
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insight into the effects of pulsed CO2 electrolysis in a zero-gap electrolyzer†
Xiao Kun
Lu
 a,
Weiyan
Ni
bc,
Adrien E.
Deberghes
a and
Linsey C.
Seitz
*a
a,
Weiyan
Ni
bc,
Adrien E.
Deberghes
a and
Linsey C.
Seitz
*a
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: linsey.seitz@northwestern.edu
bDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
cDepartment of Electrical and Computer Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 9th June 2025
Abstract
Electrochemical CO2 reduction reactions face activity, selectivity, and stability challenges as this technology moves towards commercialization. Pulsed electrolysis (PE) has been shown to improve selectivity and stability at the cost of a negligible energy increase. The effectiveness of PE was assessed in a zero-gap membrane electrode assembly with a Cu catalyst between 50 and 500 mA cm−2 with various, widely adopted binders including Nafion, Sustainion, and fluorinated ethylene propylene. PE suppresses H2 production at 50–300 mA cm−2 and nearly doubles the faradaic efficiency of multi-carbon products when this approach is combined with the use of Sustainion-incorporating electrodes. We find that the notable improvement can be accounted for via the increased local CO2 concentration observed using in situ surface enhanced Raman spectroscopy.
Of the myriad electrochemical CO2 reduction (ECR) products, selectivity for multi-carbon products (C2+) remains below the selectivity requirements (>80% faradaic efficiency (FE)1) due to complex reaction pathways and high activation barriers.2–4 Catalyst tuning has attracted much of the recent research attention to alter the binding strength of *CO,5,6 while fewer studies have focused on operational approaches to increase C2+ selectivity.
Improving FE through identification of an optimal potential/current density window is a simple technique ubiquitously performed in static electrolysis. Pulsed electrolysis (PE) is a technique where the cathode applied potential/current density is changed between two values (Ea or ja and Ec or jc, a = anodic and c = cathodic values) within time intervals, ta and tc.7 PE studies have focused on optimizing the Ea and time periods for various purposes. PE has been shown to increase selectivity towards the desired products through local reaction environment modification when Ea results in a ja close to no current.8–10 PE also modulates catalyst faceting when Ea is more anodic than the Cu oxidation redox potentials.11,12 Finally, PE prolongs the stability of both the catalyst13–15 and the gas diffusion layer (GDL)16 through regenerative processes when Ea stops the ECR. However, the majority of the previous FE enhancement results were obtained in H-type cells and flow cells. As commercialization of ECR calls for >3000 h of stability,17,18 the effect of PE in zero-gap membrane electrode assembly (MEA) devices needs to be assessed.
In this study, we explored the effects of binding agents on PE in an MEA. We further characterize the local reaction environment using a GDL-based in situ Raman cell that better represents the GDE geometry compared to commonly used H-type Raman cells. Finally, we examined the stability of anodes under PE and evaluated the stability number for catalyst precious metal dissolution.
Three types of binders were assessed for enhancement under PE in zero-gap MEA devices: (1) Nafion, a cation exchange binder that contains a fluorinated backbone and a sulfonic acid functional group,19 (2) Sustainion, an anion exchange binder with an imidazolium functional group,20 and (3) fluorinated ethylene propylene (FEP) for hydrophobic treatment (Fig. S1, ESI†).21 Binders were chosen to vary the local H2O and CO2 transport, which greatly influences the response to PE.8
Gas diffusion electrodes (GDEs) were prepared by spray coating a sonicated ink of 50 nm Cu NPs, isopropanol, and binder onto a carbon paper GDL with a microporous layer (Fig. S2–S5, ESI†). The GDEs with FEP, Nafion, and Sustainion have decreasing hydrophobicity based on water contact angle measurements, showing 132°, 128°, and 124°, respectively (Fig. S6, ESI†).
