
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidative addition of diaryldichalcogenides to the diferrocenylphosphenium ion: synthesis, structure and organocatalytic activity†
Corina
Stoian‡
 a,
Nils
Schmidt‡
a,
Thomas J.
Kuczmera
b,
Pim
Puylaert
a,
Enno
Lork
a,
Boris J.
Nachtsheim
*b,
Emanuel
Hupf
*a and
Jens
Beckmann
*a
a,
Nils
Schmidt‡
a,
Thomas J.
Kuczmera
b,
Pim
Puylaert
a,
Enno
Lork
a,
Boris J.
Nachtsheim
*b,
Emanuel
Hupf
*a and
Jens
Beckmann
*a
aInstitut für Anorganische Chemie und Kristallographie, Universität Bremen, Leobener Str. 7, 28359 Bremen, Germany. E-mail: emanuel.hupf@uni-bremen.de; j.beckmann@uni-bremen.de
bInstitut für Organische und Analytische Chemie, Universität Bremen, Leobener Str. 7, 28359 Bremen, Germany. E-mail: nachtsh@uni-bremen.de
First published on 25th April 2025
Abstract
The reaction of the phosphenium ion [Fc2P]+ with dichalcogenides gace rise to the respective phosphonium ions [Fc2P(ChR)2]+ (Ch = S, Se, Te; R = Ph, Fc, biphen), which were employed in chalcogen bond mediated Michael additions.
Bond activation and redox catalysis mediated by main group elements have the potential to replace conventional transition metal catalysis, which often suffer from serious ecological and economical drawbacks.1 There is now a plethora of examples of oxidative addition and reductive elimination reactions involving main group elements possessing various oxidation states of similar stability.2 Amongst the group 15 elements, phosphorus3 and bismuth species4 in the oxidation states +I, +III and +V are the most promising candidates for applications in bond activation and redox cycling.
In their native, unsupported form, phosphenium ions, [R2P]+ (R = H, alkyl, aryl) are fiercely reactive carbene analogues comprising an electron lone pair, but also an electron deficiency and a positive charge, which renders them as strong electrophiles. Most phosphenium ions are electronically stabilised by conjugation and/or coordination of donor atoms.5 Very recent examples of geometrically constrained phosphenium ions by Dobrovetsky's group demonstrated their utility for the activation of small molecules.6 In the preceding work, we reinvestigated the synthesis and characterisation of the diferrocenylphosphenium ion, [Fc2P]+,7 first reported by Cowley's group about 40 years ago.8 Despite the intramolecular stabilising iron atoms, [Fc2P]+ is a Lewis superacid with an unexplored potential in bond activation chemistry.7 In this work, we describe the oxidative addition reaction of [Fc2P]+ with diaryldichalcogenides to form hitherto unknown bis(arylchalcogenido)diarylphosphonium ions [Fc2P(ER)2]+ (E = S, Se, Te; R = aryl), whereby the oxidation state changes from +III to +V. These reactions complement the oxidative addition of diaryldichalcogenides to neutral arylstibinidines and arylbismuthinidenes, [2,6-[RNC(R′)]2C6H3E] (E = Sb, Bi; R = 2,6-Me3C6H3, R′ = Me), reported by Dostál and co-workers, whereby the oxidation state changes from +I to +III.9a–d In contrast, non-oxidative activations of Ch–Ch bonds by related compounds, notably the ferrocene-based diphosphane, were recently reported.9e
The reaction of [Fc2P][B(C6F5)4] with PhEEPh (E = S, Se, Te) provided [Fc2P(EPh)2][B(C6F5)4] (1, E = S; 2, E = Se; 3, E = Te) as orange crystals in 97, 80 and 80% isolated yields (Scheme 1). The reaction of PhSeSePh and PhTeTePh readily occurred at r.t., while that of PhSSPh required heating at 60 °C. At the B3PW91/6-311+G(2df,p) level of theory, the reactions are both exothermic and exergonic at 298 K. The computed enthalpy ΔH0 decreases when going from 1 (–229.5 kJ mol−1), over 2 (–212.0 kJ mol−1) to 3 (–189.5 kJ mol−1). The computed Gibbs free energy ΔG° follows the same trend, becoming less negative from compound 1 (–160.0 kJ mol−1), to 2 (–144.5 kJ mol−1), to 3 (–125.1 kJ mol−1). We also reacted [Fc2P][B(C6F5)4] with FcSeSeFc and dibenzo[1,2]diselenine (biphenSe2), which gave [Fc2P(SeFc)2][B(C6F5)4] (4) and [Fc2P(Se2biphen)][B(C6F5)4] (5) as orange crystals in 90 and 69% isolated yield (Schemes 2 and 3).
