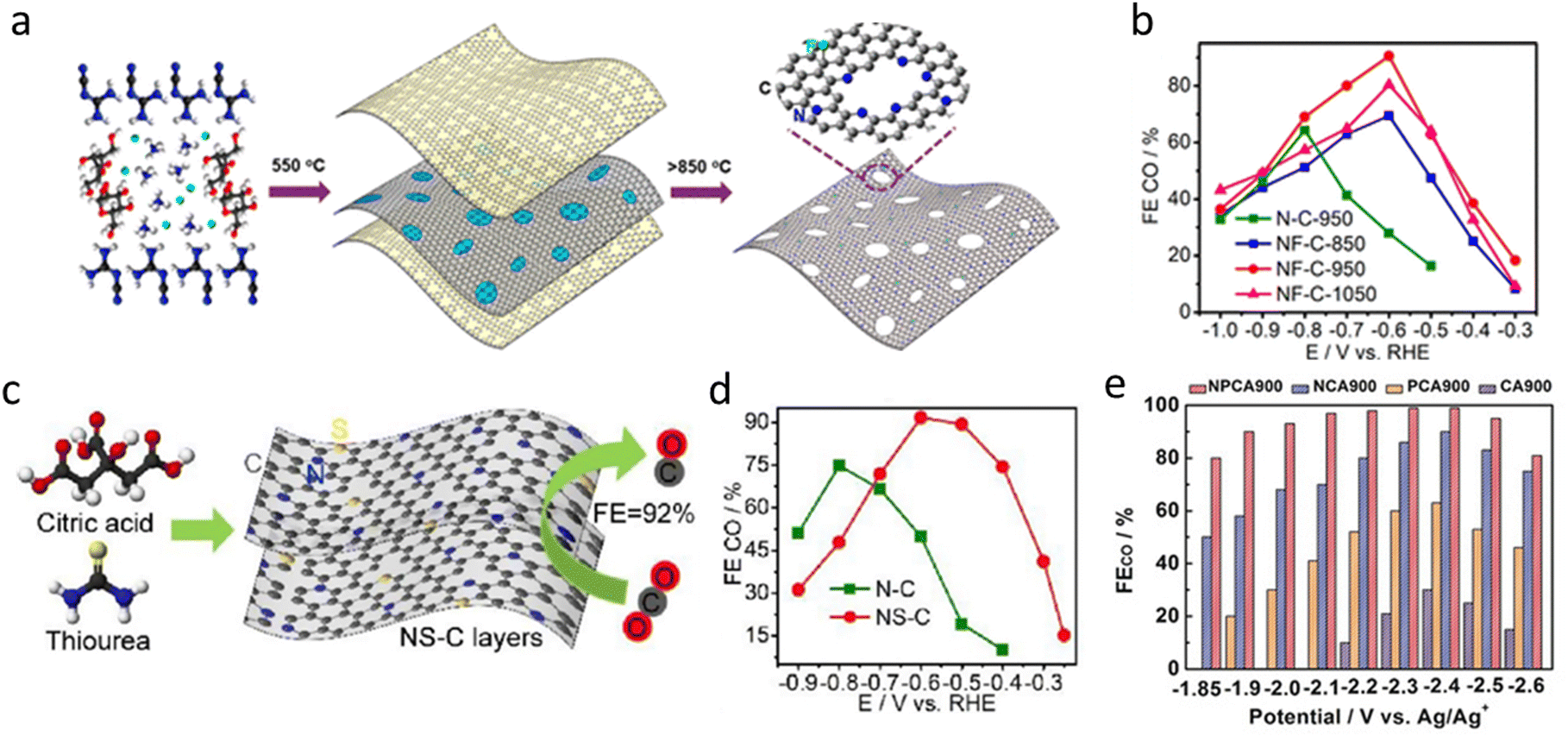 | ||
| Fig. 4 (a) and (b) Schematic illustration of the processing for the synthesis of holey NF–C layers; FECO of N–C and NF–C at different pyrolysis temperatures.97 Reprinted with permission. Copyright 2019 American Chemical Society. (c) and (d) Schematic of NS–C synthesis; CO Faraday efficiency of N–C and NS–C.98 Reproduced with permission. Copyright 2019 Elsevier. (e) The FECO for NPCA900, NCA900, PCA900 and CA900 at different applied potentials.99 Reprinted with permission. Copyright 2020 Wiley. | ||
Beyond fluorine doping, we further explored sulfur doping in N–C catalysts for the CO2RR.98 Using a thiourea–citric acid pyrolysis strategy (Fig. 4c), we synthesized sulfur–nitrogen co-doped carbon (NS–C) catalysts with enhanced catalytic performance. As shown in Fig. 4d, NS–C-900 achieved an FECO of 92% at −0.6 V vs. RHE, significantly surpassing that of N–C-900 (75%). In addition, NS–C-900 exhibited a lower onset potential (−0.4 V vs. RHE) and a reduced Tafel slope (94 mV dec−1vs. 146 mV dec−1 for N–C-900), indicating improved CO2 activation and faster reaction kinetics. Sulfur doping not only enriched Pyri-N content (52% vs. 43%) but also enhanced porosity (160 m2 g−1), facilitating mass transport and increasing active site exposure. DFT calculations further revealed that sulfur lowered the adsorption free energy of the COOH intermediate (from 0.92 eV to 0.30 eV upon S-doping), effectively reducing the CO2 activation barrier and accelerating catalytic rates. These findings highlight sulfur doping as an effective strategy for improving CO2RR selectivity and activity in metal-free catalysts.
In addition to N–F and N–S systems, other non-metal co-doping strategies have also been explored to further optimize electronic properties and catalytic performance. Chen et al. developed an N,P co-doped carbon aerogel (NPCA) catalyst using a simple gelatin-starch method, followed by carbonization at 900 °C.99 NPCA-900 exhibited an impressive FECO of 99.1% (Fig. 4e), with a CO partial current density of 143.6 mA cm−2, significantly outperforming single N- or P-doped carbon aerogels. Similarly, Yan et al. synthesized an N,P co-doped aerogel catalyst that achieved 91.44% FE of CO.100 Liu et al. explored a boron and nitrogen co-doped nanodiamond (BND) catalyst, which exhibited exceptional CO2-to-ethanol selectivity, reaching 93.2% FE at −1.0 V vs. RHE.101 N-doped diamond (NDD) preferentially reduces CO2 to the C2 product CH3COO−, whereas the major product of B-doped diamond (BDD) is HCHO. Both experimental and DFT results indicate that the synergistic effect of B and N co-doping is essential for the production of CH3CH2OH by BND.
Looking ahead, metal-free N–C catalysts are emerging as cost-effective, tunable, and highly active materials for CO2 electroreduction. Future research will focus on enhancing catalytic activity, optimizing product selectivity, and improving long-term stability. While non-metal doping has been shown to modulate electronic structures, improve CO2 adsorption, and facilitate C–C coupling, achieving stability under high current densities remains a key challenge. Expanding co-doping strategies with different elements may further optimize electron transfer pathways and reduce overpotential, while defect engineering and interfacial tuning could enhance overall catalytic performance.
Despite the promising performance of metal-free nitrogen-doped carbon (N–C) materials in CO2 electroreduction, the true nature of their active sites remains under debate. Studies by Compton, Pumera, and others have shown that many so-called “metal-free” carbon nanomaterials often contain residual transition metal (TM) impurities that are difficult to fully remove.102,103 For example, in carbon nanotubes (CNTs), even prolonged acid washing may still leave up to ∼1.0 wt% of residual metals such as Fe, Co, or Ni.104,105 These impurities have been demonstrated to dominate the electrochemical behavior of the carbon materials, masking their intrinsic activity. Notably, CNTs containing as little as 0.045 wt% Fe were found to govern the electrocatalytic performance for H2O2 reduction.106 Moreover, such metal residues can exist as encapsulated nanoparticles within graphite layers, which remain electrochemically active through diffusion or ion intercalation, even when buried deep in the carbon matrix.107
In graphene-based materials, the issue of metal impurities is even more complex. Even when using ultra-high-purity graphite (99.999%) as the precursor, metal contaminants such as Mn, Fe, and Ni are inevitably introduced during the oxidation and reduction steps used to prepare GO or rGO, some of which originate from the oxidants (e.g., KMnO4, K2FeO4) or other chemical reagents used in processing.108,109 These trace impurities, often undetectable by standard characterization techniques like XPS, can still significantly influence the electrochemical properties of the carbon framework. These findings challenge the definition of “metal-free” catalysts and highlight the necessity of careful quantification and identification of metal residues when evaluating structure–activity relationships.107
In addition to impurities inherent in carbon substrates, trace metal ions in electrolytes can also contribute to the formation of active sites during the CO2RR. Kim et al. reported that even under metal-free synthesis conditions, trace Fe2+ ions (2.5 ppm) present in the electrolyte could be electrochemically reduced and selectively deposited onto electron-rich regions of N-doped carbon during operation, forming Fe–Nx coordination structures in situ.110 These newly formed Fe–N sites significantly enhanced catalytic performance, with their NC-PPV catalyst achieving jCO > 3.8 mA cm−2 and FECO > 90% at −0.6 V vs. RHE, maintaining this performance over 120 hours of continuous operation. Structural characterization confirmed the formation of Fe–N bonds. This study revealed a “self-activation” mechanism, whereby trace metal impurities from the electrolyte can be transformed into catalytically active M–Nx sites during the CO2RR, potentially leading to the misattribution of activity to the intrinsic properties of metal-free N–C catalysts. Therefore, strict control and monitoring of metal impurities in both carbon precursors and electrolyte solutions are essential for accurate interpretation of catalytic performance in metal-free systems.
Taken together, these findings raise critical questions about the actual contribution of pyridinic-N and other heteroatoms to catalytic activity, necessitating further rigorous investigation to distinguish intrinsic activity from impurity-derived effects.
3. Towards commercialization: scaling-up M–N–C catalysts and electrolyzers
In the previous sections, we discussed the systematic optimization of the key components of the M–N–C catalysts, including metal sites, carbon substrates and heteroatom doping, and explored the catalytic performance of metal-free NC. These optimization strategies greatly enhanced the CO2RR selectivity, activity, and stability of the catalysts, resulting in excellent catalytic performance under laboratory conditions. However, practical application requires more than improved catalytic properties. Currently, the SAC preparation process is still complex, leading to low SAC catalyst yields.111 Increasing the synthesis scale to the kilogram level while maintaining performance consistency is essential for advancing their practical use. For instance, Ismail et al. reported that increasing the absolute precursor loading from 100 mg to 1 g significantly altered the product structure, leading to the formation of metal carbide particles instead of isolated M–Nx sites.112 This highlights that scalability must be carefully balanced with structural fidelity to ensure the preservation of active site configurations. In addition to catalyst production, integrating M–N–C materials into large-scale electrolyzers is a key step toward industrial implementation. Among different electrolyzer designs, membrane electrode assembly (MEA)-based systems provide an effective platform for applying M–N–C catalysts due to their compact design and high efficiency. Expanding the electrode area and stacking multiple MEAs are two commonly used strategies for increasing electrolyzer capacity.113 To advance M–N–C catalysts toward commercialization, this section discusses both the large-scale synthesis of M–N–C catalysts and their integration into expanded electrolyzer systems. These efforts are crucial for bridging the gap between laboratory research and industrial applications, paving the way for large-scale CO2 electrolysis.3.1 Scaling up catalyst synthesis
In recent years, researchers have explored various strategies to improve the scalable synthesis of M–N–C catalysts. Han et al. reported a hydrogen-bonded organic framework (HOF)-derived method, producing 1.1 g of Ni–N3 SACs through high-temperature pyrolysis with dicyandiamide (DCDA) as the nitrogen source.114 The catalyst exhibited a remarkable FECO of 95.6%, a TOF of 34000 h−1 at −0.8 V vs. RHE, and stable operation for 24 hours at −1.0 V vs. RHE. To further improve catalyst yield and simplify the synthesis, Sun et al. developed a microwave-assisted synthesis strategy, enabling the preparation of 2.5 g of Co–N3 SACs (Fig. 5a).115 This method exploits the strong microwave absorption capacity of defective carbon nanotubes (CNTs), rapidly heating them within 2 min to create a localized high-temperature environment, allowing for simultaneous Co site anchoring and nitrogen doping (Fig. 5b). This approach significantly reduces energy consumption and metal agglomeration, while achieving >90% FECO in an H-cell system and 96.0% FECO in a flow cell at a high current density of 350 mA cm−2. Further increasing the synthesis scale, Han et al. employed a polymerization-pyrolysis strategy, using commercial Vulcan carbon as the support and loading Fe through pyrrole monomer polymerization, followed by Ni precursor addition and pyrolysis at 800 °C.116 By scaling up the amount of precursors and solvents, they achieved catalyst production at the 8 g level. However, despite these advances, these synthesis strategies still rely on complex multi-step procedures, including metal precursor introduction, acid washing, and multiple high-temperature treatments.
 | ||
| Fig. 5 (a) and (b) Digital photograph of the synthesized Co-CNTs-MW at milligram-scale (insert) and at gram-scale; HAADF-STEM image of Co-CNTs-MW showing the single-atom Co site.115 Reproduced under CC BY 4.0 license. (c) M–N–C synthesis at the 10 g level and TEM of active single atomic sites.38 Reproduced with permission. Copyright 2023 American Chemical Society. (d) and (e) Scaling-up MEA: from 25 cm2 to 140 cm2 and multi-cell stacking; longtime operation of the 4-cell stack.116 Reproduced with permission. Copyright 2024 Elsevier. | ||
In 2023, our research group invented a simpler, more efficient, and scalable strategy for M–N–C catalyst synthesis, successfully achieving batch production at the 10 g level (Fig. 5c)—breaking the existing scale-up bottleneck and offering new possibilities for commercial utilization of CO2 electrocatalysts.71 Unlike conventional multi-step M–N–C synthesis, which requires additional metal precursors and complex post-treatments, our approach utilizes commercially available multi-walled carbon nanotubes (MWCNTs) as both the carbon support and metal source, relying on intrinsic Fe and Ni impurities within MWCNTs to form active sites, by directly pyrolyzing MWCNTs with nitrogen precursors (e.g., melamine) at 650 °C. This one-step method eliminates acid washing, activation, and secondary calcination processes, significantly reducing synthesis complexity and cost. In catalytic performance tests, our 10-g batch Fe–N–C catalyst exhibited >95% FECO at −0.8 V vs. RHE in H-cell tests and 97% FECO at 400 mA cm−2 in flow cells. Notably, the catalyst demonstrated excellent stability over 45 h at 100 mA cm−2, surpassing Ag-based noble metal catalysts in terms of both FECO and stability. These results demonstrate the potential of our novel catalyst and synthesis method for large-scale electrochemical CO2 conversion.
The core contribution of this work is the breakthrough of the scale-up bottleneck in the synthesis of M–N–C catalysts and the low-cost, easy-to-operate and mass-producible synthesis strategy. Our lab has currently expanded the production and successfully achieved 75 g per batch synthesis. This is at least three-orders-of-magnitude increase in yield compared to traditional milligram-scale synthesis methods. Our catalyst is commercially available for purchase at the Fuel Cell Store. Hopefully, it can serve as a benchmark M–N–C catalyst for the research community to compare with the various M–N–C catalysts prepared in individual labs. While this development brings M–N–C catalysts closer to industrial application, further efforts are still needed to scale up more complex M–N–C systems, particularly those incorporating non-metallic coordination sites (e.g., B, S, P) or multiple metal centers, to achieve high production rates and purity. Ongoing studies are addressing these challenges and will soon be available to the community.
3.2 Scaling up electrolyzers
Besides scalable synthesis of M–N–C catalysts, the scalability of CO2 electrolyzers is another important challenge for the practical application of the eCO2RR. In recent years, CO2 electrolysis has advanced from H-cells and flow cells to MEA systems, which can operate at higher current densities and larger electrode areas.44 MEA is a zero-gap structure, where the catalyst layer is directly attached to the membrane, eliminating the need for a bulk liquid catholyte. Compared to flow cells, the MEA design greatly reduces internal resistance, improves energy efficiency, and enhances mass transport. As a result, MEA systems have become the primary technology for scaling up CO2 electrolysis.113 Currently, the mainstream scaling-up strategies for MEAs include: (1) increasing the area of individual MEA cells to enhance the CO2 conversion per unit time; and (2) stacking multiple MEA cells into a stack to increase overall reaction rates and improve system integration.Sun et al. evaluated the performance of Co-CNTs-MW catalysts in MEA systems with different electrode areas (1.2 cm2 and 100 cm2).115 In the small-scale 1.2 cm2 MEA, the catalyst maintained 91.6% FECO at 200 mA cm−2, demonstrating efficient CO2 reduction. When expanded to a 100 cm2 MEA, the catalyst achieved around 85% FECO across a broad current range (1–10 A). Notably, in a 10 × 10 cm2 MEA, the catalyst sustained >90% FECO at 2 A for over 50 hours, with no significant degradation in cell voltage. This study provided the first experimental validation of M–N–C catalyst stability at high current densities over an extended operation period, supporting their feasibility for the industrial eCO2RR.
Han et al. investigated NiFe-DAC diatomic catalysts in larger MEAs and stacked systems.116 In a 25 cm2 MEA, NiFe-DAC achieved 98.7% CO selectivity at 5 A (200 mA cm−2), and maintained >98% FECO using 0.1 M KHCO3 as the anode electrolyte, demonstrating excellent stability. Further expansion to a 140 cm2 MEA showed sustained >97% CO selectivity at 28 A, with a CO production rate of 11.4 L h−1, equivalent to 1 mol CO in 1.97 h. To push scalability further, the researchers constructed a four-cell MEA stack (Fig. 5d) with a total area of 560 cm2. Operating at 200 mA cm−2 for 6 hours, the system achieved >97% CO selectivity (Fig. 5e) and a CO production rate of 45 L h−1. This work provided the demonstration of M–N–C catalysts in a multi-cell eCO2RR electrolyzer, proving their potential for both single large-area MEAs and stacked electrolyzer configurations.
However, scaling up MEA systems still faces many challenges. As the electrode area expands, spatial variations in CO2 concentration, product accumulation, and local reaction conditions become more pronounced, potentially leading to CO2 depletion zones that favour the HER over the eCO2RR.117 In large area MEAs, extended flow pathways may induce pH gradients, affecting reaction kinetics and overall system stability.118 Additionally, prolonged operation can result in back-side salinization in gas diffusion electrodes (GDEs), which reduces ion transport efficiency and increases cell resistance.119
To address these issues, further research is needed to optimize MEA structure, control catalyst layer uniformity, and refine electrolyte management strategies to mitigate the impact of GDE salt deposition.120,121 Moreover, improving CO2 mass transfer efficiency, optimizing the integration of MEA components to reduce full-cell voltage, and validating performance in larger-scale MEAs or more complex electrolyzer stacks will be crucial directions for future research.122
4. Beyond CO production: broader applications of M–N–C catalysts in the eCO2RR
While CO is the primary product of M–N–C catalysts in the eCO2RR with demonstrated high selectivity and stability, further conversion of CO into higher-value products represents an important step toward practical CO2 utilization. Recent research has explored several promising strategies to move beyond CO as the sole product, including electrocatalytic tandem systems for multi-carbon production and hybrid electro-thermal tandem catalysis.123 These approaches aim to enhance selectivity toward C2+ products or enable syngas production for thermocatalytic upgrading, offering new directions for industrial applications.4.1 M–N–C catalysts for syngas production
Syngas, a gaseous mixture of carbon monoxide (CO) and hydrogen (H2), serves as a fundamental feedstock for a variety of industrial processes, including Fischer–Tropsch (FT) synthesis and methanol production.124 Syngas has much broader industrial applications than CO alone. Conventional syngas production methods, such as coal gasification and steam methane reforming, typically operate under harsh conditions (high temperature and pressure), rely heavily on fossil resources, and emit significant amounts of CO2.125,126 In contrast, the eCO2RR offers a sustainable alternative for syngas generation under ambient conditions and eliminates the need to separate CO from H2. Hydrogen production from the HER, traditionally regarded as a competing side reaction in the eCO2RR, can contribute to syngas production as long as the H2/CO ratio can be well controlled and the long-term stability maintained. To precisely control the H2/CO ratio, a well-designed catalyst is essential. M–N–C catalysts, featuring tunable electronic structures and metal–nitrogen coordination environments, have recently emerged as promising platforms for modulating syngas output via selective CO2-to-CO and H2O-to-H2 pathways. In principle, two active sites are designed on M–N–C catalysts, one favoring CO and the other favoring H2 production.He et al. investigated Co- and Ni-based M–N–C catalysts to achieve a tunable CO/H2 product ratio.127 The synthesized Ni–N–C promotes CO production, whereas the Co–N–C mainly promotes H2 production. At −1.0 V vs. RHE, Ni–N–C exhibited high selectivity toward CO production, achieving a FE of over 90% and Jco reached 56 mA cm−2. In contrast, Co–N–C primarily facilitated H2 evolution, maintaining an FE of H2 above 80%. Based on these comparisons, the study evaluated a CoNi–N–C catalyst with equal Co and Ni content. Across a potential range of –0.5 to –1.0 V, the catalyst produced syngas mixtures (CO and H2) with FEs ranging from 45% to 55% (Fig. 6a), while maintaining high total current densities. This suggests that the coexistence of Co and Ni does not interfere with the intrinsic activity of each transition-metal-based SAC site. The CoNi–N–C bimetallic catalyst exhibited a total current density of >74 mA cm−2 at −1.0 V (vs. RHE), while offering a tunable CO/H2 ratio (0.23–2.26) by changing the Co/Ni ratio (Fig. 6b). Stability tests showed that the total current density (∼51 mA cm−2) and FE of CO (∼53%) of the CoNi–N–C system remained stable over 7 h continuous operation at −0.9 V (vs. RHE).
 | ||
| Fig. 6 (a) Faradaic efficiencies for CO and H2 evolution at different potentials. (b) Yield of syngas with tunable CO/H2 ratios.127 Reprinted with permission. Copyright 2020 Wiley. (c) Schematic of dual Co–N4 and pyridinic Co sites enabling syngas tuning.128 Copyright 2021 Elsevier. (d) A cross-sectional SEM image of a Cu/Ni–N–C tandem electrode with two catalyst layers. Scale bars: 4 μm. (e) Faraday efficiency and partial current density of C2H4 of Cu/Ag, Cu/Ni–N–C, and Cu/Au catalysts.135 Reprinted with permission. Copyright 2020 Elsevier. (f) Schematic of segmented tandem gas-diffusion electrodes. (g) Faraday efficiencies of C2H4 and C2+ liquid products of Cu/Fe–NC, Cu/Ag, and Cu/ZnO s-GDE when operated in MEA cells.136 Reprinted with permission. Copyright 2022 Springer Nature. (h) Schematic of tandem electrolyzers.137 Reproduced under CC BY 4.0 license. | ||
Ni et al. constructed a dual-site carbon-based catalyst containing two distinct single cobalt atom sites—CoN3 sites embedded in the porous carbon matrix and cobalt phthalocyanine (CoPc) molecules anchored onto the surface—enabling tunable H2/CO ratio production at industrial-scale current densities (Fig. 6c).128 The CoPc interacts with the substrate in two configurations: N–CoPc, anchored via pyridinic nitrogen and favoring CO2-to-CO conversion, and CoN3–CoPc, coordinated with embedded CoN3 sites and promoting the HER. The catalyst achieved a total current density of 225 mA cm−2 at −0.86 V (vs. RHE) in an H-cell (1.0 M KHCO3), with a tunable H2/CO ratio in the range of 1.05–3.87. When tested in a flow cell (1.0 M KOH), the total current density was further increased to 880 mA cm−2.
While these studies demonstrate the potential of M–N–C catalysts for syngas production with tunable H2/CO ratios, challenges remain regarding long-term stability under realistic operating conditions. The stability of GDEs designed for syngas production presents unique challenges compared to GDEs optimized solely for CO generation. Since syngas consists of a mixture of CO and H2, the production of H2 inherently requires water reduction at the catalyst–electrolyte interface. This implies that the GDE must be sufficiently wetted to allow for hydrogen evolution, which simultaneously increases the risk of water intrusion and initiates the early stages of electrode flooding. As water progressively permeates the porous GDE structure, it compromises gas transport pathways, leading to performance degradation and significantly shortening the operational stability of the electrode. In contrast, separating CO and H2 production into two distinct cells or electrolyzers—one optimized for CO2 reduction to CO and another for water reduction to H2—mitigates this issue. Under these conditions, the CO2 reduction GDE can maintain minimal wettability, preserving its dry interface and ensuring long-term stability without the complications associated with flooding. However, the two-electrolyzer system is more complex with a larger footprint, and thus more costly compared to the one-electrolyzer system that can produce CO and H2 simultaneously.
4.2 Tandem catalysts for direct C2+ production
Besides the production of syngas, efficient and selective production of multi-carbon (C2+) products from CO2 electroreduction remains a major challenge due to the complex reaction pathway, involving multi-step CO2 reduction, adsorption and activation of intermediates, and spatial localization for C–C coupling.129 DFT studies have indicated that the overall selectivity toward C2+ products is strongly correlated with the kinetics of *CO–CO coupling, which increases proportionally with the square of the local CO coverage.130,131 Based on this understanding, tandem catalytic strategies have been proposed to enhance C2+ production.132–134 In this approach, CO2 is first selectively reduced to CO on one catalyst, generating a high local CO concentration, which is then further converted to C2+ products on a second catalyst that promotes C–C coupling.Among various catalysts, M–N–C materials exhibit outstanding selectivity and stability for CO2-to-CO conversion, often achieving nearly 100% faradaic efficiency, making them an ideal ingredient for tandem catalysts.138 In typical tandem catalyst configurations, M–N–C and Cu-based catalysts are integrated on a single electrode to enable spatially coordinated CO production and subsequent C2+ conversion, achieving synergistic electrocatalysis.
She et al. investigated a Cu/Ni–N–C based tandem catalyst system, where the Ni–N–C layer acted as a CO generation catalyst and the Cu layer acted as a C2+ product generation catalyst (Fig. 6d).135 This Cu/Ni–N–C tandem electrode achieved a C2H4 FE of up to 62% at −0.70 V (vs. RHE), while the partial current density of C2H4 (JC2H4) reached 415 mA cm−2 (Fig. 6e). In the tandem system, different CO-selective catalysts may affect the overall catalytic performance. They further compared the effects of Ag, Au and Ni–N–C as CO-generating layers. The results showed that Cu/Ni–N–C exhibited the highest FE and production rate of C2+ products in the low-potential range (−0.20 to −0.66 V), followed by Cu/Au and Cu/Ag. This further validates the superiority of Ni–N–C as a catalyst for CO generation in the tandem system. The tandem electrode was tested in the MEA system and the FE of C2H4 reached 48% at a cell voltage of 3.0 V with a total current density of 220 mA cm2.
Unlike other studies depositing tandem catalysts uniformly across the entire electrode, Zhang et al. designed a segmented gas-diffusion electrode (s-GDE) by introducing a CO-selective catalyst layer in the inlet region and a C2+-selective Cu catalyst layer in the subsequent section (Fig. 6f).136 This structure enhanced CO coverage, improving the FE and partial current density of C2+ (JC2+) products. The Cu/Fe–N–C s-GDE achieved 90% C2+ selectivity at JC2+ above 1000 mA cm−2, demonstrating excellent catalytic performance. In a flow cell, the system reached a JC2+ of 1071.7 mA cm−2 while maintaining 89.3% C2+ selectivity. Ethylene (C2H4) was the major product, with an FE of 63.5% at a JC2H4 of 761.7 mA cm−2, indicating that the tandem system effectively promotes C2+ formation. They also compared the compatibility of different CO-selective catalysts (Ag, ZnO, and Fe–N–C) in the tandem system. The results showed that Cu/Fe–N–C s-GDE was the most effective for C2+ production at low overpotentials, achieving a maximum C2+ FE of 87.3% at 2.89 V applied voltage (Fig. 6g), with a corresponding JC2+ of 437.2 mA cm−2.
The studies discussed above demonstrate that the choice of CO-selective catalysts significantly impacts C2+ production efficiency in tandem systems. Ni–N–C exhibited higher C2+ FE than Au and Ag in the tandem system, particularly in the low potential region (−0.20 to −0.66 V).135 Similarly, Fe–N–C outperformed Ag in CO production at low overpotentials, leading to higher C2+ selectivity.136
These findings highlight the essential role of the CO-generating layer in determining the efficiency of tandem catalysts for C2+ production. With their exceptional selectivity and operational stability, M–N–C materials offer a robust and scalable platform for CO generation in such architectures. To further enhance the performance of tandem catalysts, future design efforts should consider not only the choice of active materials but also the rational integration of different catalytic components. Specifically, the reaction rates of CO production and CO consumption should be well-matched to avoid the accumulation or depletion of intermediates; the spatial arrangement of active sites should be optimized to minimize CO diffusion losses and ensure efficient intermediate transfer; and the local CO concentration should be carefully regulated through tailored catalyst architectures, such as segmented gas diffusion electrodes or spatially controlled catalyst deposition. Incorporating these design principles into M–N–C–based tandem catalysts may provide a viable pathway toward more selective, efficient, and scalable electrochemical C2+ production.
4.3 Tandem electrolyzers: sequential CO2 reduction for C2+ products
In addition to tandem catalysts integrated within a single electrode system, researchers have also explored tandem electrolyzer systems consisting of two sequential electrolysis cells. By spatially separating the CO2-to-CO and CO-to-C2+ reactions into distinct electrolyzers, the tandem electrolyzer system optimizes reaction conditions for each step, improving overall CO2 conversion efficiency. Furthermore, in alkaline conditions, CO2 reduction typically leads to carbonate formation, which can deposit on the electrode surface or flow field, hinder mass transport, or migrate across the membrane and be reoxidized at the anode, leading to energy and carbon losses.139 However, the second-step reaction in the tandem electrolyzer system uses CO instead of CO2 as the reactant, meaning that carbonate formation is no longer an issue, enabling prolonged stable operation. A low-temperature, pH-neutral tandem electrolyzer system was designed by Möller et al. (Fig. 6h), where Cell-1 used a Ni–N–C catalyst with nearly 100% CO selectivity at 200 mA cm−2, followed by Cell-2, which used commercial Cu particles as the catalyst to further reduce CO to C2+ products.137 By optimizing the CO2/CO feed ratio of Cell-2, the rate of C2H4 production was increased by 50%, and the yield of alcohols (ethanol and propanol) was doubled compared with that of the single-step Cu-catalyzed CO2RR system. This suggests that the increase in the concentration of CO effectively facilitated the generation of C2+ products. In addition, the C2+ energy efficiency (EE) of the tandem system significantly outperformed the single-cell Cu system at high current densities (>1 A). Beyond EE improvements, the tandem system also demonstrated a higher reactant utilization efficiency, as reflected by the single-pass carbon efficiency (SPCE), which nearly doubled from 17% to 30–35% compared to the single-cell Cu system. An apparent CO reduction selectivity (SCORR) was also defined to characterize the origin of the C2+ product. The results showed that at 67 mol% CO feed and a current density of 200 mA cm−2, the SCORR was close to 100%, indicating that the C2+ product was almost exclusively derived from CO reduction rather than direct CO2 reduction.Wu et al. developed a double electrolytic cell for the efficient conversion of CO2 to a C3 fuel, n-propanol.140 In the CO2 to CO reduction step, their Ni-based M–N–C catalyst achieved a FECO of 99.2% at 140 mA cm−2, maintaining >96% selectivity over a 60-min stability test. For the CO-to-n-propanol step, a Cu2O catalyst with a multi-cavity structure exhibited a 30.2% n-propanol FE at 42.5 mA cm−2 in flow-cell, corresponding to a partial current density of 12.8 mA cm−2. When integrated into the dual-electrolyzer tandem system, the CO2 to CO process maintained a 95.9% FE at 140 mA cm−2, ensuring a steady CO supply. Subsequently, the CO to n-propanol step retained an n-propanol FE of 28.0%. Overall, the system achieved an n-propanol FE of 15.9% and a power conversion efficiency (PCE) of 19.3%, outperforming previously reported CO2-to-propanol FE and PCE. These results highlight the tandem approach as a promising strategy for improving C3 fuel production efficiency.
The above studies demonstrate that M–N–C catalysts play a vital role in the first step of tandem electrolyzers by enabling efficient CO2-to-CO conversion. Optimizing the CO production rate and CO2/CO feed ratio is essential to maximize C2+ selectivity in the second electrolysis step. Moreover, decoupling the operating conditions of the two electrolyzers—for example, employing near-neutral or acidic electrolytes to improve single-pass carbon efficiency (SPCE) for CO2 reduction, and alkaline media to promote CO-to-C2+ conversion—enables independent optimization of catalysts and reaction environments. Finally, the scalability of tandem electrolyzers should be systematically evaluated using MEAs and multi-cell configurations, with a focus on overall product yield, energy efficiency, and long-term stability. Achieving a rational balance between performance, system complexity, and durability is essential for advancing this technology toward practical implementation.
Compared to tandem catalysts, which integrate CO2-to-CO and CO-to-C2+ conversion within a single electrode structure, tandem electrolyzers offer greater control over individual reaction conditions and flexibility in component optimization. This separation allows for independent tuning of catalysts, potentials, and electrolytes in each stage, facilitating higher performance in multi-carbon production. However, tandem electrolyzers also introduce additional system complexity, interstage transport challenges, and energy losses associated with multi-cell operation. In contrast, tandem catalysts provide a more compact and potentially scalable configuration but often face limitations in reaction matching and spatial control of intermediates. The choice between the two strategies ultimately depends on the application context—whether compactness, modularity, or selective control is prioritized. Continued research into both approaches will be essential for realizing practical and high-efficiency electrochemical CO2 reduction to multi-carbon products.
4.4 Electrocatalysis–thermalcatalysis tandem CO2 conversion
Due to the inherent thermodynamic stability of CO2, its direct electrochemical conversion into complex products remains challenging. While electrocatalysis is efficient for generating simple products like CO or H2 under mild conditions, it often lacks the selectivity and energy efficiency needed for multi-carbon synthesis. Thermocatalysis, in contrast, offers well-established upgrading routes but typically depends on fossil-based syngas and high-temperature conditions. To address the limitations of either method alone, electrocatalysis–thermocatalysis tandem (EC–TC) systems have emerged, wherein electrochemically generated intermediates (e.g., CO, syngas, ethylene) are thermochemically converted to more complex products such as long-chain hydrocarbons, C3 oxygenates, and aromatics.Although the EC–TC concept is still in its early stages, several recent studies pioneered by the Chen group have demonstrated its feasibility in producing value-added products from CO2. In one example, ethylene generated via CO2 electroreduction was thermochemically upgraded to BTEX aromatics using Ga- and Pt-modified ZSM-5 catalysts.141 Another report showcased the integration of an oxide-derived Cu electrocatalyst with a Rh1Co3/MCM-41 thermocatalyst to achieve C3 oxygenate synthesis via hydroformylation.142 Additionally, EC–TC systems have been used for CO2 fixation into carbon nanofibers (CNFs), highlighting their potential for both material valorization and long-term carbon sequestration.143
While most current EC–TC demonstrations utilize metal-based electrocatalysts for intermediate generation, M–N–C materials—known for their excellent CO selectivity, high stability, and metal site tunability—may offer potential as candidates for the electrochemical module. Their ability to generate high-purity CO or tunable syngas compositions makes them well-suited for coupling with downstream thermocatalysis, especially when product requirements demand precise CO/H2 ratios or intermediate purity. Incorporating M–N–C catalysts into EC–TC frameworks remain an open direction, with the potential to support modular, scalable CO2 utilization.
Despite recent advances, EC–TC systems still face notable challenges. The limited single-pass CO2 conversion in electrolysis constrains the overall efficiency, necessitating strategies to increase intermediate productivity and minimize CO2 dilution in the thermal stage.144 One potential approach is the use of bipolar or cation/proton exchange membranes in acidic electrolytes to reduce CO2 loss via (bi)carbonate crossover. Moreover, thermal catalyst design must account for residual CO2 effects—such as coke formation or reverse Boudouard reactions—as well as precise thermal integration.145 Bridging the operational gap between low-temperature electrolysis and high-temperature thermocatalysis may benefit from the use of solid oxide electrolysis cells (SOECs), which offer high CO productivity and thermal compatibility.123 Overall, as a modular platform, EC–TC provides a promising frontier for integrating M–N–C catalysts into broader CO2-to-chemical pathways.
5. Summary and outlook for future work
M–N–C catalysts have emerged as promising non-precious metal materials for electrochemical CO2 reduction (eCO2RR). The material structures can be easily manipulated, through the tuning of metal center sites, nitrogen or other non-metal heteroatom doping, and the type of carbon substrates. Those structural variations lead to the optimization of coordination environments, electronic structures, and mass transport. As a result, M–N–C catalysts have demonstrated highly competitive eCO2RR performance, often outperforming noble metal catalysts (e.g., Au and Ag) while being less expensive. Scalable synthesis of cost-effective M–N–C catalysts has been achieved by our research group, which further narrows the gap from scientific research to industrial applications. Notably, the faradaic efficiency of CO production by various M–N–C catalysts has been reported to approach nearly 100% at industrial relevant current densities (>150 mA cm−2). This advantage enables the design of tandem catalysts that consist of an M–N–C catalyst (for CO2–to–CO reduction) and a second catalyst (for C–C coupling) to produce more evaluable C2+ products at a higher activity and selectivity.To further advance the performance and applicability of M–N–C catalysts, the following three major research directions are worth exploring.
5.1 Exploration of high loading single atom systems and DACs
For SACs, increasing the density of active metal sites is essential for achieving industrially relevant current densities. However, maintaining atomic dispersion under high metal loading remains challenging, as elevated metal content often leads to aggregation or non-uniform coordination environments. Designing defect-rich or hierarchically porous carbon frameworks may offer a feasible route to stabilize dense M–Nx sites without compromising structural integrity.Meanwhile, dual-atom catalysts (DACs) with well-defined bimetallic coordination environments have attracted growing attention, especially for applications requiring precise control over syngas composition. By pairing metal centers with complementary affinities—such as one favoring CO2-to-CO conversion and the other promoting HER—DACs offer the potential to fine-tune the CO/H2 ratio at the molecular level, enabling more selective downstream transformations.
Although DACs theoretically enable cooperative effects, current studies largely report the formation of C1 products, and C–C coupling remains difficult. Their roles in intermediate tuning or tandem pathways remain open questions, offering fertile ground for future exploration in CO2 conversion strategies. Therefore, decoupling CO and H2 production on separate SAC sites embedded in a single conductive substrate may offer a practical approach, allowing each site to retain distinct coordination environments without mutual interference. In doing so, the coordination environment between both SAC identities is not affected by the other – allowing the propagation of CO and H2 on individually isolated sites.