ECR performance was evaluated in an anion exchange MEA device with a 0.1 M KHCO3 anolyte and an IrOx/Ti anode prepared by a dip-coating method (Fig. S7–S9, ESI†).22 Current densities of 50–500 mA cm−2 were applied via GS chronopotentiometric (CP) holds, or PE CPs, switching between ja = 1 and jc = 50–500 mA cm−2 at ta = 20 s “off” and tc = 40 s “on” intervals (Fig. S10, ESI†). A non-zero ja prevents returning to open circuit potential (OCP), which may cause Ti transport layer degradation.23 Studies have utilized various ta values for local reaction environment modulation (ta > 0.6 s) or catalyst regeneration (ta > 5 min).15,16 We chose a ta of 20 s in agreement with a previous report that shows mitigation of salt precipitates using PE and a tc of 40 s to match the timescales of our characterization techniques.16
Fig. 1 and Fig. S11 (ESI†) show the FE of GDEs fabricated with the three binders under GS and PE operations. Under GS operation, the FEP-GDE at <200 mA cm−2 shows hydrogen evolution reaction (HER) suppression, at just 10% FE towards H2. However, the main product at <200 mA cm−2 is CO. Ionomer-based GDEs, the Nafion-GDE and the Sustainion-GDE, achieved higher C2+ product production at <200 mA cm−2. Ion conducting groups may enable C2+ production at lower current densities.
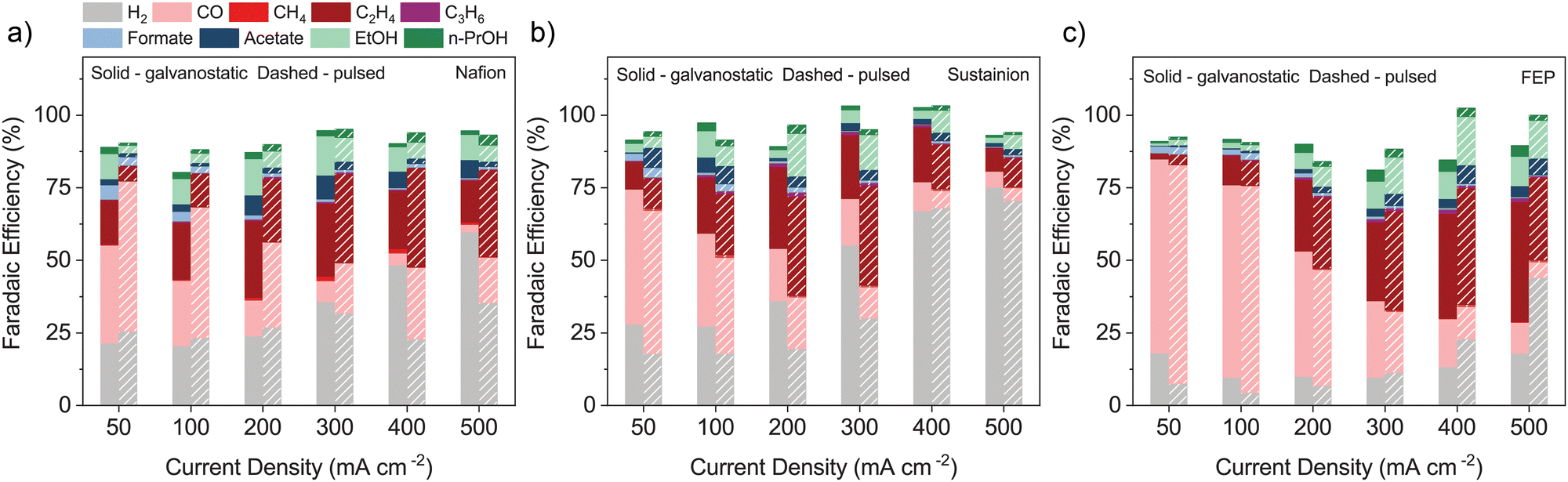 | ||
| Fig. 1 FEs of H2, C1, and C2+ products for electrodes comprising Cu NPs and (a) Nafion, (b) Sustainion, or (c) FEP binders under GS and PE operation. | ||
For GS operation between 200 and 500 mA cm−2, the C2+ FE of the FEP-GDE increases from 36% to 60%, while the Nafion-GDE and Sustainion-GDE exhibit C2+ FEs that continuously decrease as the current density increases. The Sustainion-GDE only shows an 18% FE towards ECR products at 500 mA cm−2. The ability to maintain C2+ production at a high current density can be ascribed to the hydrophobicity of the binder. Low hydrophobicity may cause flooding of the GDE and hinder CO2 transport.24,25 This is exacerbated by the applied potential due to electrowetting.26,27
The FEP-GDE in PE exhibits an increased CO FE at <200 mA cm−2, while the Nafion-GDE exhibits an increased CO FE at all current densities, compared to GS operation. The CO FE is less impacted by PE vs. GS operation for the Sustainion-GDE. For PE between 50 and 300 mA cm−2, the C2+ FE of the Nafion-GDE is lower compared to that for GS operation, while the H2 FE is lower between 300 and 500 mA cm−2. Similarly, the FEP-GDE has a higher CO FE at 50 