 | ||
| Scheme 1 Reaction of [Fc2P]+ with PhEEPh (E = S, Se, Te). | ||
 | ||
| Scheme 2 Reaction of [Fc2P]+ with FcSeSeFc. | ||
 | ||
| Scheme 3 Reaction of [Fc2P]+ with dibenzo[1,2]diselenine. | ||
The characterisation of 1–5 was conveniently achieved by heteronuclear NMR spectroscopy, mass spectrometry and X-ray crystallography. The 31P NMR chemical shift of the [Fc2P]+ ion was observed at around δ = 184 ppm, depending on the solvent and counterion.7,8 In comparison, the 31P NMR spectra (CD2Cl2) of compounds 1–3 show progressively more shielded signals as the atomic number of the chalcogen (E) increases, with chemical shifts at δ = 81.4 ppm (E = S), 57.0 ppm (E = Se), and –22.8 ppm (E = Te). Comparison of the selenium species 2, 4 and 5 reveals that the aryl substituent (R) significantly affects the 31P NMR chemical shifts at δ = 57.0 (R = Ph), 47.5 (R = Fc) and 75.0 ppm (R2 = biphen). The same species 2, 4 and 5 show 77Se NMR chemical shifts at δ = 398.6 (R = Ph), 325.7 (R = Fc) and 378.8 ppm (R2 = biphen), whereas the tellurium species 3 exhibits a 125Te NMR chemical shift at δ = 780.7 ppm. The heteronuclear NMR spectra reveal indicative coupling information. The selenium-containing compounds 2, 4, and 5 are characterized by 1J(31P–77Se) coupling constants of 475 Hz (R = Ph), 498 Hz (R = Fc), and 444 Hz (R2 = biphen), which are comparable to the value observed for [dppf(SePh)2]2+ (454 Hz) or [Ph3PSePh]+ (456 Hz).10 The tellurium species 3 is characterised by a 1J(31P–125Te) coupling of 1041 Hz that relates well with of the one of [Ph3PTeMes]+ (1148 Hz), and a 1J(31P–123Te) coupling of 870 Hz.11 These values are significantly larger in comparison to the ferrocene containing neutral telluradiphospha[3]ferrocenophane with 1J(31P–125Te) = 318 Hz, and 1J(31P–123Te) = 263 Hz.12 In electrospray mass spectrometry, the phosphonium ions show indicative singly charged mass clusters at m/z = 619.00633 (1), 714.89523 (2), 930.82771 (4) and 712.87958 (5) Da with the expected isotope patterns. An analogous mass cluster was not detected for 3. The molecular structures of 1–5 are shown in Fig. 1 and 2. The spatial arrangement of the P atoms is distorted tetrahedral and defined by C2E2 (E = S, Se, Te) donor sets.
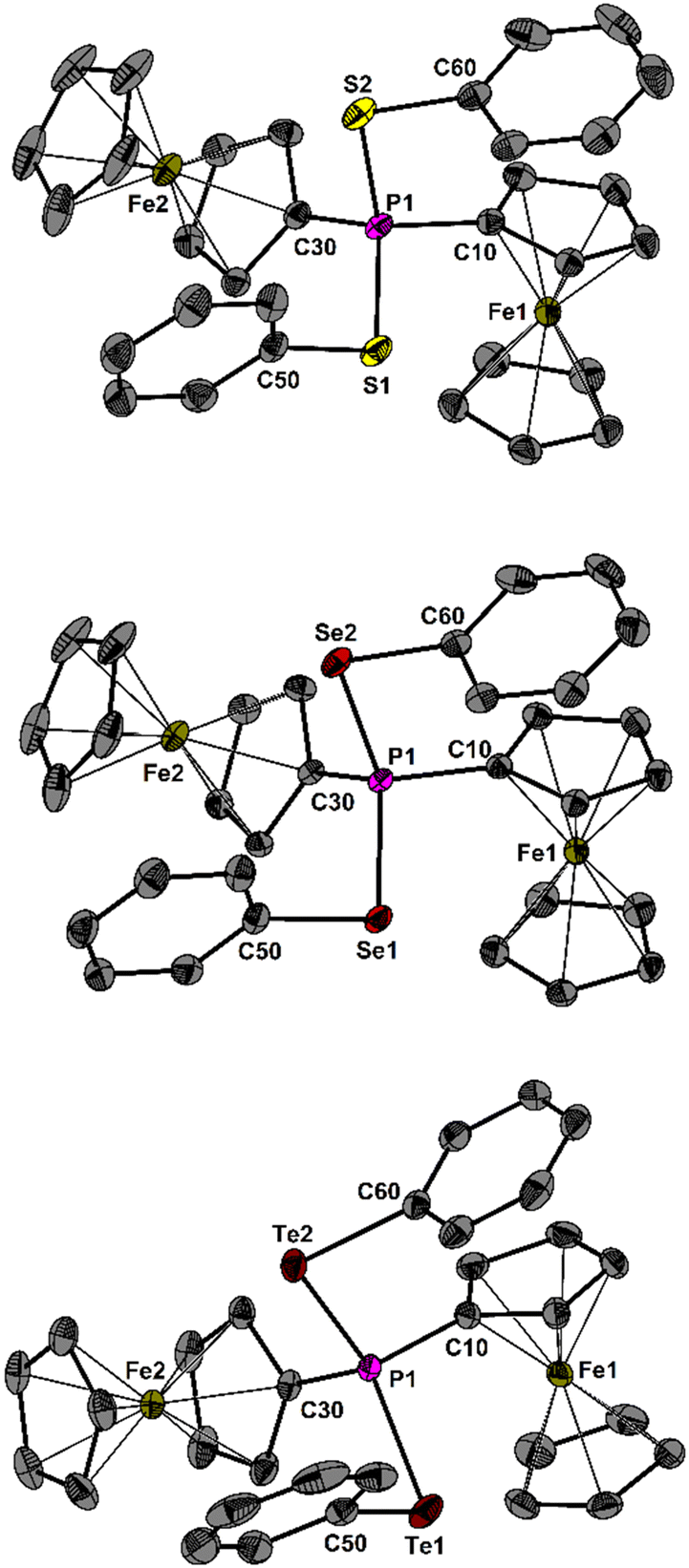 | ||
| Fig. 1 Molecular structure of 1 (top), 2 (middle) and 3 (bottom) showing 50% probability ellipsoids and the atomic numbering scheme. Hydrogen atoms and anions are omitted for clarity. | ||