5.2 Elucidation of long-term stability and deactivation mechanisms
In eCO2RR electrolyzer systems, performance degradation over time is often observed, but the underlying mechanisms—such as active site reconstruction, metal atom migration, N-site degradation, or local pH shifts—are still poorly understood. Future research should prioritize mechanistic studies that combine Operando spectroscopy, advanced electron microscopy, and computational modeling to monitor the structural and chemical evolution of M–N–C catalysts under working conditions. In particular, understanding the dynamic changes at the metal–nitrogen–carbon interface and the role of the surrounding carbon environment in anchoring metal atoms is essential for rational stabilization strategies. Additionally, exploring encapsulation techniques, hydrophobic layer integration, or electrolyte engineering may provide new ways to suppress flooding, ion crossover, and reactant depletion that often contribute to catalyst deactivation in flow cells and MEAs. Establishing a structure–stability relationship will be crucial for designing next-generation M–N–C materials capable of stable operation over thousands of hours, which is a prerequisite for commercial viability.5.3 Integration into multi-step tandem systems
Building on their excellent selectivity toward CO and syngas, M–N–C catalysts hold great potential as the upstream components in multi-step tandem systems—either through spatially integrated tandem catalysts within a single electrode for in situ intermediate conversion, or through sequential reactor setups where CO or syngas is electrochemically produced and subsequently upgraded into more complex C2+ products. In electro–thermal tandem (EC–TC) systems, the output of M–N–C catalysts can be tailored to meet the specific requirements of thermocatalytic upgrading processes—such as tuning the CO/H2 ratio for hydroformylation, maximizing C2H4 yield for aromatization, or enhancing CO purity for Fischer–Tropsch synthesis. These coupled processes demand not only rational catalyst design, but also system-level interface engineering—including gas management, thermal integration, and electrolyte compatibility—to ensure efficient operation across multiple stages. Moreover, compared to traditional CO2 utilization routes aimed at volatile liquid fuels, the integration of M–N–C catalysts into solid carbon product synthesis—such as carbon nanofibers (CNFs)—offers a pathway for long-term carbon sequestration, with added environmental and material value. Leveraging M–N–C materials as the core platform for structural tuning, functional coupling, and modular integration will be a key frontier for advancing CO2 conversion in multi-step tandem systems.Author contributions
CRediT: Shengyao Wang writing – original draft, writing – review & editing; Ahmed Badreldin writing – review & editing; Ying Li supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this feature article.Acknowledgements
The work was jointly funded by the Texas A&M University System Nuclear Security Office and Los Alamos National Laboratory as part of their Collaborative Research Program. The work represented in this publication does not expressly represent the views of either the Texas A&M University System or Los Alamos National Laboratory. The authors also acknowledge partial support from the Texas A&M University Division of Research TPT Program.References
- D. McCollum, N. Bauer, K. Calvin, A. Kitous and K. Riahi, Clim. Change, 2014, 123, 413–426 CrossRef.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- M. Höök and X. Tang, Energy Policy, 2013, 52, 797–809 CrossRef.
- S. Yadav and S. J. F. Mondal, Fuel, 2022, 308, 122057 CrossRef CAS.
- T. Wilberforce, A. Olabi, E. T. Sayed, K. Elsaid and M. A. Abdelkareem, Sci. Total Environ., 2021, 761, 143203 CrossRef CAS PubMed.
- H. Dong, M. Jung, Y. Zhang, S. Wang and S. Ding, Mol. Catal., 2024, 560, 114133 CrossRef CAS.
- T. Sun, Z. Wang, Y. Wang, Q. Xu, K. Wang and J. Jiang, Angew. Chem., Int. Ed., 2025, 64, e202422814 CrossRef CAS PubMed.
- Z. Xiao, P. Li, H. Zhang, S. Zhang, X. Tan, F. Ye, J. Gu, J.-J. Zou and D. Wang, Fuel, 2024, 362, 130906 CrossRef CAS.
- C. Xu, J. Yang, S. Gámez-Valenzuela, J.-W. Lee, J. Che, P. Chen, G. Zhang, D. Hu, Y. Wang, J. Lv, Z. Zhong, X. Chen, G. Zhang, F. Zhao, B. J. Kim, X. Guo and B. Liu, Energy Environ. Sci., 2025, 18, 5913–5925 RSC.
- X. Yan, C. Duan, S. Yu, B. Dai, C. Sun and H. Chu, Renewable Sustainable Energy Rev., 2024, 190, 114086 CrossRef CAS.
- Y. Yang, W. Zhang, G. Wu, Q. Huang, J. Wen, D. Wang and M. Liu, Angew. Chem., Int. Ed., 2025, e202504423, DOI:10.1002/anie.202504423.
- M. G. Kibria, J. P. Edwards, C. M. Gabardo, C. T. Dinh, A. Seifitokaldani, D. Sinton and E. H. Sargent, Adv. Mater., 2019, 31, 1807166 CrossRef PubMed.
- J. Ren, J. Lau, M. Lefler and S. Licht, J. Phys. Chem. C, 2015, 119, 23342–23349 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- R. Geioushy, M. M. Khaled, K. Alhooshani, A. S. Hakeem and A. Rinaldi, Electrochim. Acta, 2017, 245, 456–462 CrossRef CAS.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou and X. Dong, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS PubMed.
- B. Zhang, J. Zhang, M. Hua, Q. Wan, Z. Su, X. Tan, L. Liu, F. Zhang, G. Chen and D. Tan, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- C. Chen, J. F. K. Kotyk and S. W. Sheehan, Chem, 2018, 4, 2571–2586 CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., 2021, 133, 20795–20816 CrossRef.
- T. Gao, B. Xia, K. Yang, D. Li, T. Shao, S. Chen, Q. Li and J. Duan, Energy Fuels, 2023, 37, 17997–18008 CrossRef CAS.
- A. Badreldin and Y. Li, Chem. Sci., 2025, 16, 2483–2513 RSC.
- R. M. Navarro, M. Pena and J. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- R. Küngas, J. Electrochem. Soc., 2020, 167, 044508 CrossRef.
- Y. Hori, A. Murata, K. Kikuchi and S. Suzuki, Chem. Commun., 1987, 728–729 RSC.
- Q. Lu, J. Rosen, Y. Zhou, G. S. Hutchings, Y. C. Kimmel, J. G. Chen and F. Jiao, Nat. Commun., 2014, 5, 3242 CrossRef PubMed.
- S. Liu, X.-Z. Wang, H. Tao, T. Li, Q. Liu, Z. Xu, X.-Z. Fu and J.-L. Luo, Nano Energy, 2018, 45, 456–462 CrossRef CAS.
- H. Yang, J. Huang, H. Yang, Q. Guo, B. Jiang, J. Chen and X. Yuan, Chem. – Asian J., 2022, 17, e202200637 CrossRef CAS PubMed.
- M. Z. Iqbal, S. Imteyaz, C. Ghanty and S. Sarkar, J. Ind. Eng. Chem., 2022, 113, 15–31 CrossRef CAS.
- Y. Zhu, X. Yang, C. Peng, C. Priest, Y. Mei and G. Wu, Small, 2021, 17, e2005148 CrossRef PubMed.
- M. Inagaki, M. Toyoda, Y. Soneda and T. Morishita, Carbon, 2018, 132, 104–140 CrossRef CAS.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- X. Wang, W. Ju, L. Liang, M. Riyaz, A. Bagger, M. Filippi, J. Rossmeisl and P. Strasser, Angew. Chem., Int. Ed., 2024, 63, e202401821 CrossRef CAS PubMed.
- T. Liu, Y. Wang and Y. Li, JACS Au, 2023, 3, 943–952 CrossRef CAS PubMed.
- Y. Wang, T. Liu and Y. Li, Chem. Sci., 2022, 13, 6366–6372 RSC.
- Y. Gang, B. Li, S. Fang, J. Pellessier, L. Fang, F. Pan, Z. Du, Y. H. Hu, T. Li and G. Wang, Chem. Eng. J., 2023, 453, 139712 CrossRef CAS.
- Y. Gang, J. Pellessier, Z. Du, S. Fang, L. Fang, F. Pan, M. Suarez, K. Hambleton, F. Chen, H. C. Zhou, T. Li, Y. H. Hu and Y. Li, ACS Sustainable Chem. Eng., 2023, 11, 7231–7243 CrossRef CAS PubMed.
- Y. Gang, F. Pan, Y. Fei, Z. Du, Y. H. Hu and Y. Li, ACS Sustainable Chem. Eng., 2020, 8, 8840–8847 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef PubMed.
- H. Zhang, G. Liu, L. Shi and J. Ye, Adv. Energy Mater., 2018, 8, 1701343 CrossRef.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- R. He, N. Xu, I. M. U. Hasan, L. Peng, L. Li, H. Huang and J. Qiao, EcoMat, 2023, 5, e12346 CrossRef CAS.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- K. Liu, G. Wu and G. Wang, J. Phys. Chem. C, 2017, 121, 11319–11324 CrossRef CAS.
- F. Pan, H. Zhang, Z. Liu, D. Cullen, K. Liu, K. More, G. Wu, G. Wang and Y. Li, J. Mater. Chem. A, 2019, 7, 26231–26237 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- Q. Pan, Y. Chen, S. Jiang, X. Cui, G. Ma and T. Ma, EnergyChem, 2023, 5, 100114 CrossRef CAS.
- Y. Bao, J. Xiao, Y. Huang, Y. Li, S. Yao, M. Qiu, X. Yang, L. Lei, Z. Li, Y. Hou, G. Wu and B. Yang, Angew. Chem., Int. Ed., 2024, 63, e202406030 CAS.
- S. Huang, Z. Fang, C. Lu, J. Zhang, J. Sun, H. Ji, J. Zhu and X. Zhuang, J. Colloid Interface Sci., 2024, 675, 683–688 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., 2018, 130, 1962–1966 CrossRef.
- T.-T. Li, Y. Mei, H. Li, J. Qian, M. Wu and Y.-Q. Zheng, Inorg. Chem., 2020, 59, 14184–14192 CrossRef CAS PubMed.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun, N. Liu and J. Liu, ACS Sustainable Chem. Eng., 2021, 9, 6438–6445 CrossRef CAS.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, ACS Catal., 2020, 10, 10920–10931 CrossRef CAS.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- J. Song, Z.-X. Qian, J. Yang, X.-M. Lin, Q. Xu and J.-F. Li, ACS Energy Lett., 2024, 9, 4414–4440 CrossRef CAS.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, e2102886 CrossRef PubMed.
- S. Chen, T. Luo, X. Li, K. Chen, Q. Wang, J. Fu, K. Liu, C. Ma, Y.-R. Lu, H. Li, K. S. Menghrajani, C. Liu, S. A. Maier, T.-S. Chan and M. Liu, Nano Energy, 2024, 128, 104384 Search PubMed.
- Z. Liu, J. Cheng, Q. Niu, X. Zhang, J. Qian, C. Chu and Y. He, J. Environ. Chem. Eng., 2025, 13, 115159 CrossRef CAS.
- H. Sun, S. Hu, T. Liao, Y. Li, X. Fan and W. Peng, Electrochim. Acta, 2025, 513, 145627 CrossRef CAS.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., 2019, 131, 15013–15018 CrossRef.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- Y. Zhao, Q. Yuan, K. Sun, A. Wang, R. Xu, J. Xu, Y. Wang, M. Fan and J. Jiang, ACS Appl. Mater. Interfaces, 2023, 15, 37593–37601 CrossRef CAS PubMed.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li and D. J. N. E. Su, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- X. Yang, J. Cheng, H. Lv, X. Yang, L. Ding, Y. Xu, K. Zhang, W. Sun and J. Zhou, Chem. Eng. J., 2022, 450, 137950 CrossRef CAS.
- J. Song, X. Lei, J. Mu, J. Li, X. Song, L. Yan and Y. Ding, Small, 2023, 19, e2304423 CrossRef PubMed.
- C. Jia, X. Tan, Q. Sun, R. Liu, R. K. Hocking, S. Wang, L. Zhong, Z. Shi, S. Smith and C. Zhao, Adv. Mater., 2025, 37, e2417443 CrossRef PubMed.
- L. Shen, L. Wang, Z. Li, Y. Ye and Q. Dai, Carbon, 2025, 242, 120388 CrossRef CAS.
- Y. Zhao, Q. Yuan, R. Xu, C. Zhang, K. Sun, A. Wang, A. Zhang, Z. Wang, J. Jiang and M. Fan, Appl. Catal., B, 2024, 355, 124168 CrossRef CAS.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun, N. Liu and J. Zhou, Nanoscale, 2022, 14, 6846–6853 RSC.
- Y. Cheng, J. Chen, C. Yang, H. Wang, B. Johannessen, L. Thomsen, M. Saunders, J. Xiao, S. Yang and S. P. Jiang, Energy Environ. Mater., 2023, 6, e12278 CrossRef CAS.
- Y. Guo, S. Gao and X. Duan, Mol. Catal., 2024, 564, 114316 CrossRef CAS.
- F. Zhou, X. Fang, Y. Zhang, W. Yang, W. Zhou, H. Zhou, Q. Liu, J. Wu, F. Qi and Y. J. F. Shen, Fuel, 2023, 353, 129202 CrossRef CAS.
- F. Pan, H. Zhao, W. Deng, X. Feng and Y. Li, Electrochim. Acta, 2018, 273, 154–161 CrossRef CAS.
- F. Pan, B. Li, E. Sarnello, Y. Fei, Y. Gang, X. Xiang, Z. Du, P. Zhang, G. Wang, H. T. Nguyen, T. Li, Y. H. Hu, H. C. Zhou and Y. Li, ACS Nano, 2020, 14, 5506–5516 CrossRef CAS PubMed.
- H. Hu, J. Z. Ou, X. Xu, Y. Lin, Y. Zhang, H. Zhao, D. Chen, M. He, Y. Huang and L. Deng, Chem. Eng. J., 2021, 425, 130587 CrossRef CAS.
- S. G. Han, D. D. Ma and Q. L. Zhu, Small Methods, 2021, 5, 2100102 CrossRef CAS PubMed.
- Y. Gang, E. Sarnello, J. Pellessier, S. Fang, M. Suarez, F. Pan, Z. Du, P. Zhang, L. Fang, Y. Liu, T. Li, H.-C. Zhou, Y. H. Hu and Y. Li, ACS Catal., 2021, 11, 10333–10344 CrossRef CAS.
- W. Zhang, H. Li, D. Feng, C. Wu, C. Sun, B. Jia, X. Liu and T. Ma, Carbon Energy, 2024, 6, e461 CrossRef CAS.
- F. Pan, A. Liang, Y. Duan, Q. Liu, J. Zhang and Y. Li, J. Mater. Chem. A, 2017, 5, 13104–13111 RSC.
- F. Pan, B. Li, E. Sarnello, Y. Fei, X. Feng, Y. Gang, X. Xiang, L. Fang, T. Li, Y. H. Hu, G. Wang and Y. Li, ACS Catal., 2020, 10, 10803–10811 CrossRef CAS.
- J. Pellessier, Y. Gang and Y. Li, ES Mater. Manuf., 2021, 13, 66–75 CAS.
- T.-Y. Ma, L. Liu and Z.-Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 RSC.
- J. Liu, T. Yang, D.-W. Wang, G. Q. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 CrossRef.
- M. Inagaki, M. Toyoda, Y. Soneda, S. Tsujimura and T. Morishita, Carbon, 2016, 107, 448–473 CrossRef CAS.
- M. Dai, J. Zeng, K. Luo, W. Zhang, Z. Chen, Z. Zeng, P. Yuan, Y. Zhang, S. Zhang and W. Ni, ACS Appl. Energy Mater., 2025, 8, 5166–5173 CrossRef CAS.
- X. Han, T. Zhang, M. Biset-Peiro, A. Roldan, M. Ceccato, N. Lock, S. U. Pedersen, J. R. Morante, J. Arbiol and K. Daasbjerg, Small, 2025, 21, e2406883 CrossRef PubMed.
- Y. Zhao, Z. Shi, F. Li, C. Jia, Q. Sun, Z. Su and C. Zhao, ACS Catal., 2024, 14, 3926–3932 CrossRef CAS.
- H. Ning, Z. Guo, W. Wang, X. Wang, Z. Yang, Z. Ma, Y. Tian, C. Wu, J. Hao and M. Wu, Carbon Lett., 2022, 32, 807–814 CrossRef CAS.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- N. Sreekanth, M. A. Nazrulla, T. V. Vineesh, K. Sailaja and K. L. Phani, Chem. Commun., 2015, 51, 16061–16064 RSC.