and 100 mA cm−2 with good HER suppression. The Sustainion-GDE under PE also exhibits good HER suppression (Fig. 1d–f); at 200 mA cm−2, the HER only comprises 20% FE, while C2+ products comprise 56% FE. This is explained by the increased concentration of local CO2 due to PE, and the ability of Sustainion to stabilize ECR intermediates for further reduction. The Sustainion-GDE under PE still suffers from flooding, and the HER is the predominant reaction at >400 mA cm−2.
The Sustainion-GDE exhibiting a high C2+ FE at a lower operating current density could reduce the operating cost since the peak energy efficiency and economic benefits of ECR plummet at high current densities according to technoeconomic analysis.28 Anionic species (e.g., OH− and HCO3−) transported across the anion exchange membranes have lower ionic conductivity compared to H+ in proton exchange membranes; hence, operating anion exchange MEAs at high currents induces higher ohmic overpotential. ECR devices also drive inefficient water oxidation due to the near-neutral electrolytes.29 The improved C2+ FE at 200 mA cm−2 for the Sustainion-GDE could be of great interest as it balances faradaic and energy efficiencies.
PE enhancement effects originate from the changes in the local reaction environment, which are further corroborated by the use of ionomer binders. Nafion as a cation exchange ionomer may trap the OH− produced by the ECR or HER in the vicinity of the catalyst particles, yielding a higher local pH to suppress the HER.8,30 Sustainion allows for higher dissolution of CO2 at the catalyst interface, and its imidazolium functional group has also been shown to stabilize *CO2−, which further utilizes the high local CO2 concentration for CO2 conversion.8,31
In situ surface enhanced Raman spectroscopy (SERS) was performed in a GDE-based spectro-electrochemical cell to understand the origin of the PE enhancement with the use of a Sustainion binder (Fig. S12, ESI†). CuO was chosen as a catalyst for Raman study to ensure a focused beam onto the electrode surface at OCP (the start of the experiment) so that changes in signal intensity at various potentials are not a result of change in focal length. CuO is reduced to Cu under ECR conditions. Fig. S16 and Table S1 (ESI†) show the full SERS spectra and peak assignments of CuO/Sustainion-GDEs under GS and PE operation ranging from OCP to −1.6 VRHE. The operating potentials are chosen so that Raman spectra could reflect *CO32− adsorption rather than *CO adsorption.