 | ||
| Fig. 2 Molecular structure of 4 (top) and 5 (bottom) showing 50% probability ellipsoids and the atomic numbering scheme. Hydrogen atoms and anions are omitted for clarity. | ||
The P–S bond lengths in 1 (2.064(1) and 2.080(1) Å) are comparable to that reported for [Ph3PSPh]+ (2.077(1) Å).10 Similarly, the P–Se bond lengths of 2 (2.217(5), 2.230(5) Å), 4 (2.230(1), 2.237(1) Å) and 5 (2.228(1), 2.218(1) Å) resemble that of [Ph3PSePh]+ (2.232(1) Å).10 Finally, the P–Te bond lengths of 3 (2.450(1), 2.454(1) Å) are almost identical to that of [Ph3PTeMes]+ (2.467(1) Å).11 Additionally, heterocycle 5 contains a seven-membered ring in a twisted boat conformation. Attempts to experimentally determine the Lewis acidity of the phosphonium salts 1–5 by Gutmann–Beckett and other modified methods were unsuccessful.13 σ-Holes in acidic compounds play a pivotal role in chalcogen bonding (ChB) catalysis by interacting with electron donors to form weak intermolecular interactions, with ChB-driven organocatalysis receiving considerable attention in recent years.14
Very recently, Wang's group introduced two mono(arylchalcogenido)triorgano phosphonium ions in close proximity to each other as dual ChB catalysts.10a,15 Inspired by this work, we evaluated the catalytic activity of the bis(arylchalcogenido)diferrocenylphosphonium ions [Fc2P(ER)2]+ (1–5) in the benchmark Michael addition reaction between 1-methylindole and trans-β-crotonophenone. We employed reaction conditions similar to those used in previous studies of N-heterocyclic iod(az)olium salts as halogen bond (XB) donors in organocatalysis (Scheme 4).16 The progress of the Michael addition was monitored at room temperature by 1H-NMR after 30, 60 and 120 min of sonication, using 2 mol% catalyst loading. The conversions and yields of the desired Michael product 6 are collected in Table 1. In the homologues [Fc2P(EPh)2][B(C6F5)4] (1, E = S; 2, E = Se; 3, E = Te), both the conversion and the yield after 2h improves along with the increasing atomic number of the chalcogen. In the series of the selenium compounds, 2 shows the poorest performance. The biphen species [Fc2P(Se2biphen)][B(C6F5)4] (5) performs significantly better than 2, but slightly worse than the tetraferrocenyl species [Fc2P(SeFc)2][B(C6F5)4] (4), which reached a conversion of 93% and a yield of 86% after 2 h.
 | ||
| Scheme 4 ChB-mediated Michael addition of 1-methylindole and trans-β-crotonophenone. | ||
| Catalyst (2 mol%) | Conversion (yield) 30 min | Conversion (yield) 60 min | Conversion (yield) 120 min |
|---|---|---|---|
| Conversions and yields in %, as determined by 1H-NMR (d1 = 30 s) using tetraethylsilane as an internal standard. | |||
| 1 | 22 (10) | 27 (16) | 34 (22) |
| 2 | 19 (10) | 24 (12) | 50 (38) |
| 3 | 61 (50) | 79 (68) | 86 (74) |
| 4 | 74 (62) | 87 (78) | 93 (86) |
| 5 | 50 (36) | 70 (56) | 90 (78) |
| [Fc2P][B(C6F5)4] | 100 (90) | ||
We note that the diferrocenylphosphenium ion, [Fc2P]+, shows by far the best results; however, its utility is compromised by the sensitivity to air oxidation and hydrolysis. After 30 min, the conversion is complete yielding 6 in 90%, which can be ascribed to the greater Lewis acidity compared to 1–5. Unlike the N-heterocyclic iod(az)olium salts, 1–5 ([Fc2P]+) show lower selectivity, which is reflected in the maximum reached yield of 90%. While compounds 1–5 do not outperform the dicationic N-heterocyclic iod(az)olium salts, they exhibit comparable catalytic activity, showing promise as chalcogen bond donors and warranting comprehensive investigation in our future work.16
In an attempt to rationalize the increased catalytic activity from sulfur to tellurium within the series [Fc2P(EPh)2][B(C6F5)4] (1, E = S; 2, E = Se; 3, E = Te) we computationally investigated the electrostatic potential of the optimized gas-phase structures of all three compounds (Fig. 3). The electrostatic potential reveals an increased positively polarized area in the order sulfur (Vmax = 68.7 kcal mol−1) < selenium (Vmax = 73.3 kcal mol−1) < tellurium (Vmax = 78.5 kcal mol−1), indeed indicating a larger σ-hole for tellurium compared to sulfur. The resulting higher electrophilicity of 3 and increased dispersive interaction with the substrate, presumably, leads to the observed improved catalytic activity.
 | ||
| Fig. 3 Electrostatic potential (units in a.u.) mapped on a 0.001 a.u. electron density iso-surface of 1 (top), 2 (middle) and 3 (bottom). Orange dots refer to maxima on the ESP. | ||
In conclusion, we have successfully synthesized and characterized a series of bis(arylchalcogenido)diferrocenylphosphonium ions [Fc2P(EPh)2][B(C6F5)4] (1, E = S; 2, E = Se; 3, E = Te) through the oxidative addition of diaryldichalcogenides to the diferrocenylphosphenium ion [Fc2P]+. This transformation represents a redox process where the phosphorus centre undergoes a change in oxidation state from +III to +V. X-ray crystallographic analysis revealed similar distorted tetrahedral geometries around the phosphorus atoms in all compounds, notably without the intermolecular Fe⋯P contacts that stabilize the starting phosphenium ion. The evaluation of 1–5 as catalysts in the Michael addition reaction demonstrates their potential as organocatalysts, with the selenium derivative 4 showing particularly promising activity. These results establish bis(arylchalcogenido)diferrocenylphosphonium ions as a versatile platform for main group catalysis under mild conditions, opening avenues for the development of novel redox-active main group catalysts with tuneable electronic properties.
J. B. designed the project. C. S., N. S. and T. J. K. performed the experiments. J. B., E. H., B. J. N. and C. S. wrote the manuscript. P. P. and E. L. determined the molecular structures. B. J. N. and T. J. K. contributed to the catalysis part. E. H. performed the computations. All authors contributed to the discussion of the results. The authors acknowledge the financial support from the Deutsche Forschungsgemeinschaft (DFG).
Data availability
Spectroscopic and crystallographic data including figures of NMR spectra as well as the details of the DFT computations are given in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) P. P. Power, Nature, 2010, 463, 172–177 CrossRef PubMed; (b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- H. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- (a) A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367–382 CrossRef CAS; (b) Low-coordinate main group compounds—Group 15: D. Gudat, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. I, pp. 587–621 Search PubMed.
- (a) S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61, e202208401 CrossRef CAS PubMed; (b) D. Bawari, S. Volodarsky, Y. Ginzberg, K. Jaiswal, P. Joshi and R. Dobrovetsky, Chem. Commun., 2022, 58, 12176–12179 RSC; (c) K. Chulsky, I. Malahov, D. Bawari and R. Dobrovetsky, J. Am. Chem. Soc., 2023, 145, 3786–3794 CrossRef CAS PubMed; (d) D. Bawari, D. Tomai, K. Jalswl and R. Dobrovetsky, Nat. Chem., 2024, 16, 1261–1266 CrossRef CAS PubMed.
- (a) M. Olaru, A. Mischin, L. A. Malaspina, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2020, 59, 1581–1584 CrossRef CAS PubMed; (b) C. Stoian, M. Olaru, S. Demeshko, M. Fischer, S. Mebs, E. Hupf and J. Beckmann, Chem. – Eur. J., 2025, 31, e202403555 CrossRef CAS PubMed.
- (a) S. G. Baxter, R. L. Collins, A. H. Cowley and S. F. Sena, J. Am. Chem. Soc., 1981, 103, 714–715 CrossRef CAS; (b) S. G. Baxter, R. L. Collins, A. H. Cowley and S. F. Sena, Inorg. Chem., 1983, 22, 3475–3479 CrossRef CAS.
- (a) P. Šimon, R. Jambor, A. Růžička and L. Dostál, Organometallics, 2013, 32, 239–248 CrossRef; (b) P. Šimon, R. Jambor, A. Růžička and L. Dostál, J. Organomet. Chem., 2013, 740, 98–103 CrossRef; (c) C. Ganesamorrthy, C. Wölper, L. Dostál and S. Schulz, J. Organomet. Chem., 2017, 845, 38–43 CrossRef; (d) M. Huang, K. Li, Z. Zhang and J. Zhou, J. Am. Chem. Soc., 2024, 146, 20432–20438 CrossRef CAS PubMed; (e) R. Franz, C. Bruhn and R. Pietschnig, Molecules, 2021, 26, 1899–1914 CrossRef CAS PubMed.
- (a) X. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203671 CrossRef CAS PubMed; (b) L. C. Forfar, M. Green, M. F. Haddow, S. Hussein, J. M. Lynam, J. M. Slattery and C. A. Russell, Dalton Trans., 2015, 44, 110–118 RSC.
- J. Beckmann, J. Bolsinger, A. Duthie, P. Finke, E. Lork, C. Lüdtke, O. Mallow and S. Mebs, Inorg. Chem., 2012, 51, 12395–12406 CrossRef CAS PubMed.
- R. Franz, S. Nasemann, C. Bruhn, Z. Kelemen and R. Pietschnig, Chem. – Eur. J., 2021, 27, 641–648 CrossRef CAS PubMed.
- (a) R. Maskey, M. Schädler, C. Legler and L. Greb, Angew. Chem., Int. Ed., 2018, 57, 1717–1720 CrossRef CAS PubMed; (b) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC; (c) J. Ramler and C. Lichtenberg, Chem, 2020, 26, 10250–10258 CrossRef CAS PubMed; (d) S. Künzler, S. Rathjen, A. Merk, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2019, 25, 15123–15130 CrossRef PubMed.
- (a) G. Sekar, V. V. Nair and J. Zhu, Chem. Soc. Rev., 2024, 53, 586–605 RSC; (b) D. Jovanovic, M. Poliyodath Mohanan and S. M. Huber, Angew. Chem., Int. Ed., 2024, 63, e202404823 CrossRef CAS PubMed; (c) P. Pale and V. Mamane, Chem. – Eur. J., 2023, 29, e202302755 CrossRef CAS PubMed.
- (a) W. Wang, H. Zhu, S. Liu, Z. Zhao, L. Zhang, J. Hao and Y. Wang, J. Am. Chem. Soc., 2019, 141, 9175–9179 CrossRef CAS PubMed; (b) W. Wang, H. Zhu, L. Feng, Q. Yu, J. Hao, R. Zhu and Y. Wang, J. Am. Chem. Soc., 2020, 142, 3117–3124 CrossRef CAS PubMed; (c) X. Kong, P.-P. Zhou and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 9395–9400 CrossRef CAS PubMed; (d) H. Zhu, X. Yang, Y. Liu, H. Zhou and Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202423746 CrossRef CAS PubMed.
- (a) F. Heinen, D. L. Reinhard, E. Engelage and S. M. Huber, Angew. Chem., Int. Ed., 2021, 60, 5069–5073 CrossRef CAS PubMed; (b) A. Boelke, T. J. Kuczmera, E. Lork and B. J. Nachtsheim, Chem. – Eur. J., 2021, 27, 13128–13134 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Spectroscopic, crystallographic and DFT computational data. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01555c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |