
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot, chemoselective desulfurative functionalization of cysteine containing peptides using pyridinium salts†
Jeroen W.
van den Heuvel
,
Esther
Olaniran Håkansson
 ,
Bobo
Skillinghaug
and
Luke R.
Odell
*
,
Bobo
Skillinghaug
and
Luke R.
Odell
*
Department of Medicinal Chemistry, Uppsala University, 751 23 Uppsala, Sweden. E-mail: luke.odell@ilk.uu.se
First published on 4th June 2025
Abstract
Peptide and protein modifications, especially late-stage derivatization, are invaluable for the synthesis of new pharmaceuticals. Currently, cysteine-mediated peptide modification is mainly limited to bioconjugation and disulfide formation. Therefore, exploration of new cysteine mediated peptide modifications is of great interest. Herein, we present a practical strategy for the three-step, one-pot desulfurative functionalization of cysteine containing peptides. The use of a pyridinium salt enables diverse and selective functionalization with an array of nucleophiles such as amino acid side chains, pharmaceuticals and macrocyclizations. This method allows for easy and diverse late-stage modification of peptides enabling the discovery and synthesis of new pharmaceuticals.
Peptides and proteins are vital in all living systems, where peptides are essential for physiological and biochemical processes and proteins are responsible for cellular structure and function.1 New methods for modifying and functionalizing peptides have led to a marked increase in the number of peptide-based pharmaceuticals and drugs on the market over the last few decades.2 Out of the 20 canonical amino acids, only those bearing reactive side chains such as lysine, cysteine, tyrosine or tryptophan are routinely used for chemical modifications.3 The high nucleophilicity of the thiol group and its low natural abundance makes cysteine particularly valuable for functionalization,4 typically via S-functionalizations such as alkylations/arylations and disulfide formations.5
In contrast, transformations involving deletion of the reactive thiol group are relatively unexplored.4,6 Dehydroalanine (Dha) is a non-natural, yet biologically important, amino acid that has been widely used as a synthetic precursor for peptide/protein functionalizations via Michael additions at the electrophilic β-carbon (Scheme 1a).7,8 Dha can be accessed by several methods9 (Scheme 1b) including the reductive elimination of cysteine disulfides in the presence of electron-rich phosphines such as HMPT.10 Oxidative elimination via the formation of sulfonium salts has also been reported,11 although this approach is susceptible to side reactions with amino acid residues such as histidine, lysine, methionine, aspartate and glutamate.12 The thiol can also be bis-alkylated and eliminated using 1,4-dihalobutane derivatives13 on small peptides or proteins with a single available cysteine. However, substrates containing more than one cysteine are burdened by side-reactions due to cross-linking, intramolecular cyclization or stapling.14
 | ||
| Scheme 1 Dha functionalization via Michael addition and strategies for Dha formation. (a) Conjugate addition of nucleophiles to Dha enables diverse functionalization. (b) Reported methods for Dha formation often rely on toxic reagents, suffer from side reactions, or remain underexplored. (c) This work involves a mild, one-pot, three-step desulfurative functionalization of cysteine with broad scope and simplified purification. | ||
In 2011, Davis and co-workers disclosed a single example using Mukaiyama's reagent (2-chloro-1-methylpyridinium iodide) to promote elimination in moderate yield.12 Since then, pyridinium salts have been elegantly employed to selectively “tag” cysteines and by tuning the pyridinium salts “detagging” with thiol nucleophiles can also be achieved.15 Recognizing the potential of combining the high chemoselectivity of the cysteine-pyridinium tagging reaction, with the synthetic utility of Dha, we set out to develop a telescoped three-step one-pot desulfurative method for the straightforward, mild and selective late-stage functionalization of cysteine containing peptides (Scheme 1c). This approach enables the high-yielding modification of peptides with a variety of nucleophiles including pharmaceuticals and macrocyclizations.
Our initial experiments employed peptide 1a (Ac-Cys-Phe-Gly-NH2) and pyridinium 2a as model substrates and DBU as the base (Table 1, entry 1). Although full consumption of 1a was observed, a complex mixture of side-products was obtained. The main product was isolated and identified as thioacetate I (Fig. S1–S6, ESI†) likely formed through DBU-mediated S-arylation followed by deprotonation of the terminal acetamide and N-aryl transfer (Scheme S1, ESI†). To mitigate production of this side product, a variety of bases were tested at either 20 °C or 60 °C (Table S1, ESI†). Of these, only DABCO afforded high-selectivity towards 3a, with complete conversion after 16 h.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Peptide | Solv. | Base | Temp., Time | Conv.b |
| a Reaction conditions: 1 (0.025 mmol) and 2a (0.03 mmol) for 15 min followed by base (0.03 mmol), solvent (0.2 mL). b Conversion to 3 based on the ratio of pyridinium tagged 1 and compound 3 determined by HPLC-UV analysis. c H2O (50 mM PB, pH = 9.0). d 10 equiv. of base. | |||||
| 1 | 1a | DMF | DBU | 20 °C, <0.5 h | 3a (0) |
| 2 | 1a | DMF | DABCO | 20 °C, 16 h | 3a (>99) |
| 3 | 1a | DMF | DABCO | 37 °C, 7 h | 3a (>99) |
| 4 | 1a | H2Oc | DABCO | 37 °C, 2 h | 3a (>99) |
| 5 | 1b | H2Oc | DABCO | 37 °C, 2 h | 3b (>99) |
| 6 | 1b | H2Oc | DBU | 37 °C, 1 h | 3b (>99) |
| 7 | 1b | H2Oc | NaOHd | 37 °C, 1 h | 3b (>99) |
| 8 | 1b | H2Oc | Et3N | 37 °C, 2.5 h | 3b (>99) |
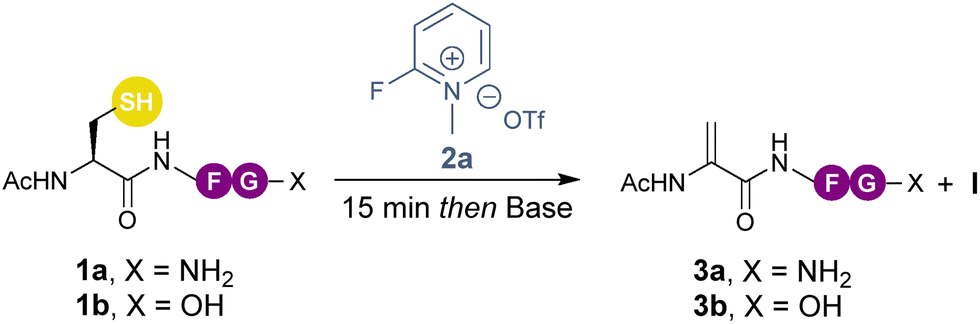
By raising the temperature to 37 °C, the reaction time could be reduced to 7 h (Table 1, entries 2 and 3). Next, different solvents were screened (Table S2, ESI†) and we were pleased to note that the elimination rate to form compound 3a was significantly enhanced in an aqueous buffer solution (Table 1, entry 4). Additionally, the solubility of the product was greatly improved. To assess the compatibility of the method with more realistic peptide substrates featuring a free C-terminus, we screened the carboxyl-containing compound 1b (Table 1, entry 5). Notably, this had no influence on the formation of 3b. Next, several bases were evaluated under aqueous conditions at 37 °C (Table 1, entries 6–8), all of which afforded full conversion (>99%). Any of these conditions could potentially be employed and NaOH was selected for further studies due to its low cost (Table 1, entry 7).
With the elimination step optimized, we turned our attention to the subsequent Michael-addition functionalization. Using piperidine as a model nucleophile, the tertiary amine containing peptide 6a was successfully isolated in 40% yield (Scheme 2 and Table S3, ESI†). However, this reaction also generated a side-product, which was isolated and characterized as the thioether dimer II (Fig. S8–S10 and Scheme S2, ESI†). This is presumably formed by competing base-mediated elimination and hydrolysis of the pyridinium-tagged intermediate, leading to formation of 1b and 3b, which then undergo a thia-Michael addition to give II. Reducing the equivalents of base did not influence formation of II, but the use of the less electron-deficient pyridinium 2b completely suppressed its formation (Table S3, ESI†) affording 6a in 56% yield. Furthermore, the yield of 6a could be increased to 69% (over three-steps) by changing the base to Et3N (Scheme 2, Table S3 and Fig. S1–S4, ESI†).
 | ||
Scheme 2 Scope of nucleophiles in the one-pot desulfurative functionalization of peptides using pyridinium salt 2b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a. aIsolated yield refers to a 1:1 mixture of diastereomers except for 6b (3:1) and 6p (7:1). bIsolated as TFA salt. c10 equiv. of the nucleophile. d74% purity based on UV (Fig. S27, ESI†). e0.5 equiv. of the nucleophile. fConditions in ESI.†gPeptide 1c was used. hPeptide 1d was used. a. aIsolated yield refers to a 1:1 mixture of diastereomers except for 6b (3:1) and 6p (7:1). bIsolated as TFA salt. c10 equiv. of the nucleophile. d74% purity based on UV (Fig. S27, ESI†). e0.5 equiv. of the nucleophile. fConditions in ESI.†gPeptide 1c was used. hPeptide 1d was used. | ||
With these optimized conditions in hand, we next investigated the nucleophile scope of for the desulfurative functionalization of 1b (Scheme 2). We began by examining the secondary amine containing amino acid proline, which afforded 64% of the branched peptide 6b, a yield comparable to the model reaction. Histidine was also found to be a viable substrate despite the lower nucleophilicity of the imidazole side chain,16 yielding 37% of 6c. The reaction could also be extended to aniline, affording a moderate yield of the N-aryl amine derivative 6d (24%). Primary amines, including the glycine N-terminus and the lysine side chain reacted efficiently to furnish 6e and 6f in 59% and 87% yield, respectively, highlighting the potential for both N-terminal and side-chain peptide conjugation. However, amino acids bearing less nucleophilic side chains (tryptophan, serine, tyrosine, and arginine), did not yield the corresponding products (Fig. S13–S16, ESI†).
Beyond amines, thiols and phosphines were readily incorporated under the same conditions delivering conjugates 6g–6i in good yields (55–83%). To explore peptide dimerization, primary and secondary diamines as well as a dithiol were investigated. Gratifyingly, all three nucleophiles produced dimeric peptides 6j–l in 41–61% yields.
To further investigate the reaction compatibility with highly functionalized substrates, three complex nucleophiles were selected for interrogation (Scheme 2). First, coenzyme A18 was used to probe reactivity with nucleotide-derived substrates, yielding 6m in 39%. Next, ciprofloxacin17 was employed to assess compatibility with drug-like structures, affording the novel antibiotic-peptide conjugate 6n in 71% yield. Then, to examine the possibility of cross-linking larger peptide-based structures, PEG2-RM26 (a GRPR-targeting antagonist19) was functionalized to give 6o in 50% yield. Employing peptide 1c, a macrocyclization strategy was explored with lysine as a linchpin motif, affording the 19-membered macrocycle 6p in a moderate 37% yield. To explore an additional challenging substrate, peptide 1d containing multiple competing nucleophilic groups and a sterically hindered cysteine residue was reacted with piperidine and glycine. In both cases, excellent conversion to the Dha derivative was observed, however the subsequent Michael addition was sluggish and the peptide derivatives 6q and 6r were both isolated in a yield of 14%. In the former case, separation of the 6q from the intermediate dehydroalanine (Dha) species was problematic. In contrast, formation of 6r was complicated by the formation of side-products III and IV (see ESI†), due to the use of an excess of 2b (1.5 equivalents), highlighting the need for careful reagent control when using multifunctional substrates.
In conclusion, we have developed a mild, three-step one-pot desulfurative functionalization of cysteine via pyridinium activation. The method exhibits broad nucleophile scope, enabling efficient conjugation with primary and secondary amines, thiols, and phosphines (14–87% yields). A range of amino acid residues including lysine, proline, cysteine, and N-terminal amines are tolerated, and the reaction proceeds smoothly with complex, functionalized substrates. Notably, the strategy supports both intermolecular conjugation and macrocyclization. This platform offers a versatile approach for selective late-stage peptide modification and holds promise for applications in peptide–drug conjugates and chemical biology. Efforts to expand this chemistry to other amino acids and peptides are ongoing and will be reported in due course.
The authors would like to thank Dr Lisa Haigh (Imperial College London, UK) and Dr Reza Shariatgorji (Uppsala University, Sweden) for assistance with high-resolution mass determination. The research was supported by Uppsala University, the Kjell and Märta Beijer Foundation, the Swedish Research Council (Vetenskapsrådet 2021-03293 and 2022-04831), the Swedish Brain Foundation (FO2024-0317-HK-70) and the Swedish Cancer Society (Cancerfonden 243889 Pj).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Shi, T. Sun and M. Yang, Org. Chem. Front., 2024, 11, 1623 RSC.
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and C. Fu, Signal Transduction Targeted Ther., 2022, 7, 1 CrossRef PubMed.
- (a) N. Naowarojna, R. Cheng, J. Lopez, C. Wong, L. Qiao and P. Liu, Synth. Syst. Biotechnol., 2021, 6, 32 CrossRef CAS PubMed; (b) S. Sakamoto and I. Hamachi, Anal. Sci., 2019, 35, 5 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- (a) E. Calce and S. De Luca, Chem. – Eur. J., 2017, 23, 224 CrossRef CAS PubMed; (b) M. H. Stenzel, ACS Macro Lett., 2013, 2, 14 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666 RSC.
- (a) J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835 CrossRef CAS PubMed; (b) A. M. Freedy, M. J. Matos, O. Boutureira, F. Corzana, A. Guerreiro, P. Akkapeddi, V. J. Somovilla, T. Rodrigues, K. Nicholls, B. Xie, G. Jiménez-Osés, K. M. Brindle, A. A. Neves and G. J. L. Bernardes, J. Am. Chem. Soc., 2017, 139, 18365 CrossRef CAS PubMed; (c) J. Dadová, K. J. Wu, P. G. Isenegger, J. C. Errey, G. J. L. Bernardes, J. M. Chalker, L. Raich, C. Rovira and B. G. Davis, ACS Cent. Sci., 2017, 3, 1168 CrossRef PubMed; (d) Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156 CrossRef CAS PubMed; (e) A. Yang, S. Ha, J. Ahn, R. Kim, S. Kim, Y. Lee, J. Kim, D. Söll, H. Y. Lee and H. S. Park, Science, 2016, 354, 623 CrossRef CAS PubMed; (f) C. Yu, E. Ruiyao, Y. An, X. Guo, G. Bao, Y. Li, J. Xie and W. Sun, Org. Lett., 2024, 26, 4767 CrossRef CAS PubMed; (g) P.-Y. He, H. Chen, H.-G. Hu, J.-J. Hu, Y.-J. Lim and Y.-M. Li, Chem. Commun., 2020, 56, 12632 RSC.
- (a) J. Dadová, S. R. Galan and B. G. Davis, Curr. Opin. Chem. Biol., 2018, 46, 71 CrossRef PubMed; (b) T. Mori, S. Sumida, K. Sakata and S. Shirakawa, Org. Biomol. Chem., 2024, 22, 4625 RSC.
- Concurrently, base-mediated Dha synthesis from cysteine was reported, though without direct functionalization. T. Mori, K. Sakatab and S. Shirakawa, Chem. Commun., 2025, 61, 4800 RSC.
- J. G. Gleason and D. N. Harpp, J. Am. Chem. Soc., 1971, 93, 2437 CrossRef.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052 CrossRef CAS PubMed.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666 RSC.
- (a) R. Okamoto, S. Souma and Y. Kajihara, J. Org. Chem., 2009, 74, 2494 CrossRef CAS PubMed; (b) M. Liu, M. Sovrovic, H. Suga and S. A. K. Jongkees, Org. Biomol. Chem., 2022, 20, 3081 RSC.
- P. M. Morrison, P. J. Foley, S. L. Warriner and M. E. Webb, Chem. Commun., 2015, 51, 13470 RSC.
- (a) B. M. Lipka, V. M. Betti, D. S. Honeycutt, D. L. Zelmanovich, M. Adamczyk, R. Wu, H. S. Blume, C. A. Mendina, J. M. Goldberg and F. Wang, Bioconjugate Chem., 2022, 33, 2189 CrossRef CAS PubMed; (b) B. M. Lipka, D. S. Honeycutt, G. M. Bassett, T. N. Kowal, M. Adamczyk, Z. C. Cartnick, V. M. Betti, J. M. Goldberg and F. Wang, J. Am. Chem. Soc., 2023, 145, 23427 CrossRef CAS PubMed; (c) S. Y. Yap, T. Butcher, R. J. Spears, C. McMahon, I. A. Thanasi, J. R. Baker and V. Chudasama, Chem. Sci., 2024, 15, 8557 RSC; (d) C. Wan, Y. Zhang, J. Wang, Y. Xing, D. Yang, Q. Luo, J. Liu, Y. Ye, Z. Liu, F. Yin, R. Wang and Z. Li, J. Am. Chem. Soc., 2024, 146, 2624 CrossRef CAS PubMed.
- M. Baidya, F. Brotzel and H. Mayr, Org. Biomol. Chem., 2010, 8, 1929 RSC.
- R. S. Vardanyan and V. J. Hruby, Synth. Essential Drugs, 2006, 1, 499 Search PubMed.
- I. Papet, D. Rémond, D. Dardevet, L. Mosoni, S. Polakof, M. A. Peyron and I. Savary-Auzeloux, Nutrition and skeletal muscle, Academic Press, Cambridge, 2019, p. 335 Search PubMed.
- A. Abouzayed, H. Tano, Á. Nagy, S. S. Rinne, F. Wadeea, S. Kumar, K. Westerlund, V. Tolmachev, A. E. Karlström and A. Orlova, Pharmaceutics, 2020, 12, 977 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01813g |
| This journal is © The Royal Society of Chemistry 2025 |