The two main findings from in situ SERS experiments are that the local CO2 concentration and local pH are increased during PE compared to GS operation. Fig. 2a shows that the Cu–*CO2− peak intensity (at 362 cm−1) increases as the applied potential is stepped cathodically, from −1.0 to −1.6 VRHE under GS operations.32,33 The Cu–*CO2− peak intensity under PE is higher than that of their GS operated counterparts at −1.0 and −1.3 VRHE with the maximum intensity at −1.3 V vs. RHE for PE rather than at −1.6 VRHE for GS operation. Local CO2 regeneration during the “off” time of PE is the reason for the higher *CO2−, which leads to enhanced ECR performance and HER suppression.
 | ||
| Fig. 2 In situ SERS spectra for (a) Cu–*CO2− stretching and (b) *CO32− peaks during GS, CP and PE of the CuO/Sustainion-GDE in a GDE-based Raman cell with 0.1 M KHCO3. | ||
Fig. 2b shows the *CO32− peak intensity (at 1073 cm−1). Previous studies have used HCO3−/CO3− peak ratios to calculate the local pH, but in the present system, CO32− is the only dominant species and would not lead to accurate values of local pH.34 The *CO32− peak intensity during GS operation increases as the potential becomes more negative due to the production of OH− as a side ECR or HER product. The *CO32− peak intensities during the “on” periods of all three PE conditions were higher than those of their GS counterparts. This may arise from the higher local CO2 concentration due to PE. Switching of electrical potential during PE allows for accumulation of the produced OH− and CO2-neutralized CO32− rather than repulsion from the cathode.10
It is noteworthy that Ir-based anode catalysts contribute to a significant amount of capital expenditure of CO2 electrolyzers. Due to the (bi)carbonate equilibrium, even when alkaline electrolytes are used, the anodic local pH is neutral-acidic and can cause dissolution of 3d-transition metal-based catalysts (e.g., Ni), making Ir indispensable in CO2 MEA electrolyzers for their stability under acidic OER conditions.35,36 PE may benefit ECR from multiple perspectives as described previously, but the stability of Ir-based anodes under PE remains unclear.
The stability of the IrOx/Ti anode under PE was examined over ∼5700 cycles (1 min per cycle totaling to ∼4 days of continuous testing) between jc = 200 mA cm−2 and ja = 1 mA cm−2. The HER was chosen as the cathodic reaction since the overpotential and degradation rate are hypothesized to be much lower than those for the OER or ECR; therefore, the changes in potential response for this system primarily reflect the stability of the anode. The local pH of the anode when operating in neutral buffer is predicted to be ∼3 due to consumption of OH−.29
Fig. 3a shows the potential response of the water electrolyzer under PE. We took average full cell potentials (over 10 cycles) at 0 cycles, 500 cycles, and each time before and after the electrolyte was replaced (Fig. S20–S22, ESI†). The full cell potentials increased after each electrolyte replacement, from ∼3.10 V to ∼3.36 V, with the exception of the initial electrolyte replacement after 30 minutes of conditioning (Fig. 3b). The number of cycles that the system requires to reach stabilized operation also increases with the number of times the electrolyte is replaced. Changes in full cell potential are attributed to the pH change at the cathode after electrolyte replacement. The catholyte reaches a pH of 12–13 after 1400 cycles due to the lack of CO2 in the system to buffer the cathodic pH. In an actual CO2 MEA, electrolyte replacement would not have drastic impacts on the cathodic half-cell potential. The anolyte pH remains at ∼7, yielding a similar OER environment to an actual CO2 electrolyzer. In contrast, the steady operation full cell potential does not increase starting from the ∼900th cycle to the ∼5660th cycle, suggesting that the anode catalyst does not undergo significant degradation due to pulsing over the timescale that we examined.
 | ||
| Fig. 3 (a) Potential response of near-neutral water electrolysis under PE at 200 mA cm−2 with the Pt/C cathode and the IrOx/Ti anode in the 0.1 M KHCO3 anolyte. (b) Average full cell potential (over 10 cycles) before (“steady state” under PE) and after electrolyte replacement periods. | ||
The stability number (S-number) is defined as the ratio of the O2 molecules produced to Ir atoms dissolved. The S-number of the IrOx/Ti anode under PE post-testing is 5 × 105, determined by inductively coupled plasma mass spectrometry of Ir in the anolyte and the digested AEM. The S-numbers of the aqueous media system and MEA are 104–105 and 106–109, respectively.37,38 In a CO2 electrolyzer, the IrOx anode forms interfaces with both a liquid electrolyte and an AEM, which yields a unique system with a combination of MEA and aqueous media.
In summary, ECR in a zero-gap MEA can be enhanced by PE between 50 and 300 mA cm−2, which is likely due to the increased CO2 concentration and local pH, as verified by in situ SERS. With the Sustainion binder, a 56% FE for C2+ species was achieved using commercial Cu NP under PE, almost double that of its GS counterpart. HER suppression and enhancement to CO formation were also observed for electrodes with FEP or Nafion binders. The Ir anode stability of near-neutral water oxidation in MEA in PE can be predicted via the S-number, which falls in between the aqueous media and MEA due to the presence of the 0.1 M KHCO3 anolyte. PE provides a promising pathway to enhance the ECR with just ∼1% of energy consumption; however, the dilution of the product stream due to down time requires technoeconomic assessment to identify the optimal balance of stability and performance.16,39
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- J. H. Montoya, A. A. Peterson and J. K. Nørskov, ChemCatChem, 2013, 5, 737–742 CrossRef CAS.
- T.-C. Kuo, J.-W. Chou, M.-H. Shen, Z.-S. Hong, T.-H. Chao, Q. Lu and M.-J. Cheng, J. Phys. Chem. C, 2021, 125, 2464–2476 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119(12), 7610–7672, DOI:10.1021/acs.chemrev.8b00705.
- S. Du, P. Yang, M. Li, L. Tao, S. Wang and Z. Liu, Chem. Commun., 2024, 60, 1207–1221 RSC.
- R. Casebolt, K. Levine, J. Suntivich and T. Hanrath, Joule, 2021, 5, 1987–2026 CrossRef CAS.
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS.
- C. Kim, L.-C. Weng and A. T. Bell, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- A. Herzog, M. Lopez Luna, H. S. Jeon, C. Rettenmaier, P. Grosse, A. Bergmann and B. Roldan Cuenya, Nat. Commun., 2024, 15, 3986 CrossRef CAS.
- J. Timoshenko, A. Bergmann, C. Rettenmaier, A. Herzog, R. M. Arán-Ais, H. S. Jeon, F. T. Haase, U. Hejral, P. Grosse, S. Kühl, E. M. Davis, J. Tian, O. Magnussen and B. Roldan Cuenya, Nat. Catal., 2022, 5, 259–267 CrossRef CAS.
- H. S. Jeon, J. Timoshenko, C. Rettenmaier, A. Herzog, A. Yoon, S. W. Chee, S. Oener, U. Hejral, F. T. Haase and B. Roldan Cuenya, J. Am. Chem. Soc., 2021, 143, 7578–7587 CrossRef CAS.
- Y. Jännsch, J. J. Leung, M. Hämmerle, E. Magori, K. Wiesner-Fleischer, E. Simon, M. Fleischer and R. Moos, Electrochem. Commun., 2020, 121, 106861 CrossRef.
- T. N. Nguyen, Z. Chen, A. S. Zeraati, H. S. Shiran, S. M. Sadaf, M. G. Kibria, E. H. Sargent and C. T. Dinh, J. Am. Chem. Soc., 2022, 144, 13254–13265 CrossRef CAS PubMed.
- J. Kok, J. de Ruiter, W. van der Stam and T. Burdyny, J. Am. Chem. Soc., 2024, 146, 19509–19520 CrossRef CAS.
- Y. Xu, J. P. Edwards, S. Liu, R. K. Miao, J. E. Huang, C. M. Gabardo, C. P. O’Brien, J. Li, E. H. Sargent and D. Sinton, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- E. W. Lees, B. A. W. Mowbray, F. G. L. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2021, 7, 55–64 CrossRef.
- C. P. O’Brien, D. McLaughlin, T. Böhm, Y. C. Xiao, J. P. Edwards, C. M. Gabardo, M. Bierling, J. Wicks, A. Sedighian Rasouli, J. Abed, D. Young, C.-T. Dinh, E. H. Sargent, S. Thiele and D. Sinton, Joule, 2024, 8, 2903–2919 CrossRef.
- P. Ding, H. An, P. Zellner, T. Guan, J. Gao, P. Müller-Buschbaum, B. M. Weckhuysen, W. van der Stam and I. D. Sharp, ACS Catal., 2023, 13, 5336–5347 CrossRef CAS PubMed.
- Z. Liu, H. Yang, R. Kutz and R. I. Masel, J. Electrochem. Soc., 2018, 165, J3371 CrossRef CAS.
- T. H. M. Pham, J. Zhang, M. Li, T. H. Shen, Y. Ko, V. Tileli, W. Luo and A. Züttel, Adv. Energy Mater., 2022, 12, 2103663 CrossRef CAS.
- W. Luc, J. Rosen and F. Jiao, Catal. Today, 2017, 288, 79–84 CrossRef CAS.
- A. Weiß, A. Siebel, M. Bernt, T. H. Shen, V. Tileli and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, F487 CrossRef.
- Y. Wu, L. Charlesworth, I. Maglaya, M. N. Idros, M. Li, T. Burdyny, G. Wang and T. E. Rufford, ACS Energy Lett., 2022, 7, 2884–2892 CrossRef CAS.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS.
- K. Yang, R. Kas, W. A. Smith and T. Burdyny, ACS Energy Lett., 2020, 6, 33–40 CrossRef.
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC.
- S. C. da Cunha and J. Resasco, Nat. Commun., 2023, 14, 5513 CrossRef CAS.
- C. P. O’Brien, R. K. Miao, A. Shayesteh Zeraati, G. Lee, E. H. Sargent and D. Sinton, Chem. Rev., 2024, 124(7), 3648–3693, DOI:10.1021/acs.chemrev.3c00206.
- M. Li, E. W. Lees, W. Ju, S. Subramanian, K. Yang, J. C. Bui, H.-P. Iglesias van Montfort, M. Abdinejad, J. Middelkoop, P. Strasser, A. Z. Weber, A. T. Bell and T. Burdyny, Nat. Commun., 2024, 15, 8222 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS.
- Y. Zhao, X.-G. Zhang, B. Nataraju, W. Yang, Q. Liang, P. Radjenovic, Y.-H. Wang, Y.-J. Zhang, J.-C. Dong, Z.-Q. Tian and J.-F. Li, Energy Environ. Sci., 2022, 15, 3968–3977, 10.1039/d2ee01334g.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261–E9270 CrossRef CAS.
- D. A. Henckel, M. J. Counihan, H. E. Holmes, X. Chen, U. O. Nwabara, S. Verma, J. Rodríguez-López, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2020, 11, 255–263 CrossRef.
- Á. Vass, B. Endrődi, G. F. Samu, Á. Balog, A. Kormányos, S. Cherevko and C. Janáky, ACS Energy Lett., 2021, 6, 3801–3808 CrossRef.
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef.
- J. Knöppel, M. Möckl, D. Escalera-López, K. Stojanovski, M. Bierling, T. Böhm, S. Thiele, M. Rzepka and S. Cherevko, Nat. Commun., 2021, 12, 2231 CrossRef.
- J. Edgington and L. C. Seitz, ACS Catal., 2023, 13, 3379–3394 CrossRef CAS.
- Y. L. Chung, S. Kim, Y. Lee, D. T. Wijaya, C. W. Lee, K. Jin and J. Na, iScience, 2024, 27, 110383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01463h |
| This journal is © The Royal Society of Chemistry 2025 |