- J. Wu, R. M. Yadav, M. Liu, P. P. Sharma, C. S. Tiwary, L. Ma, X. Zou, X.-D. Zhou, B. I. Yakobson and J. Lou, ACS Nano, 2015, 9, 5364–5371 CrossRef CAS PubMed.
- J. Wu, M. Liu, P. P. Sharma, R. M. Yadav, L. Ma, Y. Yang, X. Zou, X.-D. Zhou, R. Vajtai and B. I. Yakobson, Nano Lett., 2016, 16, 466–470 CrossRef CAS PubMed.
- F. Pan, B. Li, X. Xiang, G. Wang and Y. Li, ACS Catal., 2019, 9, 2124–2133 CrossRef CAS.
- F. Pan, B. Li, W. Deng, Z. Du, Y. Gang, G. Wang and Y. Li, Appl. Catal., B, 2019, 252, 240–249 CrossRef CAS.
- C. Chen, X. Sun, X. Yan, Y. Wu, H. Liu, Q. Zhu, B. B. A. Bediako and B. Han, Angew. Chem., 2020, 132, 11216–11222 CrossRef.
- Y. Yan, H. Wang, X. Bi, Y. Zhao and M. Wu, Chem. Commun., 2024, 60, 6439–6442 RSC.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang and H. Yu, Angew. Chem., 2017, 129, 15813–15817 CrossRef.
- C. E. Banks, A. Crossley, C. Salter, S. J. Wilkins and R. G. Compton, Angew. Chem., Int. Ed., 2006, 45, 2533–2537 CrossRef CAS PubMed.
- K. Jurkschat, X. Ji, A. Crossley, R. G. Compton and C. E. Banks, Analyst, 2007, 132, 21–23 RSC.
- M. Pumera, Chem. Rec., 2012, 12, 201–213 CrossRef CAS PubMed.
- M. Pumera, Langmuir, 2007, 23, 6453–6458 CrossRef CAS PubMed.
- M. Pumera and H. Iwai, J. Phys. Chem. C, 2009, 113, 4401–4405 CrossRef CAS.
- W. Kiciński and S. Dyjak, Carbon, 2020, 168, 748–845 CrossRef.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12899–12904 CrossRef CAS PubMed.
- C. H. A. Wong, Z. Sofer, M. Kubešová, J. Kučera, S. Matějková and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS PubMed.
- C. Kim, Y.-K. Choe, D. H. Won, U. Lee, H.-S. Oh, D. K. Lee, C. H. Choi, S. Yoon, W. Kim, Y. J. Hwang and B. K. Min, ACS Energy Lett., 2019, 4, 2343–2350 CrossRef CAS.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- F. Ismail, W. Alnoush, A. Abdellah, S. Tan, K. E. Salem, A. Rakhsha, N. Noor, M. Fefer, Y. Terazono, N. Chen and D. C. Higgins, ACS Electrochem., 2025 DOI:10.1021/acselectrochem.5c00006.
- J. W. Sun, H. Q. Fu, P. F. Liu, A. Chen, P. Liu, H. G. Yang and H. Zhao, EES Catal., 2023, 1, 934–949 RSC.
- S. Han, S. Jia, W. Xia, X. Xing, R. Qi, H. Wu, M. He and B. Han, Chem. Eng. J., 2023, 455, 140595 CrossRef CAS.
- J. W. Sun, X. Wu, P. F. Liu, J. Chen, Y. Liu, Z. X. Lou, J. Y. Zhao, H. Y. Yuan, A. Chen, X. L. Wang, M. Zhu, S. Dai and H. G. Yang, Nat. Commun., 2023, 14, 1599 CrossRef CAS PubMed.
- H. Han, S. Lee, J. Im, M. Lee, T. Lee, S. T. Hyun, J. Hong, T. Seok and D. Choo, Chem. Eng. J., 2024, 479, 147603 CrossRef CAS.
- S. Subramanian, J. Middelkoop and T. Burdyny, Sustainable Energy Fuels, 2021, 5, 6040–6048 RSC.
- J. W. Blake, V. Konderla, L. M. Baumgartner, D. A. Vermaas, J. T. Padding and J. W. Haverkort, ACS Sustainable Chem. Eng., 2023, 11, 2840–2852 CrossRef CAS PubMed.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- Z. Zhang, X. Huang, Z. Chen, J. Zhu, B. Endrodi, C. Janaky and D. Deng, Angew. Chem., Int. Ed., 2023, 62, e202302789 CrossRef CAS PubMed.
- X. She, L. Zhai, Y. Wang, P. Xiong, M. M.-J. Li, T.-S. Wu, M. C. Wong, X. Guo, Z. Xu, H. Li, H. Xu, Y. Zhu, S. C. E. Tsang and S. P. Lau, Nat. Energy, 2024, 9, 81–91 CrossRef CAS.
- V. E. Nelson, C. P. O'Brien, J. P. Edwards, S. Liu, C. M. Gabardo, E. H. Sargent and D. Sinton, ACS Appl. Mater. Interfaces, 2024, 16, 50818–50825 CrossRef CAS PubMed.
- S. Garg, Z. Xie and J. G. Chen, Nat. Chem. Eng., 2024, 1, 139–148 CrossRef.
- Y. Hua, J. Wang, T. Min and Z. Gao, J. Power Sources, 2022, 535, 231453 CrossRef CAS.
- S. Lu, Y. Shi, N. Meng, S. Lu, Y. Yu and B. Zhang, Cell Rep. Phys. Sci., 2020, 1, 100237 CrossRef.
- A. Abdulrasheed, A. A. Jalil, Y. Gambo, M. Ibrahim, H. U. Hambali and M. Y. Shahul Hamid, Renewable Sustainable Energy Rev., 2019, 108, 175–193 CrossRef CAS.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS PubMed.
- W. Ni, Z. Liu, X. Guo, Y. Zhang, C. Ma, Y. Deng and S. Zhang, Appl. Catal., B, 2021, 291, 120092 CrossRef CAS.
- T. Ahmad, S. Liu, M. Sajid, K. Li, M. Ali, L. Liu and W. Chen, Nano Res. Energy, 2022, 1, e9120021 CrossRef.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang and H. Xie, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed.
- B. Chen, L. Gong, N. Li, H. Pan, Y. Liu, K. Wang and J. Jiang, Adv. Funct. Mater., 2023, 34, 2310029 CrossRef.
- Y. Chen, X. Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B. H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2024, 19, 311–318 CrossRef CAS PubMed.
- Q. Qin, H. Suo, L. Chen, Y. X. Wang, J. Z. Wang, H. K. Liu, S. X. Dou, M. Lao and W. H. Lai, Adv. Mater. Interfaces, 2024, 11, 2301049 CrossRef CAS.
- X. She, T. Zhang, Z. Li, H. Li, H. Xu and J. Wu, Cell Rep. Phys. Sci., 2020, 1, 100051 CrossRef.
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS.
- T. Moller, M. Filippi, S. Bruckner, W. Ju and P. Strasser, Nat. Commun., 2023, 14, 5680 CrossRef PubMed.
- F. Pan, X. Yang, T. O'Carroll, H. Li, K. J. Chen and G. Wu, Adv. Energy Mater., 2022, 12, 2200586 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- G. Wu, Y. Song, Q. Zheng, C. Long, T. Fan, Z. Yang, X. Huang, Q. Li, Y. Sun, L. Zuo, S. Lei and Z. Tang, Adv. Energy Mater., 2022, 12, 2202054 CrossRef CAS.
- S. Garg, Z. Xie, A. X. Lam and J. G. Chen, ACS Energy Lett., 2024, 9, 2990–2996 CrossRef CAS.
- A. N. Biswas, Z. Xie, R. Xia, S. Overa, F. Jiao and J. G. Chen, ACS Energy Lett., 2022, 7, 2904–2910 CrossRef CAS.
- Z. Xie, E. Huang, S. Garg, S. Hwang, P. Liu and J. G. Chen, Nat. Catal., 2024, 7, 98–109 CrossRef CAS.
- S. Garg, A. N. Biswas and J. G. Chen, Carbon Future, 2024, 1, 9200002 CrossRef.
- Z. Xie and J. G. Chen, CCS Chem., 2024, 6, 2855–2865 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |