
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotomodulation of dinuclear europium(III) complex luminescence using thermally reversible photochromism of diarylethene†
Yoann
Fréroux
 a,
Salauat
Kiraev
b,
Olivier
Galangau
a,
Tuan-Anh
Phan
a,
Thierry
Roisnel
a,
Olivier
Maury
b,
Stéphane
Rigaut
*a and
Lucie
Norel
*ac
a,
Salauat
Kiraev
b,
Olivier
Galangau
a,
Tuan-Anh
Phan
a,
Thierry
Roisnel
a,
Olivier
Maury
b,
Stéphane
Rigaut
*a and
Lucie
Norel
*ac
aUniv Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: lucie.norel@univ-rennes1.fr
bENS de Lyon, CNRS, UMR 5182, Université Claude Bernard Lyon 1, Laboratoire de Chimie Lyon, F69342, France
cInstitut Universitaire de France, France
First published on 28th April 2025
Abstract
With the use of an original chelating T-type photochromic diarylethene unit, we describe the efficient switching of visible luminescence of a dinuclear europium(III) complex possessing a minute-scale thermal back reaction.
Photochromism, characterized by a reversible isomerization in response to light irradiation, resulting in two different isomers with two different absorption spectra, is extensively studied as a way to photocontrol a large range of properties.1 Diarylethene (DAE) molecules are popular photochromic compounds because of their good fatigue resistance and photochromic behaviour retained in the solid state.2 Although most of the DAE molecules are P-type, meaning that both the open and closed isomers are thermodynamically stable, some modifications of the central ethene bridge and of the aryl groups result in a less stable closed isomer. The resulting T-type photochromes exhibit thermally activated back reaction with timescales ranging from hours to milliseconds.3 In parallel, the modulation of the photophysical properties of lanthanide complexes with DAE is a rich area of research.4 Compared to other photochromic units, the precise design of tailored switchable optical properties is facilitated in the case of DAE by the consequent body of structure–property studies available in the literature.2,5 In particular, DAE molecules in their open state are compatible with europium, as well as ytterbium sensitization, while efficient luminescence quenching occurs in the closed state.6 The resulting well-defined luminescence, slowly fading upon continuous UV excitation, is interesting in the context of anticounterfeiting for instance.6,7 However, all the investigated complexes are P-type and the reverse isomerization reaction so far requires the use of a second irradiation, in the closed DAE absorption band located in the visible range. Moreover, the quantum yields for this back-isomerization reaction are usually quite low, resulting in long irradiation times.2 Thus, for practical reasons, it appears very appealing to rely on spontaneous return to the open emissive state. To date, only a few combinations of lanthanide visible emitters with T-photoswitches other than DAE like spiropyrans,8 viologens,9 anthracene10 or a peculiar photochromic organoboron unit11 have been reported, with various quenching efficiencies and some fatigability issues.4 In this article, we report the first example of a T-type DAE photochromic europium(III) complex exhibiting a rapid thermally activated back-isomerisation process with good fatigability and suited to control red emission.
Recently, we described the behaviour of a pyrazine-based bis-tridentate dinuclear dysprosium complex in the context of single-molecule magnet photoswitching (Fig. 1).12 This complex is a T-type photochrome with minute-scale back isomerisation kinetics at room temperature. Thus, to demonstrate the concept of emission switching with spontaneous signal recovery, we decided to investigate the corresponding analogues with red-emitting europium(III) and NIR-emitting ytterbium(III) ions. Hence, the ligand 2,3-bis(5-methyl-2-(pyridine-2-yl)thiazol-4-yl)pyrazine (BTPo)8 reacted with two equivalents of [M(hfac)3·2H2O] (M = Yb or Eu, hfac = hexafluoroacetylacetonate) in refluxed dichloromethane in order to obtain 1Mo as single crystals after slow cooling. These complexes have been fully characterized by paramagnetic 1H, 13C and 19F NMR, HR/MS and elemental analysis (see the ESI†). X-ray structure analyses revealed that only 1Euo was isostructural to the previously investigated 1Dyo and 1Yo complexes12 as it crystallized in the space group C2/c with an asymmetric unit composed of half a complex and the C2 axis generating the whole dinuclear species. The coordination sphere around the Eu(III) center is composed of three nitrogen atoms from the BTPo ligand and six oxygen atoms from the three hfac ligands, resulting in a coordination number of 9 (Fig. 2). The europium-nitrogen bond lengths are 2.517(17) Å, 2.634(18) Å and 2.669(19) Å with the thiazole, pyridine and pyrazine rings, respectively. The europium–oxygen bond distances are much shorter and more homogeneous in the range of 2.374(15)–2.436(15) Å. In contrast, 1Ybo crystallized in the space group P21/n where only one of the two Yb(III) centers keeps the same coordination environment with slightly shorter distances in the range of 2.426(3)–2.581(3) Å with the N-rings and 2.295(3)–2.362(3) Å with the hfac ligand. The second Yb(III) center is only coordinated by two nitrogen atoms and three hfac ligands giving a coordination number of 8. The ytterbium-nitrogen distance with the pyrazine core is indeed drastically longer (3.360(3) Å) and cannot be considered as a bond (Fig. 2). As the ionic radius of Yb(III) is smaller than those of the other studied lanthanide complexes (Dy, Y and Eu),13 the N–Yb–N bite angles within the tridentate pocket are larger (65.07 and 64.00° vs. 62.8 and 62.4° for 1Euo). This effect causes a distortion of the central pyrazine bridge toward one of the metal centers and explains the two distinct metal coordination environments.
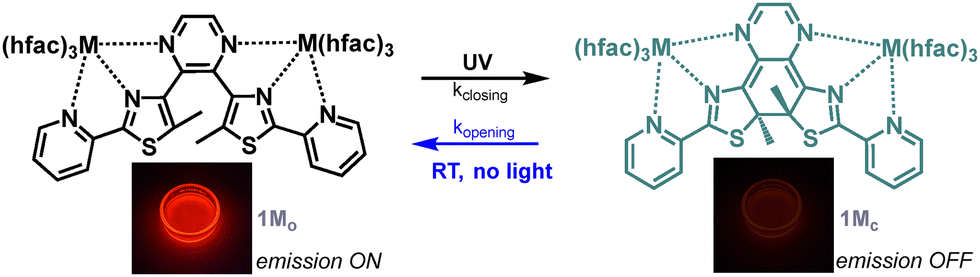 | ||
| Fig. 1 Isomerisation reactions of 1M (M = Eu, Dy,12 Yb, Y12) with images of the luminescence of a 1Eu solution before and after DAE isomerisation. | ||
 | ||
| Fig. 2 Single-crystal XRD structures of 1Euo (top) and 1Ybo (bottom). Grey, blue, yellow, pink and purple ellipsoids represent C, N, S, Eu and Yb atoms, respectively. H atoms are omitted, and the hfac ligands are shown in wire style. | ||
Complementary insights were obtained from the 1H NMR spectra of 1Euo and 1Ybo in CD2Cl2 (Fig. S1 and S6, ESI†). The spectra showed strongly deshielded signals up to 45 ppm for the aromatic proton of the BTP core, as expected from the paramagnetism of both the complexes. A strongly shielded signal was also observed at −15.05 ppm (1Ybo) or at 1.69 ppm (1Euo), characteristic of the hfac protons. In solution, the apparent symmetries of both complexes were the same, with a single set of signals corresponding to two equivalent half-complex units. This means that a rapid exchange faster than the NMR time scale (10−5 s)14 occurs between the 8-coordinated and 9-coordinated sites of 1Ybo in solution. Titration experiments were conducted and clearly showed the stepwise formation of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 complexes (Fig. S8 and S9, ESI†). We also investigated the stability of the dinuclear species upon dilution in a range of concentrations (2 10−3 to 4 10−6 M) by 1H NMR and witnessed very different behaviours between 1Ybo and 1Euo. Indeed, the europium(III) complex showed the same spectrum whatever the dilution (Fig. S10, ESI†), while for 1Ybo, dissociation into the mononuclear species was already apparent at the highest concentration (Fig. S11, ESI†). This weaker stability of 1Ybo may be correlated with the presence of a bidentate site as observed in the X-ray structure, but further thermodynamic studies will be needed to fully clarify this point.15
:1 and 1:2 complexes (Fig. S8 and S9, ESI†). We also investigated the stability of the dinuclear species upon dilution in a range of concentrations (2 10−3 to 4 10−6 M) by 1H NMR and witnessed very different behaviours between 1Ybo and 1Euo. Indeed, the europium(III) complex showed the same spectrum whatever the dilution (Fig. S10, ESI†), while for 1Ybo, dissociation into the mononuclear species was already apparent at the highest concentration (Fig. S11, ESI†). This weaker stability of 1Ybo may be correlated with the presence of a bidentate site as observed in the X-ray structure, but further thermodynamic studies will be needed to fully clarify this point.15
Since only 1Euo was shown to be stable at spectroscopic dilutions, we only report here the photophysical properties of this complex. The electronic absorption spectra measured in dichloromethane solutions showed an intense band at λmax = 309 nm (ε ∼ 80000 M−1 cm−1). This band has contributions from the hfac ligands and from the HOMO → LUMO π–π* transition centered on the BTP ligand (λmax = 315 nm with ε ∼ 36000 M−1 cm−1).12 Upon irradiation with UV light, the solution turns green and a new absorption band appears at λmax = 722 nm as a signature of the isomerization to 1Euc. The corresponding electronic transition is associated with the HOMO → LUMO π−π* characterized transition centered on the closed and more conjugated BTP fragment (Fig. 3). The closed 1Euc isomer is thermally unstable and undergoes a retrocyclisation reaction at room temperature. As a result, the green color of the closed complex fades spontaneously. This absorbance decrease was followed for solutions thermalized at different temperatures as shown in Fig. 4 and fitted according to a first order law (for details, see the ESI†). The kinetic parameters extracted from the fits are in the range of what was previously observed on the corresponding yttrium(III) complex by 1H NMR with activation energies of 62.5 kJ mol−1 for 1Euo and 66.7 kJ mol−1 for 1Yo.12
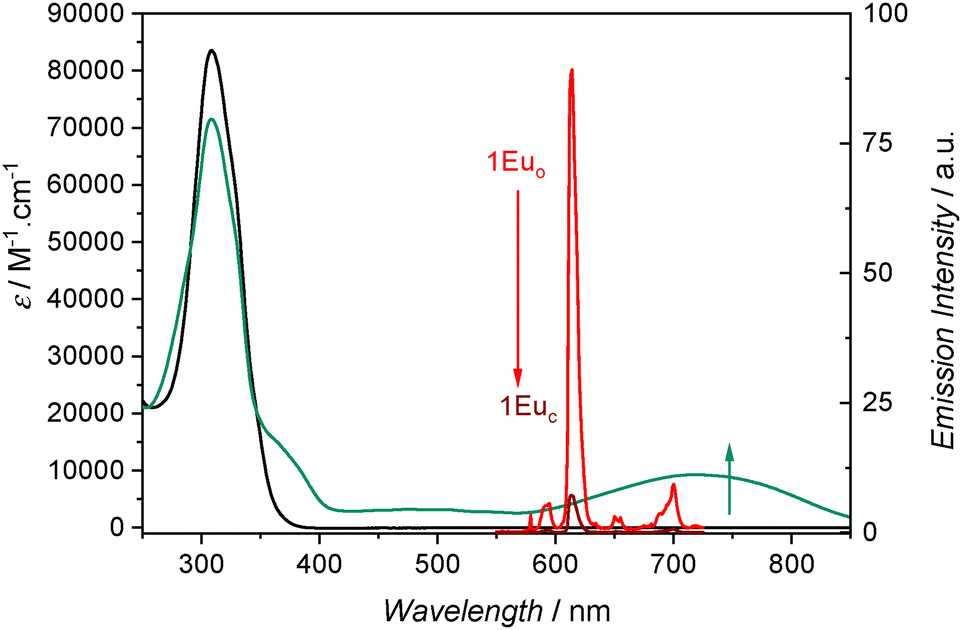 | ||
| Fig. 3 Electronic absorption spectra of 1Euo (black line) and the photostationary state obtained upon 365 nm irradiation (green line) in dichloromethane. Corresponding emission spectra under 305 nm excitation of 1Euo (red line) and the photostationary state containing 1Euc (dark red line). 10 °C, C ∼ 2.5 × 10−6 M. | ||
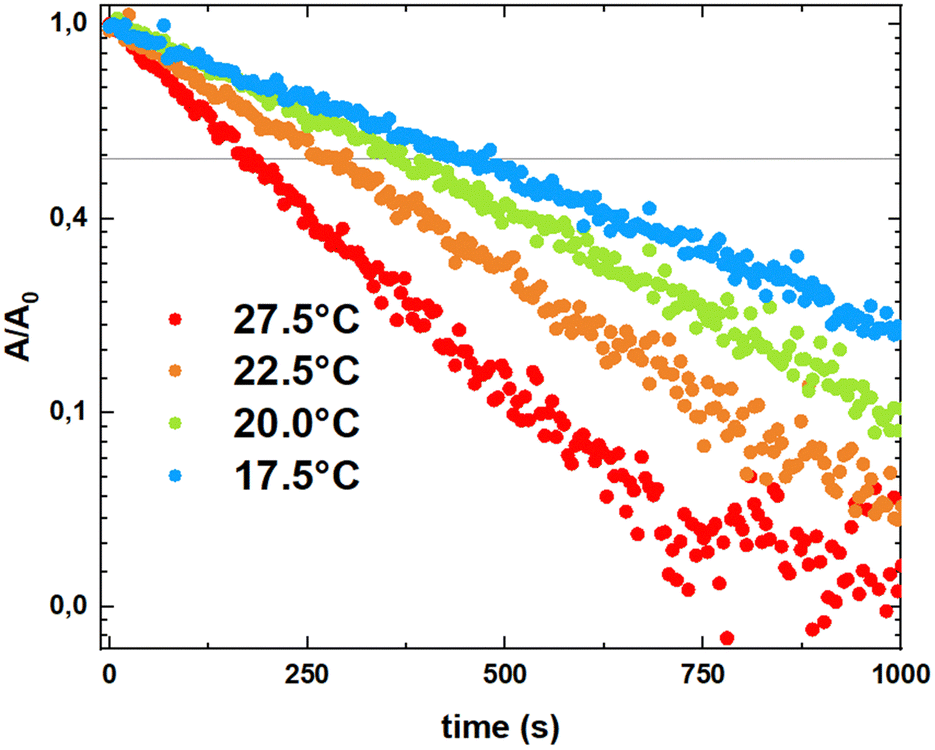 | ||
| Fig. 4 Arrhenius plot showing first order kinetics associated with the retrocyclisation reaction at different temperatures for 1Euc in dichloromethane. The rate constant dependence with temperature can be fitted with an Arrhenius law (k = Aexp(−Ea/RT), with Ea = 62.5 kJ mol−1 and A = 2.8 × 109 s−1). C = 1.3 × 10−5 M. | ||
Emission spectra have then been studied in dichloromethane solution. We observed for 1Euo the characteristic Eu(III) emission profile assigned to 5D0 → 7FJ transitions (J = 0–4) with an intense sharp band located at λ = 615 nm for the hypersensitive 5D0 → 7F2 transition (Fig. 3 and Fig. S13, ESI†). This luminescence is easily distinguished by the naked eye, as well as the progressive emission quenching upon prolonged UV excitation (see Fig. 1 and the video provided in the ESI†). Qualitatively, we noticed that the emission intensity is much higher than that of our previously investigated systems6 although quantum yield measurements are experimentally challenging to carry out because of the fast evolution of the complex under light excitation. The improved sensitisation of the europium emission is due to the well adapted triplet state of the open BTP form, located at 21900 cm−1. This triplet state was energetically lower than that of hfac ancillary ligands (22200 cm−1)16 and was measured at 77 K from a dichloromethane solution of the 1Dyo complex, exhibiting BTP-centred emissions and very weak Dy centred emission (Fig. S15, ESI†). The 50-μs delayed spectral acquisition allowed recording of the phosphorescence of the triplet state, which had a long lifetime of 320 ms at 77 K (Fig. S15, ESI†). This ligand-based emission could also be detected in 1Yo although with a lower intensity, probably because of improved intersystem crossing in the paramagnetic dysprosium(III) complex.17
To better characterize the quenching of luminescence upon DAE isomerisation, we measured the emission decrease over time upon continuous irradiation at λex = 350 nm and with large slit opening to maximize the irradiated volume. This is because the specific features of the emission loss upon UV irradiation were strongly dependent on the exact irradiation configuration and in particular on the proportion of the cuvette volume that could efficiently be irradiated. Indeed, diffusion of open species from non-irradiated areas has to be minimized to obtain the best photoconversions. As a result, for 1Euo, the maximum quenching of luminescence can be reached in 30 seconds and leads to a residual emission of less than 10% of the initial intensity (Fig. 5). The corresponding emission decrease fits with a mono-exponential curve, providing a characteristic time of 3.5 seconds at room temperature. Additionally, luminescence quenching has been recorded upon three successive closing events: in each case the same behaviour is observed, which underlies the stability and reversibility of the process.
 | ||
| Fig. 5 Monitoring of emission intensity (λem = 615 nm) of 1Euc with time upon irradiation at λex = 350 nm followed by spontaneous reopening (for 300 s, not shown) for three successive cycles. Dichloromethane, 20 °C, C = 4.2 × 10−6 M. | ||
While this quenching of europium(III) emission upon DAE isomerisation has previously been observed in various complexes, the spontaneous return to the luminescent ON state has not been investigated with DAE systems. This experiment is tricky to perform since any attempt to read the luminescence of the system should induce an evolution of the ratio of closed isomers. However, by measuring the emission intensity at 615 nm once the 350 nm excitation has been turned off, and using the anti-photobleaching mode of the spectrometer that acquires experimental points only every 6 seconds, we could indeed follow the 1Euc → 1Euo reaction and the corresponding increase in emission intensity (Fig. 6). This increase is much slower than the quenching and shows a characteristic time of 65 s (T = 293 K). Several quenching/recovery curves with reproducible profiles could be measured, successively providing solid evidence of thermal recovery of the luminescent ON state, at room temperature, with a fast response. This feature is well-suited for multiple successive luminescence readouts in practical situations.
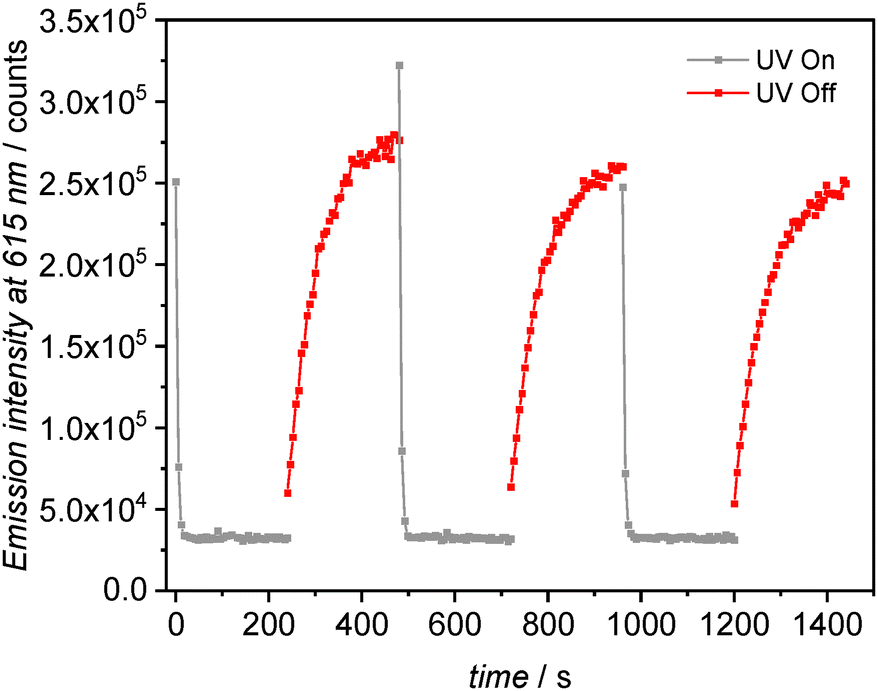 | ||
| Fig. 6 Monitoring of emission intensity of 1Euo (λem = 615 nm) upon irradiation at λex = 350 nm followed by spontaneous re-opening for three successive cycles. Dichloromethane, 20 °C, C = 4.2 × 10−6 M. | ||
To conclude, we report the synthesis of two new dinuclear lanthanide(III) complexes with fast T-type photochromism. Only the europium(III) complex was stable upon dilution. It exhibits intense red luminescence in only one of its forms. The closing reaction causes an important quenching of this emission, and the initial intensity can be fully recovered spontaneously, thanks to the thermal back isomerisation of the ligand. This switching cycle can open the door to dynamic systems displaying a fine emission signature and temporal response with only one irradiation wavelength in the UV region to operate emission control, leading to a simplification of the system towards real life applications. We are currently investigating how the kinetics of the thermal back reaction can be tuned by chemical design to suit different applications.
We thank the Université de Rennes, the ENS de Lyon, the CNRS and the Agence Nationale de la Recherche (2LCDOR – ANR-21-CE07-0063) for support. L. N. thanks Institut Universitaire de France. T. A. P. thanks Région Bretagne for support (SAD19048). We thank Akos Banyasz and Bogdan Marekha for their help in implementing optical measurements.
Data availability
The data that support this article have been included as a part of the ESI.† Crystallographic data for 1Ybo and 1Euo have been deposited at the CCDC under 2423937 and 2423938† and can be obtained from https://www.ccdc.cam.ac.uk/.Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Nakatani, J. Piard, P. Yu and R. Métivier, Photochromic materials: preparation, properties and applications, ed. H. Tian and J. Zhang, Wiley-VCH, 2016, pp. 1–45 Search PubMed.
- M. Irie, T. Fulcaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- (a) M. Walko and B. L. Feringa, Chem. Commun., 2007, 1745–1747 RSC; (b) D. Kitagawa, T. Nakahama, Y. Nakai and S. Kobatake, J. Mater. Chem., 2019, 7, 2865–2870 CAS; (c) J. V. Milić, C. Schaack, N. Hellou, F. Isenrich, R. Gershoni-Poranne, D. Neshchadin, S. Egloff, N. Trapp, L. Ruhlmann, C. Boudon, G. Gescheidt, J. Crassous and F. Diederich, J. Phys. Chem. C, 2018, 122, 19100–19109 CrossRef; (d) D. Kolarski, P. Steinbach, C. Bannwarth, K. Klaue and S. Hecht, Angew. Chem., Int. Ed., 2024, 63, e202318015 CrossRef CAS PubMed; (e) M. Yamada, T. Sawazaki, M. Fujita, F. Asanoma, Y. Nishikawa and T. Kawai, Chem. – Eur. J., 2022, 28, e202201286 CrossRef CAS PubMed; (f) C. Zhang, K. Morinaka, M. Kose, T. Ubukata and Y. Yokoyama, Beilstein J. Org. Chem., 2019, 15, 2161–2169 CrossRef CAS PubMed.
- Y. Fréroux, L. Caussin, N. El Beyrouti, S. Rigaut and L. Norel, Control of 4f complexes luminescence and magnetism with organic photochromic units, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and S. M. Kauzlarich, Elsevier, 2024, ch. 338, vol. 65, pp. 35–91 Search PubMed.
- Z. Li, X. Zeng, C. Gao, J. Song, F. He, T. He, H. Guo and J. Yin, Coord. Chem. Rev., 2023, 497, 215451 CrossRef CAS.
- H. Al Sabea, L. Norel, O. Galangau, T. Roisnel, O. Maury, F. Riobé and S. Rigaut, Adv. Funct. Mater., 2020, 2002943 CrossRef CAS.
- (a) T. Nakagawa, Y. Hasegawa and T. Kawai, Chem. Commun., 2009, 5630–5632 RSC; (b) Z. Li, X. Liu, G. Wang, B. Li, H. Chen, H. Li and Y. Zhao, Nat. Commun., 2021, 12, 1363 CrossRef CAS PubMed.
- M. Koji, N. Yoshio and K. Keiichi, Bull. Chem. Soc. Jpn., 2009, 82, 472–474 CrossRef.
- (a) H.-Y. Li, H. Xu, S.-Q. Zang and T. C. W. Mak, Chem. Commun., 2016, 52, 525–528 RSC; (b) H.-Y. Li, Y.-L. Wei, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, Chem. Mater., 2015, 27, 1327–1331 CrossRef CAS; (c) T. Zhou, J. Chen, T. Wang, H. Yan, Y. Xu, Y. Li and W. Sun, ACS Appl. Mater. Interfaces, 2022, 14, 57037–57046 CrossRef CAS PubMed.
- Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS PubMed.
- N. Wang, J. Wang, D. Zhao, S. K. Mellerup, T. Peng, H. Wang and S. Wang, Inorg. Chem., 2018, 57, 10040–10049 CrossRef CAS PubMed.
- L. Norel, K. Bernot, F. Gendron, C. A. Gould, T. Roisnel, S. Delbaere, B. Le Guennic, D. Jacquemin and S. Rigaut, Chem. Squared, 2020, 4, 2 Search PubMed.
- R. D. Shannon, Acta Cryst. A, 1976, 32, 751–767 CrossRef.
- L. G. Nielsen and T. J. Sørensen, Inorg. Chem., 2020, 59, 94–105 CrossRef CAS PubMed.
- K. Baudet, V. Kale, M. Mirzakhani, L. Babel, S. Naseri, C. Besnard, H. Nozary and C. Piguet, Inorg. Chem., 2020, 59, 62–75 CrossRef CAS PubMed.
- D. A. Gálico, R. Marin, G. Brunet, D. Errulat, E. Hemmer, F. A. Sigoli, J. O. Moilanen and M. Murugesu, Chem. – Eur. J., 2019, 25, 14625–14637 CrossRef PubMed.
- S. Tobita, M. Arakawa and I. Tanaka, J. Phys. Chem., 1984, 88, 2697–2702 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2423937 and 2423938. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01334h |
| This journal is © The Royal Society of Chemistry 2025 |