DOI:
10.1039/D5CC01305D
(Highlight)
Chem. Commun., 2025,
61, 6226-6245
Spin-controlled electrocatalysis: an out-of-the-box strategy for the advancement of electrochemical water splitting
Received
10th March 2025
, Accepted 27th March 2025
First published on 27th March 2025
Abstract
Spin-polarized catalysts have garnered significant interest in electrocatalysis, namely in the electrocatalytic oxidation of water, which has very sluggish kinetics due to its high overpotential. After the groundbreaking discovery that the electron's spin employing the chiral-induced spin selectivity (CISS) effect can control the kinetics of the oxygen evolution reaction (OER), numerous studies have been carried out to demonstrate the impact of electron's spin on reducing the overpotential of the OER. Apart from CISS, various magnetic materials have been explored as OER catalysts, and the outcomes are found to be very promising for the development of spin-based OER catalyst materials. This review highlights the remarkable journey of the evolution of the spin-polarized catalyst, starting from chiral materials to magnetic materials, which has happened in the last decade and its contribution toward the enhancement of OER kinetics, which is a very essential process for the advancement of renewable energy technologies.
1. Introduction
Electrochemical water splitting is considered as the central process for renewable energy technologies, though the high overpotential of the oxygen evolution reaction (OER) restricts its extensive application. The history of the OER in electrocatalysis is rich and spans several centuries, reflecting its importance in scientific research and practical applications. However, the precise mechanism of the oxygen evolution reaction is still considered as a “great enigma”.1 The first proposed OER mechanisms identified the metal centers as the reaction's active sites. Afterward, several studies were carried out to explore the OER process at different pH levels and using different precious metals, such as Pt, Ir, Ru, and their alloys.2 Subsequent research showed that the presence of oxygen in the catalyst in the oxide form positively impacts the production of oxygen, whether it is formed during the OER or an oxide catalyst is used.3 After evaluating all of the potential processes, it was demonstrated that in the OER, molecular oxygen is generated by several proton/electron-coupled mechanisms. The process is significantly pH-dependent, and it follows different pathways in different electrolyte solutions having different pH as shown in Scheme 1a and b. Hence, it was revealed that the formation of the O2 molecule needs the transfer of four electrons, and the kinetically advantageous oxygen evolution reaction (OER) happens via multi-step processes involving single-electron transfers at each level as depicted in Scheme 1c. Thus, the accumulation of energy at each step makes OER dynamics incredibly slow and leads to enormous overpotentials. Therefore, a highly active electrocatalyst is necessary to break the energy barrier, and to reduce the overpotential of the OER, which is considered as the bottleneck of the electrochemical water-splitting process.
 |
| | Scheme 1 OER mechanism. (a) In acidic medium, and (b) in basic medium. (c) The OER process occurs on the catalyst surface under alkaline (red line) and acidic (blue line) conditions. The black line shows oxygen evolution through the peroxide (M–OOH) intermediate and the green line represents oxygen evolution by direct reaction between two neighboring oxo (M–O) intermediates. Reproduced from ref. 4 with permission from The Royal Society of Chemistry copyright 2017. | |
However, the fundamental concept in the design of OER catalysts primarily emphasizes the electronic and crystal structure of the materials. The effectiveness of a catalyst is dictated by the binding energies between the catalyst and the intermediate. According to traditional methods, as outlined by the Sabatier principle, the ideal catalyst should preferentially and strongly bind to reaction intermediates. In this regard, the catalyst's electronic arrangement and crystal structure are given foremost priority. The efficacy of an electrocatalyst is determined by its binding energy to reaction intermediates, whereas scaling relationships inhibit enhancements in catalytic systems beyond their volcano-plot thresholds. However, in all these investigations, spin-polarization is not taken into account, though the ground state of oxygen is inherently triplet. To circumvent these constraints, unconventional strategies that are not governed by the surface binding energy might be beneficial. However, in recent years, mainly in the last decade, numerous novel approaches that polarize the spin in the electrocatalysts, employing chirality and magnetic control, are investigated to increase electrocatalytic efficiency above the volcanic limit, and these strategies have sparked considerable interest. A roadmap of the evolution of the spin-polarized catalyst is given in Scheme 2. Specifically, it is found that a spin-specific anodic current employing a chiral anode or a spin-aligned magnetic electrode can produce spin-polarized hydroxyl species during the oxidation of water, which increases the rate of OER and decreases the overpotential. The phenomenon known as chiral-induced spin selectivity (CISS) explains the rationale of using a chiral catalyst as a spin-filtering electrode. Hence, spin-controlled electrocatalysis can be achieved in two ways, namely, by employing the CISS effect, and the second way is using ferromagnetic electrodes in the presence of a magnetic field, as shown in Scheme 3. In this highlight, we will review the evolution of spin-polarized OER catalysts that happened in the last decade and discuss the prospects of these spin-polarized catalysts for advancing electrocatalytic oxygen evolution.
 |
| | Scheme 2 Schematic representation of the road map of the advancement in oxygen evolution reaction. | |
 |
| | Scheme 3 Spin-polarized electrodes. Spin-polarization in the catalyst can be incorporated either by (a) functionalization of the catalyst using chiral molecules or, (b) using magnetic materials in the presence of a magnetic field. | |
2. Rationale behind the enhancement of the OER employing a spin-polarized catalyst
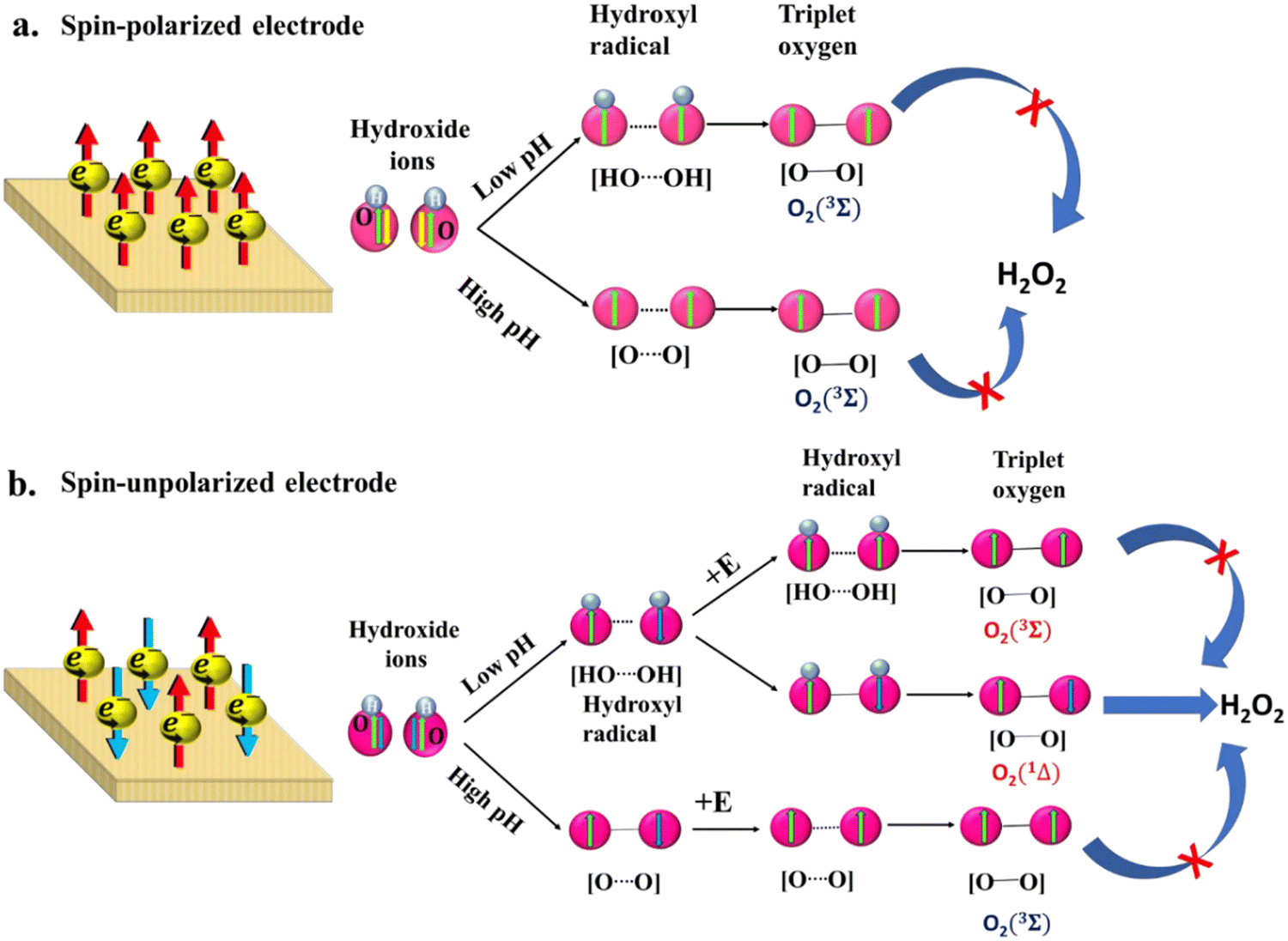
The polarization of electrons in the electrocatalyst plays an important role in generating spin-aligned intermediates that drive the oxygen evolution via a low-energy path as depicted in Scheme 4. Briefly, as oxygen has a triplet ground state, it has been demonstrated that spin selectivity during catalysis may alter the transition state energies for the reaction coordinate and reaction intermediates.5 Typically, during the water oxidation, when the hydroxide ions eject electrons to the anode, if the anode accepts the electron, showing its selectivity for spin of the electrons, hydroxyl radicals or the subsequent oxygen species that will be generated at the adjacent catalytic centers will have parallel spin-orientations, which help in triplet oxygen evolution. However, if the anode does not exhibit spin-selectivity, the hydroxyl radicals produced at the adjacent centers will have opposite spin orientations, which retards the generation of oxygen and produces hydrogen peroxide as a byproduct in the neutral medium, whereas it increases the overpotential at higher pH.
 |
| | Scheme 4 Effect of electron's spin in the OER. (a) In chiral or magnetic catalysts, the generation of triplet oxygen is promoted at higher as well as lower pH levels. (b) For achiral catalysts at lower pH, the generation of H2O2 may compete with the production of triplet oxygen, while at higher pH, the spin disorder can hinder the generation of triplet oxygen. Reproduced from ref. 6 with permission from American Chemical Society © 2020. | |
3. CISS in the OER
During the OER, the effect of spin-polarization was first accomplished using the chiral induced spin-selectivity (CISS) effect.7,8 According to this effect, when an electron transfer occurs through the chiral molecules, it happens in a spin-selective way. Typically, if one enantiomer prefers to transfer one spin of an electron, the opposite enantiomer prefers to transfer the opposite spin, as shown in Scheme 5.9 In electrocatalysis, CISS creates novel opportunities for spin-selective responses.10–12 Various studies manifest that the performance of OER electrocatalysts is greatly enhanced by utilizing the CISS effect.9,13 To incorporate CISS, the catalyst has been modified using chiral molecules employing different techniques. The first few studies that explored the effect of CISS in the OER were carried out by adsorbing the chiral molecules on the catalyst-coated surface.7,8 Later, chiral molecule-chelated metal oxide-based nanoparticles were synthesized, and then they were coated on the electrode surface. OER study has also been carried out by incorporating chiral molecules in between the catalyst and the substrate as shown. Besides, electrochemical deposition has also been explored to incorporate chiral catalysts as another route to fabricate chiral catalyst-coated electrodes.14 Apart from this, chiral organic–inorganic hybrid perovskite, chiral metal–organic frameworks (MOFs), chiral covalent organic frameworks (COFs), and chiral semimetals have also been explored to exhibit CISS-enhanced OER activity.
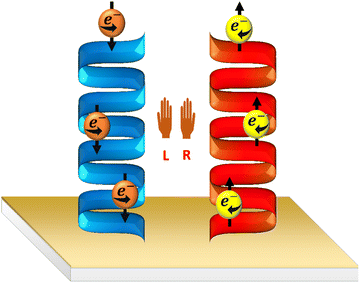 |
| | Scheme 5 Schematic diagram presenting the spin selectivity of the enantiomers due to the chiral induced spin selectivity (CISS) effect. | |
3.1. Demonstration of CISS-enhanced OER
3.1.1. Adsorbing chiral molecules on the catalyst-coated electrode surface.
In 2015, Naaman's group first demonstrated that chiral molecules can effectively be used to catalyze the oxygen evolution reaction because of their spin-polarization ability that results from the CISS effect.7 Initial efforts indicate that coating the catalyst-coated anode with chiral molecules or chiral molecular assembly enhances the overall cell performance.8 TiO2 nanoparticulate films were deposited adopting the electrophoretic deposition (EPD) technique on fluorine-doped tin oxide (FTO)-coated glass and then those films were functionalized with the chiral as well as achiral organic linker molecules to probe the effect of chirality. Chiral peptide molecule coated anodes perform better than the achiral molecules such as 3-mercaptobenzoic acid (3 MBA), 4-mercaptobenzoic acid (4 MBA) etc. coated anodes.7 A similar effect was demonstrated in the photocatalytic oxidation of water while using a Zn–porphyrin-based chiral supramolecular assembly-coated anode.8
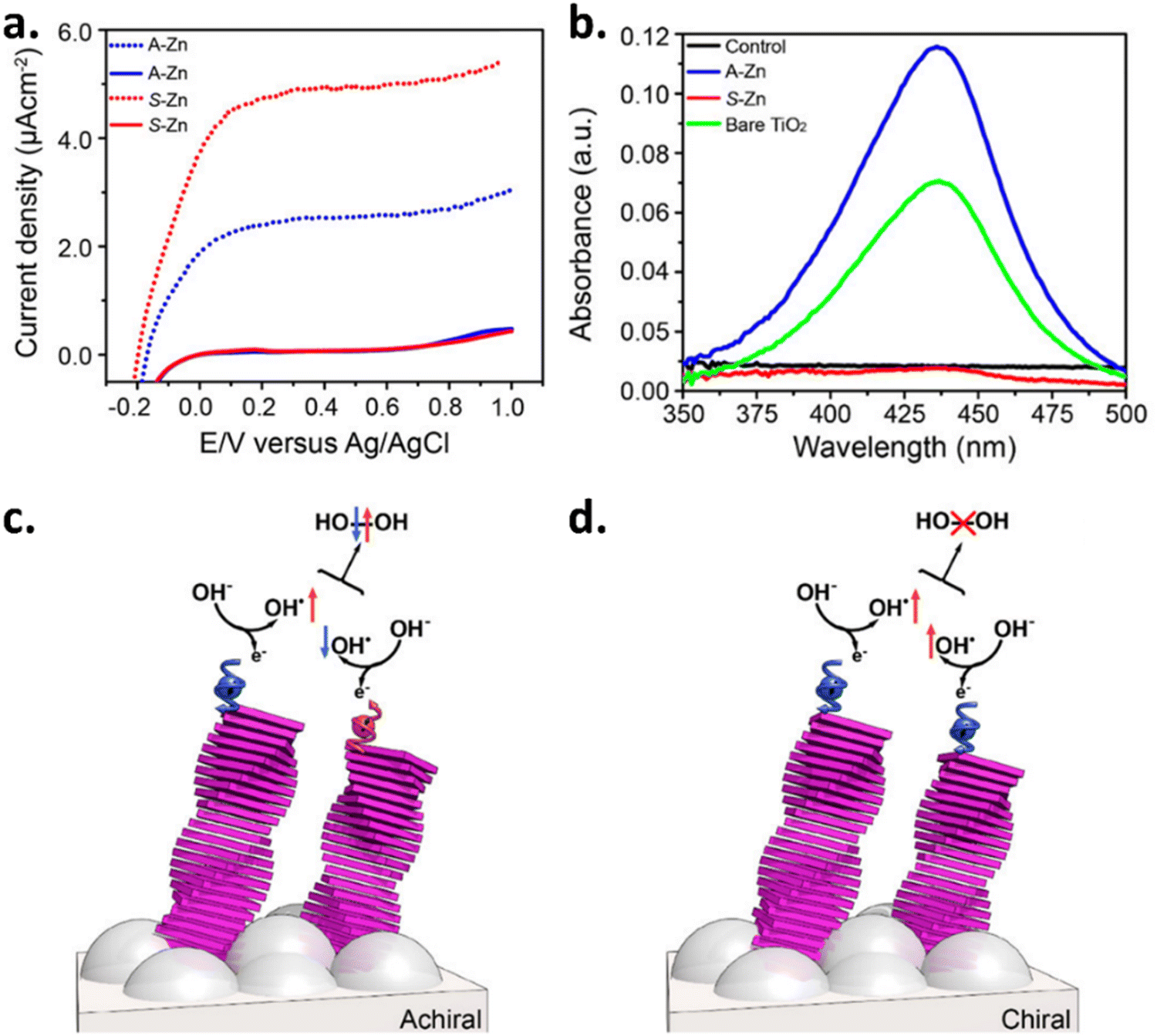
The data shown in Fig. 1a demonstrate that the current density acquired from the chiral anode is higher than the achiral assembly. Besides, the production of the byproduct, hydrogen peroxide, which is stable at lower pH was reduced due to the formation of a spin-polarized intermediate that drives the oxygen formation, as shown in Fig. 1(b–d). In a related study, Tassinari et al. reported chiral functionalization of titania (TiO2) coated-FTO with a chiral poly(fluorene-co-thiophene) conducting polymer and compared its activity with the corresponding achiral polymer (AP), which served as the ref. 15. Later Bhartiya et al. prepared a thin Au–Ni bilayer substrate for use as an electrode by forming a self-assembled monolayer of L-cysteine molecules on the top of it by the chemisorption method and reported a 390 mV decrease in overpotential with a 5-fold higher current density in comparison with the bare bilayer Ni/Au thin film during the OER.16
 |
| | Fig. 1 Spin-specific electron transfer and its impact on photocurrent density and peroxide formation. (a) Photocurrent density comparison of TiO2 electrodes coated with self-assembled molecules. Achirality is presented by the blue line, whereas chirality is represented by the red line. (b) The UV-vis absorption spectra of the neutral electrolyte solution (0.1 M Na2SO4) titrated with o-tolidine while using bare TiO2 and Zn–porphyrin-coated TiO2 (achiral A-Zn or chiral S-Zn) working electrodes. (c) When the anode does not exhibit spin-selectivity, OH˙ radicals have antiparallel spins, interacting on a singlet surface that produces H2O2. (d) Conversely, spin-specific electron transfer aligns the spins parallelly, leading to interaction on a triplet surface that prevents H2O2 formation and promotes ground-state oxygen production. Figures adopted from ref. 8 with permission from American Chemical Society © 2017. | |
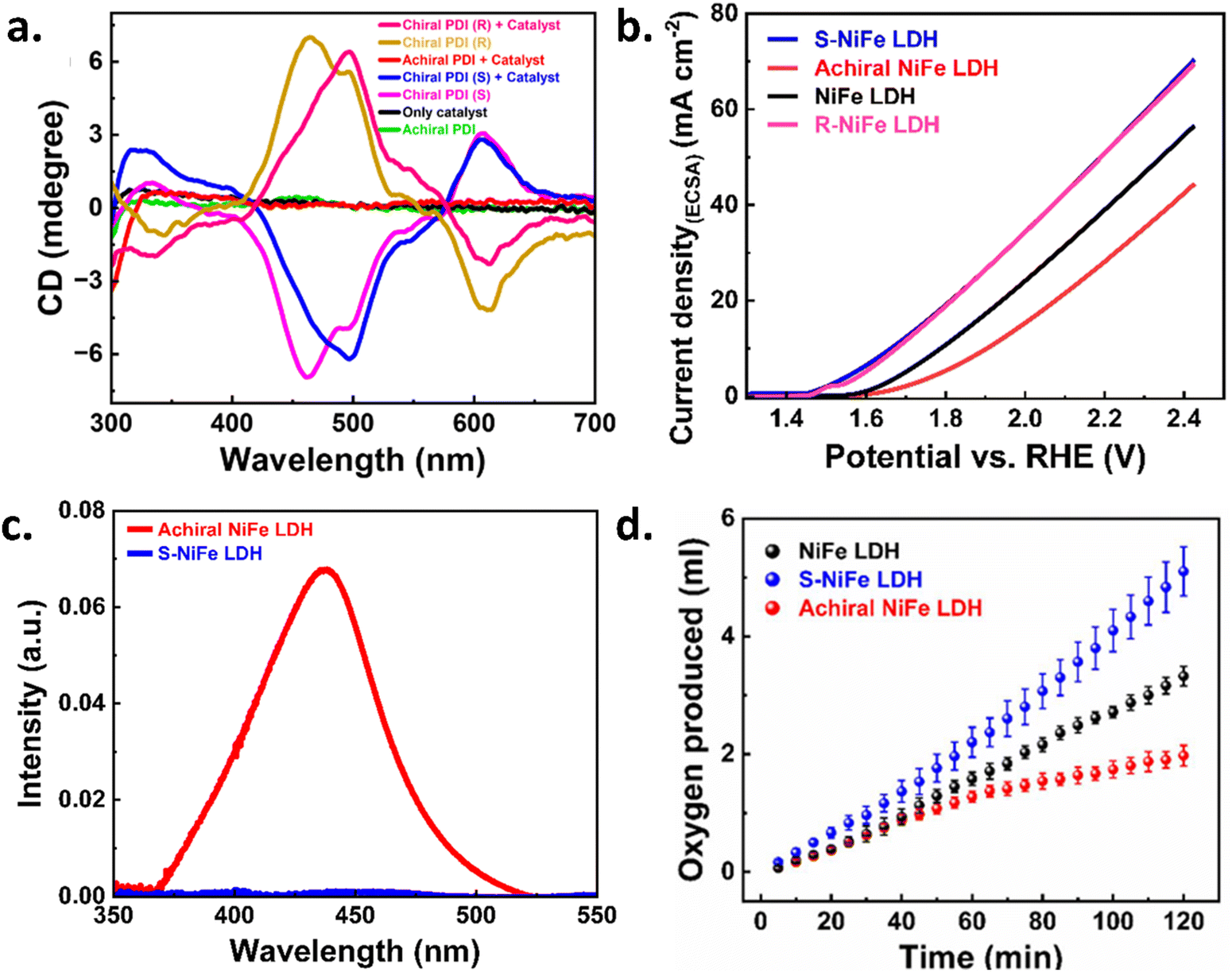
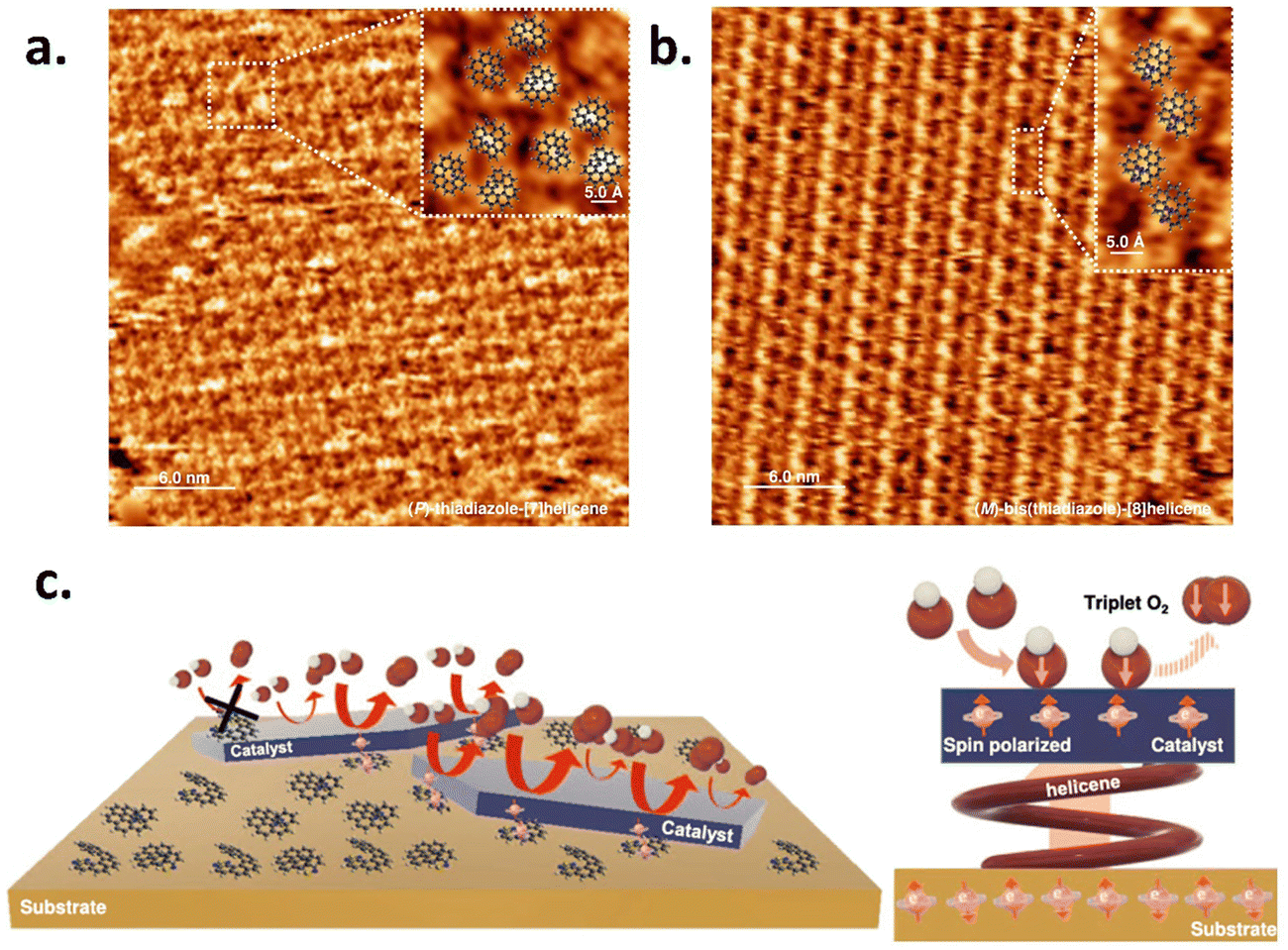
Recently, Utkarsh et al. explored the effect of a perylene diimide (PDI) based chiral supramolecular polymer functionalized layered double hydroxide (LDH)-based catalyst for the oxygen evolution reaction.17Fig. 2a shows the optical activity of chiral and achiral PDI with a catalyst and without a catalyst. The data is shown in Fig. 2b and c, which demonstrates that the helical supramolecular polymer results in higher current density with lowering of the overpotential for the OER process and hydrogen peroxide production. The rate of oxygen evolution was also found to be 1.7 times greater for the chiral functionalized catalyst compared to only the catalyst, as shown in Fig. 2d. Liang et al. used different electrode configurations to incorporate the CISS effect in the layered double hydroxide catalyst in the anode.18 They adsorbed two types of chiral hetero-helicene molecules as spin polarizers: (M, P)-thiadiazole-[7] helicene and (M)-bis(thiadiazole)-[8] helicene on a gold-coated surface and observed that the helicene molecules formed self-assembled monolayer (SAM) structures with the forming of trimeric structures with ∼0.75 molecules per nm2, while the latter formed rows of dimers with ∼1.1 molecules per nm2. The STM images of both the helicenes are shown in Fig. 3a and b. The correlation between the improvement of OER activity and the molecule-catalyst arrangement is seen in Fig. 3c. Helicene molecules on the catalyst surface obstruct the active sites, hence diminishing the total catalytic activity. However, when the chiral helicene is in between the catalyst and the substrate, spin polarization is transferred through the two-dimensional catalyst layer without blocking the active sites that enhance the rate of the OER. In 2024, Vensaus et al. prepared mesoporous TiO2 on Au(111) substrates by evaporation-induced self-assembly having a pore diameter of ∼8 nm and to induce chirality, they used enantiopure (M)-bis(thiadiazole)-[8] helicene, which can bind to the gold substrate, and reported a decrease in H2O2 formation.19
 |
| | Fig. 2 Spectroscopic and electrochemical characterization of chiral supramolecular polymer functionalized catalyst-coated surfaces for the OER. (a) CD spectra were analyzed for thin films of NiFe-LDH catalyst, chiral (S) and (R) isomers, and achiral PDI polymer, both with and without coating of the catalyst. (b) The OER activity of the fabricated electrodes was evaluated by measuring current density against applied potential in 1 M KOH, using a scan rate of 20 mV s−1. (c) UV-visible absorption spectra were obtained after the titration of the electrolyte (0.1 M Na2SO4) with o-tolidine to analyze the reaction dynamics. (d) Oxygen evolution was monitored over time while maintaining a constant anode potential in 1 M KOH. The gas volume was normalized to the geometrical area of the electrodes. Figures are adopted from ref. 17 with permission from the Royal Society of Chemistry © 2024. | |
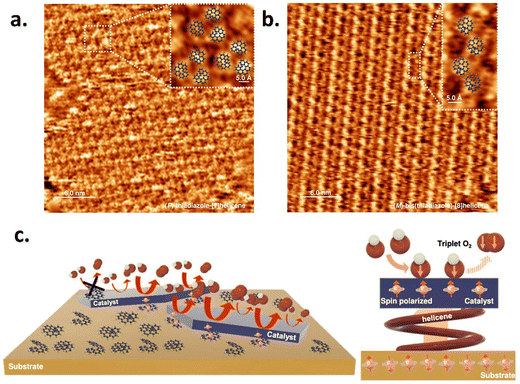 |
| | Fig. 3 Chirality-driven modulation of NiOx electrocatalysts: STM image of SAM of (a) (P)-thiadiazole-[7] helicene and (b) (M)-bis(thiadiazole)-[8] helicene. (c) Illustration of the effect of spin polarization employing the catalyst-chiral molecule-Au substrate configuration. The figure is adopted from ref. 18 with permission from Nature © 2022. | |
3.1.2. Electrodepositing chiral films.
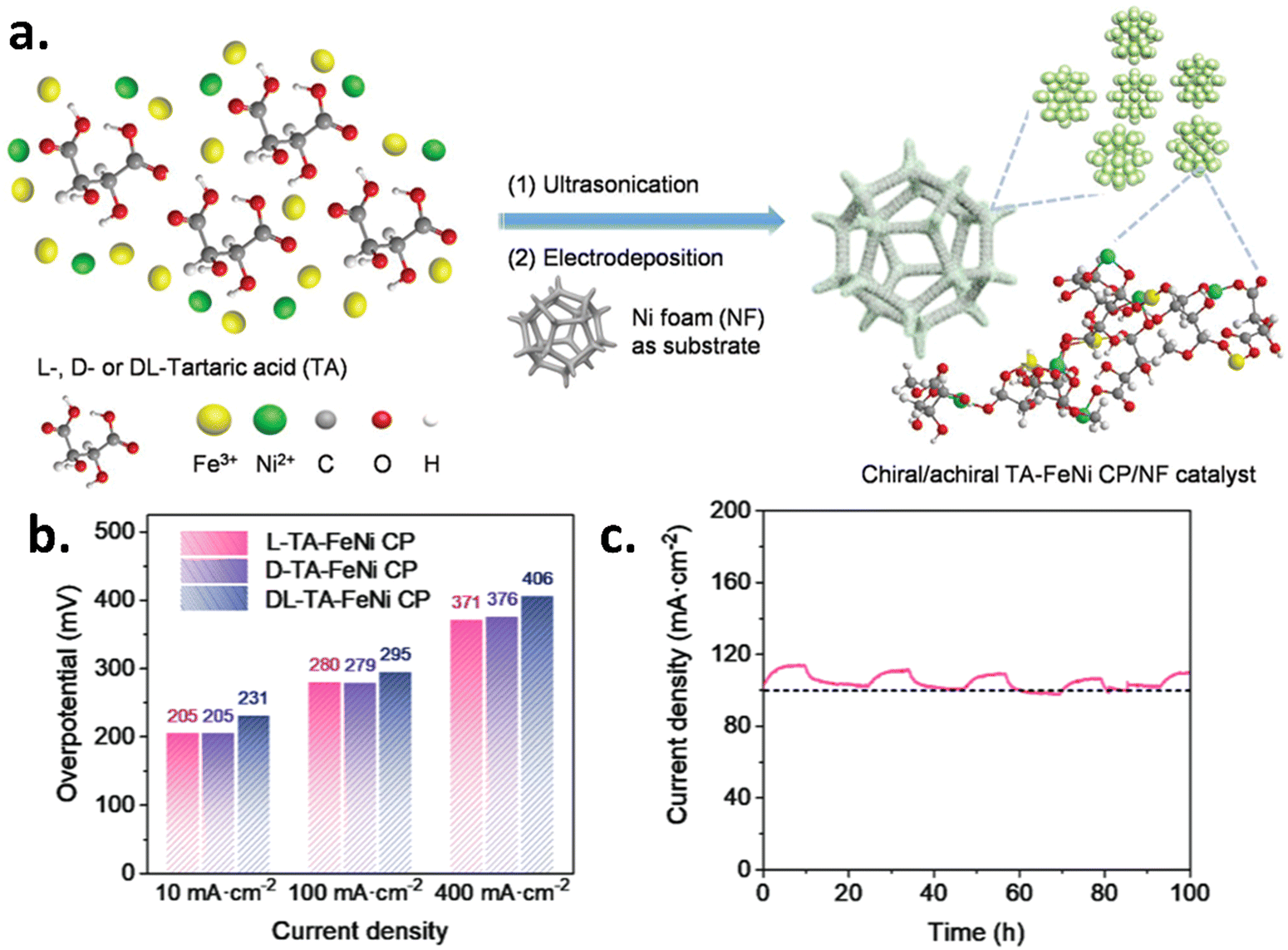
Chiral catalysts were also fabricated using the electrodeposition method. Banerjee Ghosh et al. reported that the chiral CuO films electrodeposited on polycrystalline gold and TiO2/FTO-coated glass substrates result in higher current density and selectivity towards O2 formation over H2O2 with respect to the achiral one, as depicted in Fig. 4a and b.20 Later, Waldeck and his team also investigated the oxygen evolution reaction using chiral cobalt oxide-based thin film electrocatalysts.6 They investigated how the spin-polarization characteristics could be used to control intermediates generated during the electrochemical reactions using various electrolyte solutions with different pH. These studies revealed that chiral cobalt oxide reduced hydrogen peroxide production and increased the oxygen yield with respect to the meso-CoOx analogs, which are achiral. The data is shown in Fig. 4c and d. Gazzotti et al. investigated the water-splitting process in relation to various chiral additives/catalyzers on nickel and stainless-steel electrodes, the latter of which was deposited directly on a magnetic surface, to determine which was the most promising option for an effective spin filtering that would lower the production of H2O2.21 Feng et al. synthesized an amorphous chiral tartaric acid–FeNi coordination polymer (TA–FeNi CP) on a nickel foam substrate utilizing an electrochemical deposition method to demonstrate chiral-induced spin selectivity effect for enhancement of the electrochemical oxygen evolution reaction, as shown in Fig. 5a.22 This study found that chiral Fe–Ni electrocatalysts having superior spin polarization, which results in excellent OER activity with a durability of around 100 hours. The data is shown in Fig. 5b and c. The chiral L-TA–FeNi CP catalyst has low overpotentials of 205 and 280 mV, delivering 10 and 100 mA cm−2, respectively.22 Recently, Utkarsh et al. reported that the amorphous chiral iron-doped cobalt oxide electrodeposited on the FTO substrate performs better than the achiral one. The chiral catalyst shows a high oxygen evolution rate and a lower Tafel slope compared to the achiral catalyst.23
 |
| | Fig. 4 Comparative analysis of electrodeposited chiral and achiral metal oxide films. (a) LSV plots of 500 nm thick L-CuO (shown as red line) and D-CuO (shown in blue line) film electrodes, with achiral glycine-CuO films (light green) showing reduced activity with respect to the chiral films. (b) UV-vis absorption spectra of the 0.1 M Na2SO4 electrolyte after titration with o-tolidine using chiral CuO (presented as red line) and achiral CuO (presented as a black line) as the working electrode. (c) LSV plots showing OER performance of L-CoOx (shown as black line), meso-CoOx (shown as blue line), and bare FTO (green) in 0.1 M KOH. (d) Oxygen production measured with a Hoffman apparatus for L-CoOx (black) and meso-CoOx (blue) catalysts. Figures are adopted from ref. 6 and 20 with permission from American Chemical Society © 2019 and American Chemical Society ©2020. | |
 |
| | Fig. 5 Design, performance, and stability of electrochemically deposited chiral and achiral TA–FeNi CP/NF catalysts for the OER. (a) Schematic illustration of the synthesis of chiral and achiral catalysts conducting polymer-based catalysts. (b) Comparative analysis of the overpotential required for the OER across different catalyst configurations. (c) Long-term stability assessment of the L-TA–FeNi CP electrocatalyst. Figures are adopted from ref. 22 with permission from Willey-VCH GmbH ©2023. | |
3.1.3. Chiral functionalization of metal oxide nanoparticles.
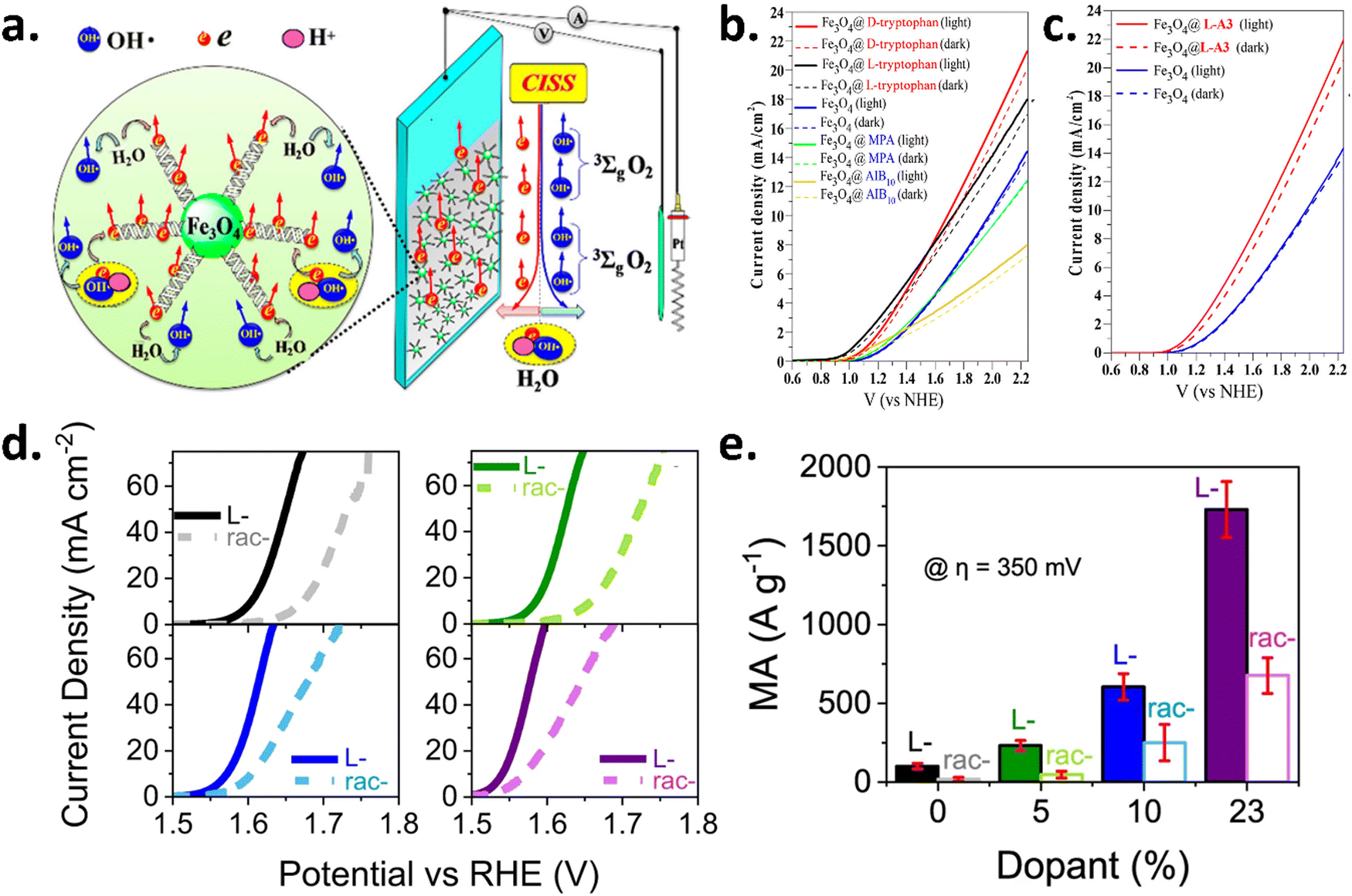
Employing the chiral functionalization of metal oxide nanoparticle-based catalysts, the CISS effect can also be utilized in the electrocatalytic water splitting process. Zhang et al. showed that chiral molecules integrating Fe3O4 nanoparticles exhibited higher electrocatalytic activity than the achiral catalyst, as shown in Fig. 6(a–c).24 Furthermore, in neutral environments, the chiral molecular coating inhibited the generation of H2O2. This discovery prompted numerous other efforts to harness the CISS effect to improve the OER. Later, Zhang et al. synthesized a hollow porous Fe3O4 microsphere (MFe3O4) functionalized with a self-assembled hemoglobin monolayer and reported that it reduced the OER onset potential by 100 mV and enhanced the current density by 2-fold.25
 |
| | Fig. 6 Chiral functionalization of Fe3O4 and CoxOx-based nanoparticles and enhancement of spin-selectivity for the OER. (a) Schematic of chiral-functionalized electrodes, where the CISS effect enhances the OER via triplet oxygen production. (b) Photocurrent and dark current densities for Fe3O4 NPs linked to chiral (D- and L-tryptophan) and achiral (mercaptopropionic acid, azo isobutyric acid) molecules, compared to pure Fe3O4 NPs. (c) Photocurrent density of Fe3O4 NPs linked to chiral L-A3 versus pure Fe3O4 NPs. (d) LSV plot showing undoped (black) and iron-doped Rac- (dashed) and L-cobalt oxide (solid) nanoparticle catalysts in Nafion, with doping levels of 5% (green), 10% (blue), and 23% (purple), measured in 1 M NaOH. (e) Mass activity (MA) comparison of Rac- (open symbols) and L-cobalt oxide (filled symbols) nanoparticle catalysts in Nafion. Figures are adopted from ref. 24 and 26 with permission from American Chemical Society © 2018 and Nature © 2023. | |
Vadakkayil et al. synthesized Co(3−x)·FexO4 nanoparticles utilizing chiral cysteine as a capping ligand and also demonstrated that iron doping into the chiral catalyst decreases the overpotential required for oxygen evolution and also increases the mass activity and specific activity of the catalyst.26 The enhancement of activity of the chiral catalyst over the achiral analogs has been shown in Fig. 6d and e. Recently, Sobrido's group synthesized iridium nanoparticles with a diameter of ∼2.1 nm functionalized with chiral L- and D-cysteine, which registered a marked enhancement in the activity in comparison with the unfunctionalized iridium nanoparticles and achiral functionalized iridium nanoparticles.27 Chiral nanoparticles showed 85% increase in activity at a potential of 1.55 V vs. reversible hydrogen electrode (RHE), while achiral functionalized iridium nanoparticles showed an average 13% increase to the unmodified catalyst.
Jin et al. developed a simple wet chemical synthesis to achieve intrinsic chiroptical activity and consequent spin-polarization within colloidal CuO nanostructures to control spin-dependent electrocatalysis for the OER.28 They probed the relationship between the chiroptical magnitude that is the g-factor with the electrocatalytic activity in the OER. Fig. 7a shows the relation between g values of chiral (L and D) and achiral (DL) CuO nano spindles at the CD band at the wavelength of 380 nm and the current density obtained from those catalysts, as shown in Fig. 7(b–d).
 |
| | Fig. 7 Relation between the g-value and the OER activity. (a) Relative g values of the chiral copper oxide-based nanospindles synthesized following different conditions measured at the 380 nm CD band. LSV plots of (b) L- and D-CuO having higher g-value and DL-CuO, and (c) L- and D-CuO having lower g-value and DL-CuO in alkaline electrolyte (0.1 M KOH). (d) Linear fit showing the relationship between g values and current density, with an inset displaying current density at 1.9 V for different samples. Figures are adopted from ref. 28 with permission from American Chemical Society © 2023. | |
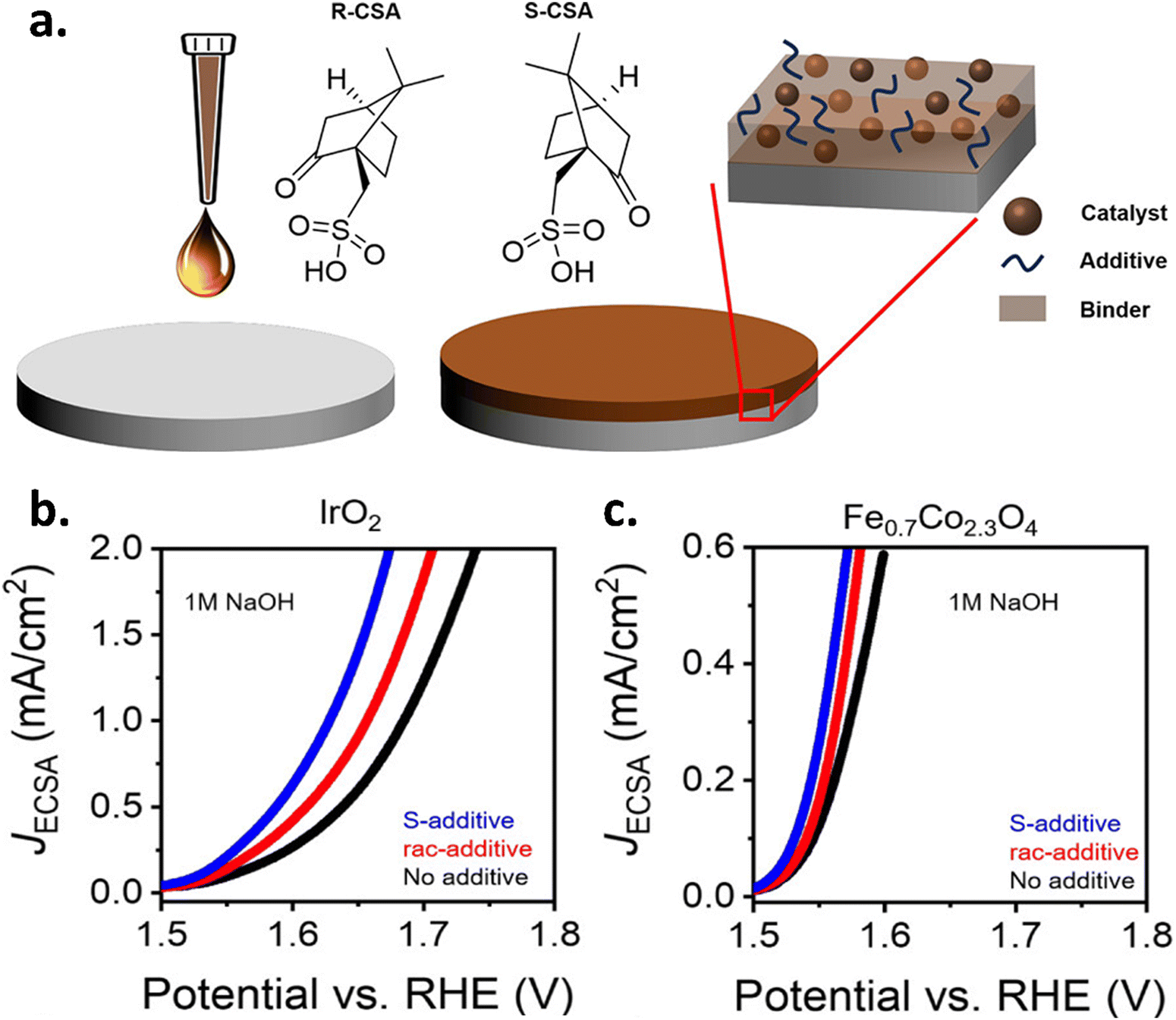
Very recently, Ran et al. fabricated a chiral L-type Co@CoO catalyst (Co@CoO-L) and compared the catalyst's activity with that of achiral racemic substances and an unmodified catalyst.29 They found that Co@CoO-L catalysts performed better than the pristine and racemic ones in alkaline media. They explained this outcome by pointing to the chiral catalyst's spin polarization effect on the OH* species, the oxygen vacancies it created in the structure, and the minor structural distortion it brought about. These factors probably contributed to the increased number of active sites, which improved the reaction kinetics and sped up the process. Recently, Li et al. synthesized chiral copper oxide nanoflowers using a straightforward self-assembly approach, achieving an unprecedented spin polarization of 96%.30 As a result, the chiral nanoflower exhibits a low overpotential and a lower Tafel slope of 93.5 mV dec−1, surpassing that of commercial RuO2. Additionally, they performed DFT calculation and discovered that changing the potential-determining step from *O → *OOH to *OH → *O decreased the energy barrier from 1.81 eV to 1.62 eV. A different approach was also demonstrated by Vadakkayil et al. where it was explored that the use of chemical additives to provide a chiral bias promotes the creation of spin-polarized intermediates.31 These chiral molecular additives show no redox activity at OER potentials and are incorporated with standard catalytic binder supports. They have fabricated IrO2 and RuO2 nanoparticle catalyst films using Nafion binder and employed S-camphor sulfonic acid as a chiral additive and a racemic combination of camphor sulfonic acid (rac-CSA, red) as the achiral additive for reference. The findings depicted in Fig. 8(a–c) reveal that S-CSA additives had a lower reaction over potential than the rac-CSA additives and the additive-free samples.
 |
| | Fig. 8 Chiral binder-assisted OER enhancement. (a) Schematic of thin-film electrocatalyst formation and the impact of chiral additives on the OER. LSV curves for (b) IrO2 and (c) Fe0.7Co2.3O4. Figures are adopted from ref. 31 with permission from American Chemical Society © 2024. | |
3.1.4. Chiral organic–inorganic hybrid perovskite electrocatalysts.
Chiral organic–inorganic hybrid 2D perovskites (OIHPs) have recently been recognized as excellent spin polarizers, thanks to the synergistic impact of the chiral organic molecules and the substantial spin–orbit coupling of the inorganic layers. Lee and colleagues synthesized chiral two dimensional OIHPs to act as a spin-filtering layer on the top of three dimensional OIHP containing electrode and showed that the 2D OIHP-based electrode achieved better oxygen evolution performance, with a lower overpotential, a higher fill factor, and a higher photocurrent with respect to an electrode without a spin-filtering layer.32
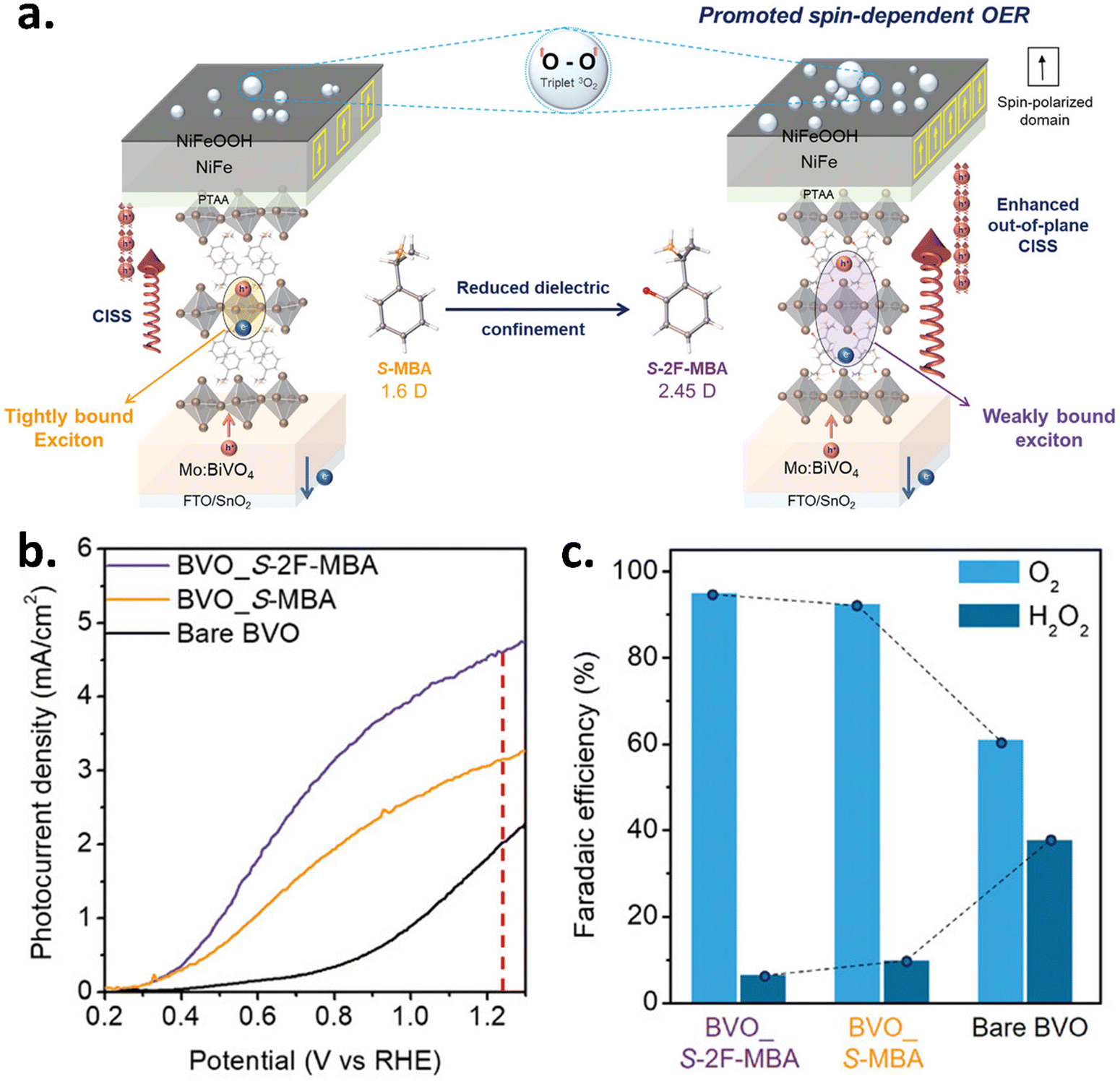
In 2024, Son et al. developed a stable and effective chiral perovskite-based spin polarizer by using a fluorinated chiral cation.33 They transitioned to (S)-ortho-fluorinated-methyl-benzylamine (S-2F-MBA) from the traditional (S)-methyl-benzylamine (S-MBA). Using S-2F-MBA chiral OIHP as an implanted spin polarizer in the BiVO4 (BVO) photoanode, they decreased H2O2 byproduct generation while attaining a much higher photocurrent for the OER compared to the S-MBA-BVO counterpart. The data is shown in Fig. 9(a–c). In a related study, Lee et al. created a 2D chiral OIHP spin polarizer using naphthyl ethylamine (NEA). This polarizer is placed on top of the 3D OIHP photoanode, which helps to realign the spin-polarized orientation of charge carriers in the catalyst layer before the oxygen evolution reaction (OER) process. Consequently, a secondary spin filter on the catalyst surface was enabled through the utilization of an enantioselective NiFeOOH catalyst, which operates as a chiral spin-selective catalyst. Hence, both effects result in an outstanding oxygen evolution performance, showing a high photocurrent, a high fill factor, and a lower onset potential.34
 |
| | Fig. 9 Chiral organic–inorganic hybrid perovskite (cOIHP) for enhancing the OER carried out photoelectrochemically. (a) Schematic of the underlying principle of enhancement of out-of-plane conductivity as well as the CISS effect in S-2F-MBA cOIHP. (b) LSV curves of bare BVO and BVO in conjunction with cOIHP in 1 M K-Bi (pH 9.0) under AM 1.5 G illumination. (c) Bar chart of the faradaic efficiency of O2 and H2O2 generation while using BVO_S-2F-MBA, BVO_S-MBA, and bare BVO devices. Figures are adopted from ref. 33 with permission from Willey-VCH GmbH © 2024. | |
3.1.5. Chiral metal–organic framework (MOF) and covalent organic framework (COF) for the OER.
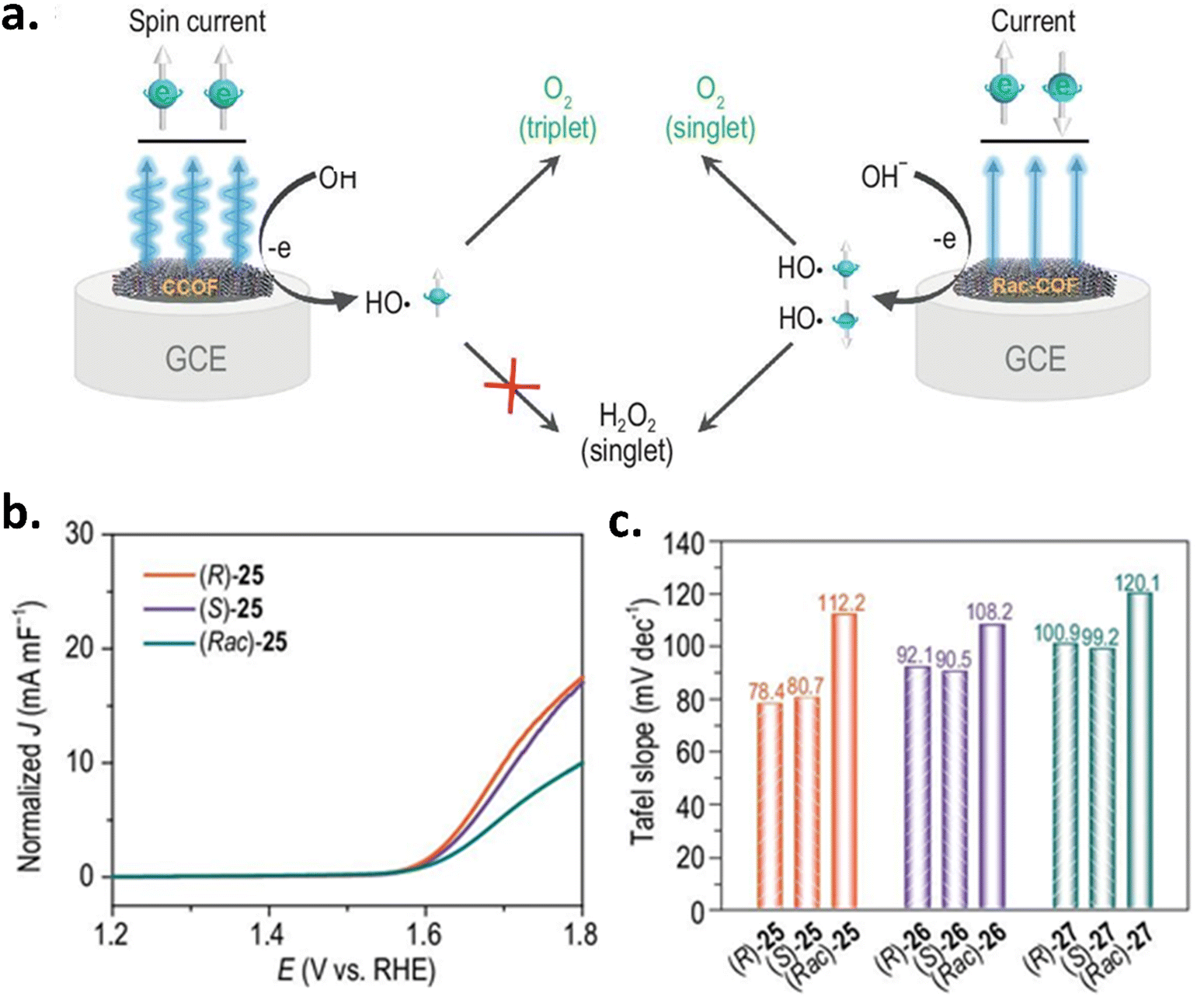
Chiral metal–organic frameworks (MOFs) have also been explored for use as OER catalysts for CISS-controlled spin-polarized charge transfer. Zhang et al. synthesized a novel class of chiral 3D MOFs with a hierarchical rod-like morphology that displayed remarkable electrocatalytic activity for both the ORR and OER and surpassed the activity of Pt/C for the ORR and RuO2 for the OER.35 In 2024, Li and colleagues synthesized and structurally characterized three electroactive three-dimensional (3D) chiral COFs by incorporating three redox-active organic monomers and optically pure 1,1-binaphthol (BINOL) as a chiral site.36 The schematic representation of the spin-polarized oxygen evolution mechanism in chiral and achiral COFs is shown in Fig. 10a. When the electrochemical performance is compared to their achiral counterparts, these materials exhibit a lower Tafel slope and overpotential. The data is displayed in Fig. 10(b and c).
 |
| | Fig. 10 Exploring the spin-selective OER in covalent organic frameworks. (a) Schematic representation of the spin-polarized oxygen evolution mechanism in chiral and achiral covalent organic frameworks (COFs). (b) Normalized polarization curves plotted against the double-layer capacitance (Cdl) for (R)-25, (S)-25, and racemic (Rac)-25 COFs. (c) Tafel slope analysis of the chiral and racemic COF systems. Figures are adopted from ref. 36 with permission from Oxford University Press © 2024. | |
3.1.6. Chiral semimetal for the OER.
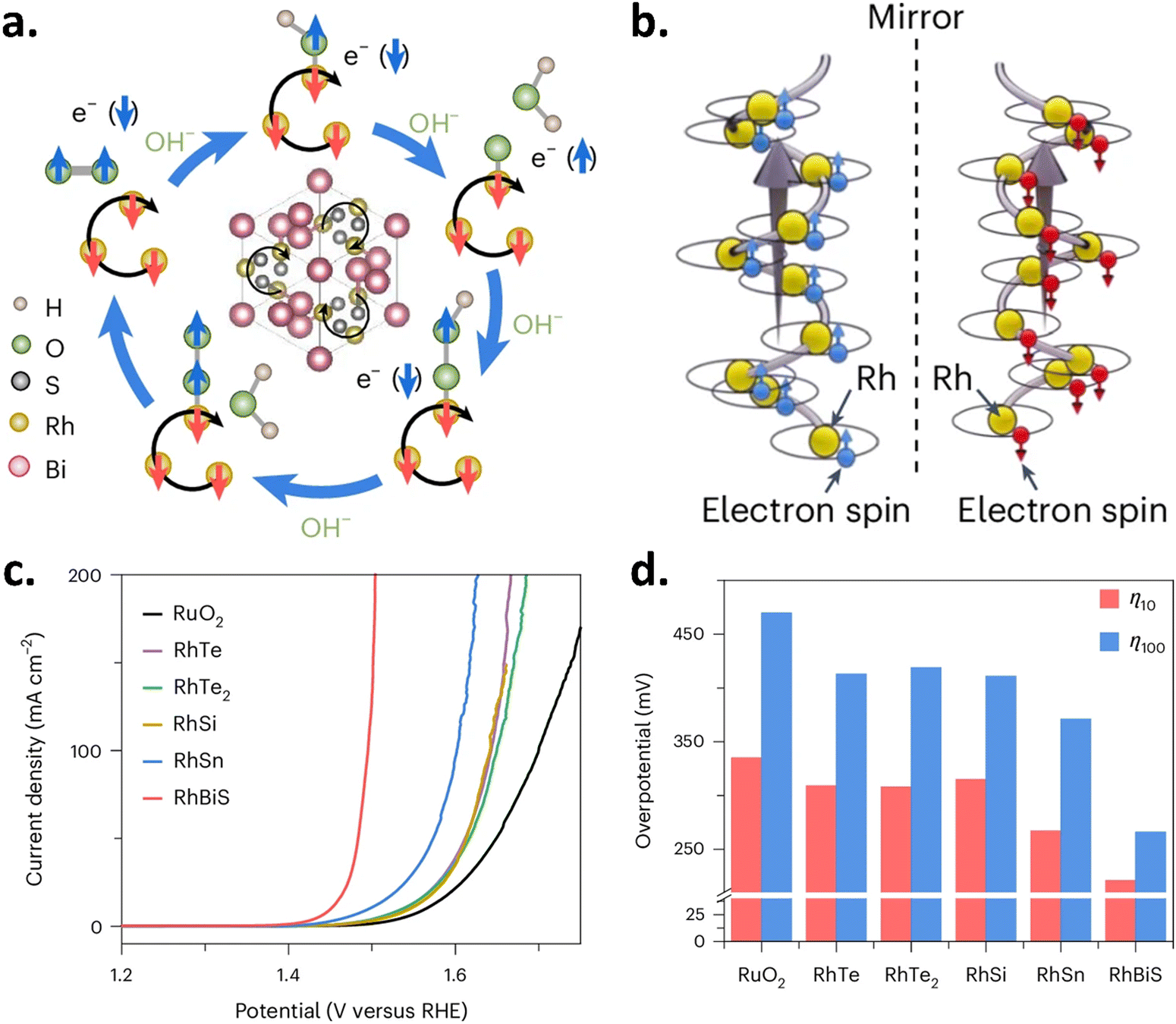
Recently, an interesting study showed that the spin-polarized Fermi surfaces of chiral semimetals (RhSi, RhSn, and RhBiS) and their potential to improve spin-dependent electron transport in the OER were used by Wang et al. to overcome the traditional volcano-plot constraints. These Rh-based chiral crystals have significantly greater OER activity than RuO2 when employed as OER catalysts in alkaline electrolytes. The spin polarization of the charge carriers in these crystals further promotes the creation of spin-aligned oxygen intermediates, as shown in Fig. 11a. These crystals' chirality results from the Rh atoms arranging themselves in a left- or right-handed spiral, as shown in Fig. 11b. Experimental investigations and quantum transport simulations show that spin polarization and spin–orbit coupling are related to the OER activity of these crystals. Thus, RhBiS with the largest SOC exhibits the lowest overpotential among all.37 The data is shown in Fig. 11(c and d).
 |
| | Fig. 11 Unraveling the spin-dependent OER activity on intrinsically chiral Rh catalysts. (a) Schematic of spin polarization during the OER on a chiral catalytic surface. (b) Representation of intrinsic chirality, where Rh atom helicity determines handedness. Grey arrows show current flow, while blue and red arrows indicate induced spin-polarization. (c) LSV plots recorded in 1 M KOH. (d) Corresponding overpotential at 10 mA cm−2 (red) and 100 mA cm−2 (blue) derived from the polarization curves. Figures adopted from ref. 37 with permission from Nature energy © 2025. | |
4. Magnetic substrate for the OER
As we already discussed, spin polarization can also be utilized in the OER by employing a magnetic surface as an electrode. In recent studies, transition metal-based magnetic materials are evaluated for magnetic field-assisted electrocatalysts.38 The external magnetic field promotes the spin alignment of the electrons in the catalysts. The polarized magnetic moment of the ferromagnetic materials can act as an inherent spin filter.39 However, due to weaker or opposing spin alignments, this effect is less pronounced in antiferromagnetic or paramagnetic materials.40 Hence, the ordered spin orientation of the ferromagnetic materials boosts the spin-selective electron transfer during the OER, particularly at the first step of electron transfer where spin-exchange interaction takes place between the catalyst and adsorbed intermediate oxygen species. The spin-polarized environment in ferromagnetic materials stabilizes high-spin states of reaction intermediates (*OH, *O, *OOH), lowering the energy barriers for the OER. Garcés-Pineda et al. reported that by employing an external magnetic field, the OER activity of a magnetic oxide can be augmented. This finding also paves a new way to manipulate the spin alignment of the catalyst for application in the OER.41 To explore the electrocatalytic activity of magnetic anodes under an external magnetic field in alkaline conditions, they investigated a range of OER catalysts placed on two-dimensional Ni–foil anodes under the same operating conditions.41 They studied some cutting-edge OER catalysts (e.g. Raney Ni, NiFe2Ox, FeNi4Ox, and Ni2Cr2FeOx) and some catalysts with very different magnetic properties like the antiferromagnetic NiO, the non-magnetic IrO2, spinel ZnFe2Ox and the highly magnetic ferrites NiZnFe4Ox and NiZnFeOx. They demonstrated that NiZnFe4Ox showed the greatest relative effect, with current doubling from 24 to 40 mA cm−2 at 1.65 V and above. Later, Ren et al. prepared electrocatalysts with different magnetic properties and compared their OER activity. The as-prepared CoFe2O4 shows ferromagnetic behavior and gives a wider hysteresis loop, indicating higher coercivity and remanence at room temperature. The Co3O4 and IrO2 samples with much less susceptibility, as shown in Fig. 12a, at 300 K exhibit poor spin polarization owing to their paramagnetic and anti-ferromagnetic properties.5 They documented a study on the significant kinetic alterations in the ferromagnetic CoFe2O4 catalyst under a magnetic field where CoFe2O4 functions as a spin polarizer in the presence of an external magnetic field. They reported that with the increase of the magnetic field, the magnetic moment of the CoFe2O4 ferromagnetic catalyst became more orderly before reaching the saturation field as shown in Fig. 12a. They carried out a chronoamperometry experiment for ferromagnetic CoFe2O4, non-ferromagnetic Co3O4 and IrO2 in 1 M KOH under different magnetic field strengths (0, 500, 1000, 3000, 5000, 7500, and 10 000 Oe) at a constant potential of 1.66, 1.66, and 1.56 V (vs. RHE), respectively, that is summarized in Fig. 12b. They reported that the current density of the ferromagnetic catalyst CoFe2O4 increases with the increase of the magnetic field strength; however, for non-ferromagnetic Co3O4 and IrO2, there is almost no change when changing the field strength, as shown in Fig. 12c. The linear sweep voltammetry plots shown in Fig. 12d present the effect of a magnetic field on the OER performance of the ferromagnetic catalyst CoFe2O4 before, after and under the application of a magnetic field and a similar trend of Tafel slope is found that is shown in Fig. 12e. An interesting finding from Fig. 12f is that even after the removal of the magnetic field, the magnetic moment is still aligned in magnetized CoFe2O4 that confirms the role of indirect quantum spin-exchange interaction in enhancing the OER performance rather than the weak direct spin–spin interactions from the external field. Fig. 12g demonstrates the demagnetization of the magnetized CoFe2O4 carried out using an oscillating magnetic field.
 |
| | Fig. 12 Effect of magnetic field on the catalyst having different magnetic properties. (a) Magnetic moment measurement with respect to the magnetic field. (b) Chronoamperometry measurements of CoFe2O4 (at 1.66 V vs. RHE), Co3O4 (at 1.66 V vs. RHE), and IrO2 at (1.67 V vs. RHE) in 1 M KOH under varying magnetic field strengths. (c) The increase in the current density of IrO2, non-ferromagnetic Co3O4, and ferromagnetic CoFe2O4 catalysts at varying magnetic field intensities. (d) The LSV curves of CoFe2O4 measured in oxygen saturated 1 M KOH electrolyte solution using a scan rate of 10 mV s−1 with and without a constant magnetic field of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe, after demagnetization, and after the magnetic field was withdrawn after magnetization and (e) the corresponding Tafel plots. (f) The magnetization of CoFe2O4 following the removal of a continuous magnetic field of 10000 Oe. (g) Demagnetization curve for CoFe2O4 using an oscillating magnetic field. Figures are adopted from ref. 5 with permission from Nature © 2021. 000 Oe, after demagnetization, and after the magnetic field was withdrawn after magnetization and (e) the corresponding Tafel plots. (f) The magnetization of CoFe2O4 following the removal of a continuous magnetic field of 10000 Oe. (g) Demagnetization curve for CoFe2O4 using an oscillating magnetic field. Figures are adopted from ref. 5 with permission from Nature © 2021. | |
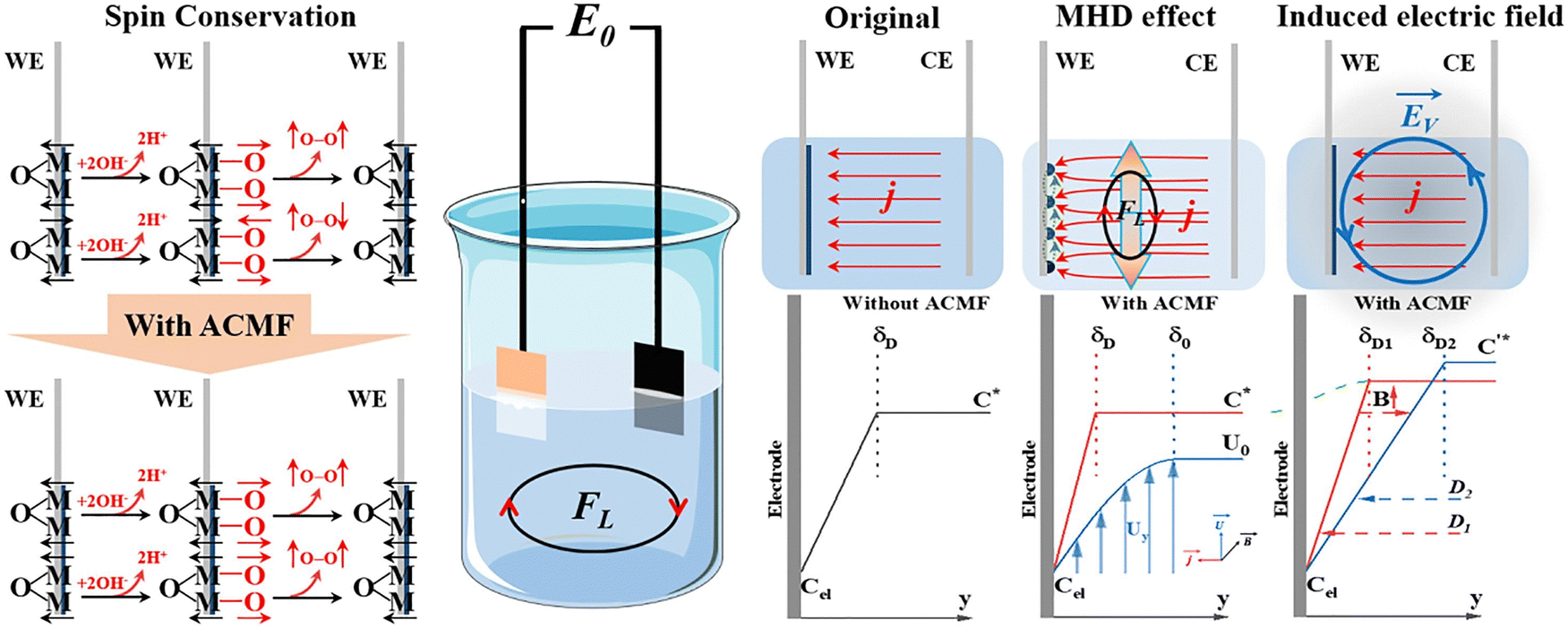
Besides, Yan et al. synthesized a carbon-based bifunctional catalyst consisting of magnetic catalytic nanocages (MCNs) consisting of metallic cobalt nanodots doped into macroporous carbon nanofibers that could directly boost oxygen catalytic activity by applying a modest magnetic field (350 mT).42 An external magnetic field induced magnetization in cobalt, transforming it into nanomagnets with elevated spin polarization, enhancing oxygen intermediates' adsorption and facilitating electron transfer, greatly augmenting catalytic efficiency. Zheng et al. synthesized a series of NiCoFe-MOF-7 and found that the spin conservation and magnetohydrodynamic (MHD) effect of an AC magnetic field can greatly enhance the OER activity at lower current densities while reducing it at higher current densities, as depicted in Fig. 13.43
 |
| | Fig. 13 Effect of AC magnetic field on magnetic catalyst in controlling the OER. Schematic illustration showing the influence of an AC magnetic field on the OER in an alkaline electrolyte. The diagram highlights how the AC magnetic field interacts with the catalyst surface and potentially enhances spin polarization and consequently modifies electron transfer dynamics. Figures are adopted from ref. 43 with permission from Elsevier B.V. © 2021. | |
A ferromagnetically ordered electrocatalyst was reported by the same group, capable of functioning as both a heater and a spin polarizer under an external AC magnetic field, thereby significantly enhancing the oxygen evolution reaction in an alkaline medium at low current density.44 Later Qin et al. reported that the electrocatalytic performance of the NiFe-LDH catalyst for the OER in alkaline conditions was greatly enhanced by the applied magnetic field.45 The proposed mechanism is given in the data shown in Fig. 14a. According to the proposed mechanism, the magnetohydrodynamic effect and charge transfer effect at the electrode/electrolyte interface driven by the applied magnetic field may be the primary reason for promoting the OER. The OER activity of NiFe LDH is considerably increased by the proper external magnetic field, as shown in Fig. 14b. In the presence of a 200 mT magnetic field, the sample exhibits negligible attenuation after 12 hours; however, in the absence of a magnetic field, the attenuation is around 5%, as shown in Fig. 14c.
 |
| | Fig. 14 Controlling the stability and performance of NiFe LDH for the OER using an external magnetic field. (a) Proposed mechanisms for enhanced OER performance of NiFe LDH catalysts under an applied magnetic field. (b) LSV curves of NiFe LDH electrodes before and after 2000 cycles, with and without a 200 mT magnetic field. (c) Chronoamperometry curves of NiFe LDH at 0 mT and 200 mT magnetic field strengths. Figures are adopted from ref. 45 with permission from Springer Nature © 2022. | |
NiFe/NiFeOOH core/shell nanoparticles in the highly conductive carbon matrix were found to exhibit higher activity when an AC magnetic field was applied as reported by Peng et al.46 Recently, Li et al. prepared a super hydrophilic γ-Fe2O3 hydrosol and dip-coated it onto several electrode substrates to alter their surface wettability and magnetism and found to enhance OER performance.47 A different mechanism of enhancing OER using a magnetic field is proposed by the study reported in the literature by Vensaus et al.48 They proposed that the primary factor influencing the kinetics is the alteration in mass transport inside a magnetic field. Based on the spin coupling of iron group elements, Lin et al. developed three typical iron-layered double hydroxides (LDHs) and investigated the connection between OER activity, spin polarization, and magnetic field.49 Tao Sun demonstrated that magnetic Ni/MoS2 facilitates spin-specific charge transfer that drives the production of triplet O2.50 Zhang et al. discovered that the geometric positions of metal cations can be efficiently stimulated by a magnetic field that also strengthens the OER.51 In a similar study, Lyu et al. found that an external magnetic field can cause the Ni2+ cation in mixed NiFe2O4 (NFO) to transfer from the octahedral (Oh) to the tetrahedral (Td) sites.52 Under alkaline conditions, the current density of NFO affected by the applied magnetic field is twice as high as the original NFO. They propose that the primary causes of OER enhancement are the modification of the active units at spinel tetrahedral sites and the alteration of electron geometry by the external magnetic field. Wang et al. synthesized NiFe2O4 @ (Ni, Fe) S/P materials with a heterogeneous core–shell structure and reported that the potential of NiFe2O4 @ (Ni, Fe) S/P at 10 mA cm−2 drops by 45.5% under an alternating magnetic field, from 323 mV (0 mT) to 176 mV (4.320 mT).53 Zhang et al. discovered that when an external magnetic field was applied, the exchange bias effect between the active species reacting with the OER and the magnetic matrix NF takes place which lowers the electron spin scattering and magnetoresistance.54 They developed an effective OER catalyst by supporting Co nanowires and NiFe-layered double hydroxide (NiFe-LDH) nanosheets on NF to form a heterojunction. They suggest that the magnetic field-induced PN junction can alter the heterojunction's electron distribution and Fermi level position, improving the adsorptive capacity of the *OOH intermediate on the catalyst surface and facilitating more effective OER. Li et al. showed that the electrodeposited highly active magnetic nanosheets with Ni–S–CoFe2O4/NF as a magnetic carrier significantly enhanced the OER performance.55 Recently Niar et al. synthesized ferrimagnetic (f-Fe3O4) and superparamagnetic iron oxide (s-Fe3O4) catalysts, which exhibit OER enhancement on applying an external magnetic field (Hext). Additionally, the CISS effect was employed in synergy with the external magnetic field to produce a maximal increase of up to 89% improvement in current density (at 1.8 V vs. RHE) with a low onset potential of 270 mV in s-Fe3O4 catalysts. The data is shown in Fig. 15(a and b).56
 |
| | Fig. 15 Triplet pathway of water oxidation via spin-polarized hole accumulation on magnetic catalysts. (a) Influence of stirring and external magnetic field (Hext) on s-Fe3O4. (b) Spin-selective OER mechanism under a magnetic field. (c) O2 current density for pristine magnetic electrodes, hematite, and YIG. (d) Schematic of the formation of polarons and electron–hole recombination at Oh sites, resulting in spin-polarized hole accumulation on the YIG surface, which facilitates triplet-pathway water oxidation and O2 generation. Figures are adopted from ref. 56 and 57 with permission from American Chemical Society © 2023 and Royal Society of Chemistry © 2024. | |
Li et al. introduced a ferrimagnet by altering the chiral molecule on Ni-entrapped carbon nanotubes (chiral-CNT@Ni). This prevented the formation of nonmagnetic singlet oxygen and the H2O2 by-products. The synthesized chiral-CNT@Ni can attain a current density of 100 mA cm−2 for the OER at a low overpotential of 479 mV, which is nearly 100 mV lower than that of bare CNT@Ni.58 A spectroscopic and photocatalytic comparison of water splitting employing yttrium iron garnet (Y3Fe5O12, YIG) and hematite (α-Fe2O3) photoanodes was published by Gajapathy et al. very recently.57 With a photocurrent density increase of more than an order of magnitude, YIG performs noticeably better than the well-researched hematite while having identical electrical structures. Because of site-dependent electron–phonon interaction, ultrafast XUV observations reveal a decrease in the production of surface electron polarons in comparison to hematite. In contrast to quick trapping and electron/hole pair recombination in the Oh sub-lattice, effective charge separation takes place on the Td sub-lattice in YIG, resulting in spin-polarized photocurrents. Spin-aligned holes are hybridized between the O 2p and Fe 3d valence band states, and they are responsible for YIG's extremely efficient, spin-selective water oxidation process.57 The data is shown in Fig. 15(c and d).
Recent studies demonstrate that the catalytic property of magnetic materials depends on different parameters, like facets, phase, dopants etc. Fe3O4(001) and Fe3O4(110) surfaces were examined by Muelner et al.,59 who discovered that the latter needed a lower OER onset potential. The OER reactivities of NiO films with various preferred orientations deposited on the MgO substrate exposing (001), (110), and (111) facets were investigated by Poulain et al.60 They discovered a reactivity trend (110) > (111) > (001). In contrast to the majority of previous investigations, Liu et al.61 Recently examined the single-particle activities of Co3O4(001)-terminated nanocubes and spheroids and concluded that the (001) surface should be more active than the (111) surface. This was explained by the fact that the two surfaces had varying numbers of octahedral and tetrahedral cation reaction sites. Davis et al. have examined the structure–composition–reactivity correlations for epitaxial thin-film electrocatalysts that expose either the (001) or (111) surface facets of Co3O4, CoFe2O4, and Fe3O4. According to their findings, the (111) aspect of Co3O4 is more active than the (001) facet, which is more reactive.62
On the other side, the number of layers in 2D materials significantly influences their oxygen evolution reaction (OER) performance due to changes in electronic structure, active site availability, and interlayer interactions. Moreover, ultrathin structures are prone to the formation of more defects to generate more active edges and coordination-unsaturated metal sites, which can reduce the reactive energy barrier by superior interfacial charge transfer and rapid mass transport.63 Song et al. were the first to exfoliate the bulk NiFe, NiCo, and CoCo LDH materials by exchanging the intercalated water molecules by smaller anions (Br−, and CO32−) followed by larger ones (NO32−, and ClO4−) which led to stable colloidal solutions.64 Coronado and his group members developed another method of exfoliating bulk LDH materials. Alkoxide intercalation and ultrasonic treatment in two steps were used here for exfoliating CoFe and NiFe LDH.65 As expected, they too observed significant enhancement in the electrocatalytic OER performance of exfoliated versions of both CoFe and NiFe LDH. Munonde et al. used the ultrasonic exfoliation process on the NiFe LDH/CB materials in water without any additional conditions.66 The exfoliated NiFe LDH/CB nanosheets display significantly higher OER activity than their corresponding bulk NiFe LDH/CB in an alkaline solution, with an overpotential of 220 mV at a current density of 10 mA cm−2, which is 60 mV lower than the 280 mV of the bulk NiFe LDH/CB.
5.
Operando studies to evaluate spin-polarized OER kinetics
Spectro-electrochemical (SEC) data were found to be a promising tool to investigate the reaction kinetics during electrocatalysis. Liu and coworkers reported enhancement of urea oxidation reaction by controlling the cation vacancy with higher valent Ni4+ species and demonstrated the origin of the high activity and catalytic mechanism during the UOR by utilizing X-ray absorption near edge structure (XANES) and operando FTIR techniques.67 In a related study, to explore the influence of electron delocalization on the intermediates of the UOR, in situ attenuated total reflection ATR-FTIR spectra were employed to carry out quantitative analysis of the molecular-level changes during the electrochemical process.68
Recently, Garcés-Pineda et al. explored the relationship between the increased OER kinetics and the circular dichroism signal of enantiopure Fe–Ni metal oxides by in situ SEC characterization.69 The SEC was carried out in transmission mode using FTO as a support to study the development of the catalytic sites during the slowest step or rate-determining step of the OER. The circular dichroism (CD) spectra exhibited by the chiral catalyst during the OER exhibit a broad range of absorption (400–600 nm) in the potential range of 1.125 and 1.325 V vs. RHE. The reaction dynamics analysis reveals a similar slope corresponding to 1st order kinetics with respect to the density of catalytic sites, which suggests that the OER occurs through the same mechanism irrespective of the chirality of the catalyst (Fig. 16). However, the kinetics are revealed to be slower for the achiral catalyst. Similarly, Sun et al. also reported a room-temperature ferromagnetic material Ni1–MoS2, and when it was used as an OER catalyst, X-ray absorption near edge structure (XANES) operando characterizations were employed to study its spin-polarization with and without applying a magnetic field.50 It was found that in the presence of a magnetic field, Ni1/MoS2 exhibits superior OER activity performance due to a magnetic field-directed spin-alignment. The spin-density optimization over S active sites also results from the magnetic field-induced S(p)–Ni(d) hybridization that controls the free energy of adsorption of the oxygen-containing intermediates and reduces the overall activation energy of the reaction.
 |
| | Fig. 16 Spectro-electrochemical study of chiral NiFe oxides in alkaline OER systems: (a) and (b) circular dichroism spectro-electrochemical (CD-SEC) analysis data of D-NiFe and L-NiFe chiral oxides at an applied potential in the 0.9–1.8 V vs. RHE; (c) and (d) CD difference as a function of the applied potential during the OER in a 1 M KOH electrolyte, showing a sharp increase in the slope after a threshold of 1.2 V vs. RHE. The figures are adopted from ref. 69 with permission from the Royal Society of Chemistry © 2025. | |
6. Theoretical study
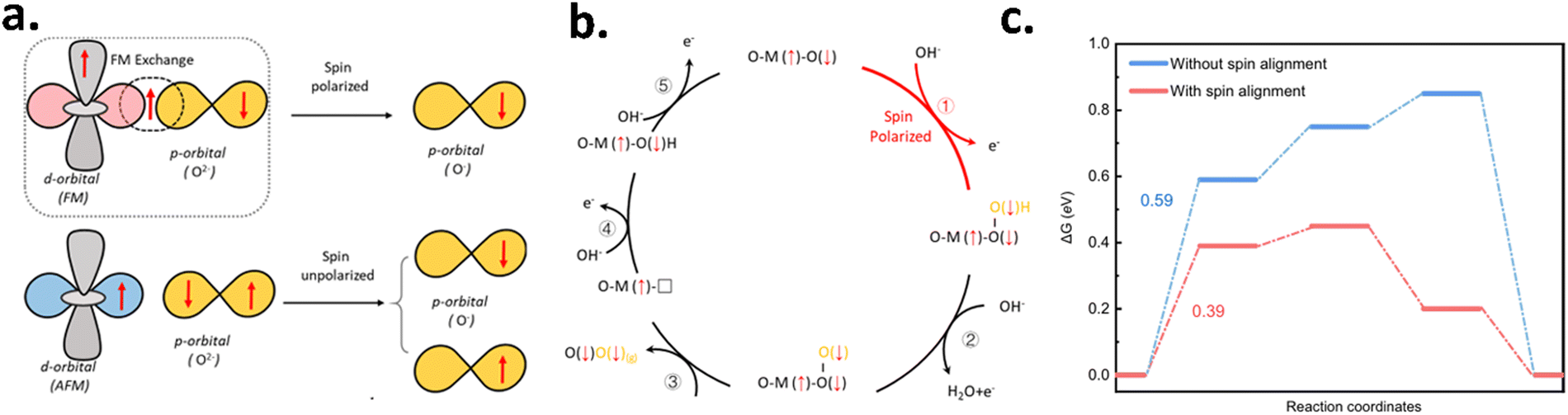
Recently, Gracia et al. have suggested that the spin states or magnetic structures of the active centers in electrocatalysts can directly influence the reaction barriers of catalytic reactions by adjusting the bonding of the reaction intermediate species with catalytic sites.70–72 This, in turn, affects the binding strength between the catalyst and the reaction intermediates (e.g., *OH, *O, *OOH). A change in binding strength alters the Gibbs free energy of adsorption, which is a critical factor in determining the reaction pathway and overpotential. Theoretically, it has been found that spin-polarization occurs in the oxygen evolution reaction at the first electron transfer step where coherent spin-interaction occurs between the spin-polarized catalyst and the adsorbed oxygen species.5 However, in the following steps, the OER is mainly dictated by the Hund rule and Pauling exclusion principle as all the adsorbed O species possess a fixed spin alignment. Hence, the anodic catalyst having spin-polarization leads to the generation of triplet oxygen with lower activation energy in comparison with the anodic catalyst having no spin-polarization, as shown in Fig. 17.
 |
| | Fig. 17 Gibb's free energy calculation to demonstrate the effect of spin-polarization in the OER. (a) Schematic of the spin-exchange mechanism for the OER, where spin polarization via FM exchange (QSEI) reduces electron repulsion and aligns adsorbed O species. (b) Mechanism of spin-polarized OER. (c) Illustration of the free energy diagram calculated for the OER at an applied potential of 1.23 V (vs. RHE) on the CoFe2O4(111) surface with and without spin alignment for triplet oxygen formation. The figures are adopted from ref. 5 with permission from Nature © 2021. | |
Future outlook
Chiral catalysts transmit spin-polarized electrons due to the CISS effect. Highly ordered conjugated organic molecules and polymers exhibit much higher spin polarization than randomly oriented analogs. Similarly, organic–inorganic hybrid materials exhibit high spin-polarization due to confined crystal structure. The spin-polarization of the different chiral systems measured using magnetic conducting atomic force microscopy or magnetoresistance measurement is presented in Table 1.73 The establishment of a correlation between spin-polarization and electrocatalytic activity has been attempted for the chiral catalysts. A chiral catalyst's spin-aligned electron transfer can be compared with a paramagnetic one under an external magnetic field. Although both chiral molecule functionalization and an applied external magnetic field can result in an activity augmentation by spin-polarization, the degree of enhancement may vary depending on the materials, external magnetic field etc. It has been reported by Liang et al. that utilizing (P)-thiadiazole-[7] helicene on the NiOx-based catalyst, the OER activity was boosted by around 61% in 0.1 M KOH at an applied potential of 1.65 V vs. RHE, which was found to be higher than the activity enhancement by applying an external magnetic field (≤ 450 mT) to the NiOx-based catalyst in 1 M KOH.41 However, optimization of the external magnetic field by measuring the magnetic moment of the catalyst varying the magnetic field may result in better activity. Besides, external magnetic fields can result in the enhancement of the mass transfer process due to the magnetohydrodynamic effect and also magnetic heating at the locale of the catalyst due to an external magnetic field results in the enhancement of the OER.38,44 Beyond this, magnetic field-induced structure regulation may also positively affect the kinetics.42,74,75
Table 1 Correlation between spin-polarization and the OER activity of the chiral catalyst
| Catalyst |
Chiral components |
Electrolyte |
Tafel's slope (mV dec−1) |
Spin-polarization |
Ref. |
| Ni–Au/glass substrate |
L-Cysteine |
0.1 M KOH |
114 |
6.6% |
16
|
| CuO on TiO2/FTO |
L-Tartaric acid |
0.1 M Na2SO4 |
52.7 |
10% |
20
|
|
D-Tartaric acid |
0.2 M borate buffer solution |
80.9 |
5% |
| FeNi/nickel foam |
L-Tartaric acid |
1 M KOH |
45.4 |
68% |
22
|
|
D-Tartaric acid |
46.5 |
74% |
| Copper oxide nanoparticles |
L-Cysteine |
0.1 M KOH |
117 |
80% |
28
|
|
D-Cysteine |
122 |
| CuO nanoflower |
Amino phenylpropanoic (APP) |
1 M KOH |
93.5 |
96% |
30
|
| Perovskite |
| BiVO4 |
(S)-methyl-benzylamine (S-MBA) |
1 M potassium borate (K-Bi) solution (pH 9.0) |
12676 |
86.6% |
33
|
| (S)-ortho-fluorinated-methylbenzylamine (S-2F-MBA) |
88.9% |
| MOF/COF |
| (R)- or (S)-2,2′-dihydroxy-1,1′-binaphthyl-3,3′,6,6′-tetrakis(4-benzaldehyde) (BTBA) |
N,N,N′,N′-Tetrakis(4-aminophenyl)-1,4-benzenediamine (TPDA) [R-25] |
1 M KOH |
78.4 |
57% ± 5% |
36
|
| 1,3,6,8-Tetrakis(4-aminophenyl)pyrene (TTA-Py) [R-26] |
92.1 |
52% ± 4% |
| 4,4′,4′′,4′′′-([2,2′-bi(1,3-dithiolylidene)]-4,4′,5,5′-tetrayl)tetraaniline (TTF-NH2) [R-27] |
100.9 |
41% ± 4% |
| Chiral semimetal |
| RhBiS |
|
1 M KOH |
52.4 |
5% |
37
|
Although chiral molecules exhibit superior catalytic activity, their poor long-term stability under the harsh oxidative conditions of the OER leads to a loss of catalytic activity over time. Hence, the integration of chiral materials into a commercial electrochemical system requires additional modifications to enhance their durability and compatibility with electrolytes. The functionalizing chiral organic molecules should be designed in such a way that they become stable in the electrolyte solution within the potential range. Secondly, the bonding strength among the elements of the chiral catalysts can be increased by doping heteroatoms or controlling the electronic structure of the catalysts.77–79 On the other side, the use of magnetic materials requires an external magnetic field for their application. Therefore, the evolution of ferromagnetic materials with high coercivity is essential so that the magnetic moment is retained for a longer time in the catalyst even after the removal of the external magnetic field.
Hence, over the last decade, substantial research has been carried out on the spin-controlled electrocatalytic oxidation of water. Numerous directions have been explored to generate a spin-polarized anodic current for water oxidation. Besides exploring CISS, new directions have been investigated to develop spin-polarized materials. Namely, magnetic materials and materials containing intrinsic spin have been found to show superior catalytic activity for the OER, proving that an electron's spin has a controlling role in the oxygen evolution reaction. Several fundamental studies have been executed to get an enhanced understanding of the OER mechanism so that the underlying principle of designing OER catalysts can be modified. However, stability and cost restrict their commercial application. Therefore, more studies are required to unravel new avenues and investigate and comprehend the full potential of spin-polarized catalysts in the electrocatalytic oxidation of water. Also, more regulatory strategies have to be implemented to develop the synergistic effect from the spin-polarized catalyst, as shown in Scheme 6.
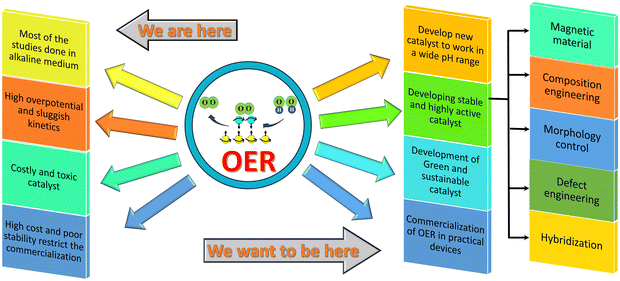 |
| | Scheme 6 Current status and prospect of spin-polarized catalysts for application in the oxygen evolution reaction. | |
Author contributions
Conceptualization supervision: KBG; writing – original draft: DB and UU; writing, review and editing: KBG, DB and UU. All authors have read and agreed to the published version of the manuscript.
Data availability
No primary research results, software, or code have been included, and no new data was generated or analyzed as part of this review article.
Conflicts of interest
The authors declare no competing interests related to this work.
Acknowledgements
DB acknowledges CSIR-Aspire (01/WS(023)/2023-24/EMR-II/ASPIRE) for his fellowship. UU acknowledges the University Grant Commission (UGC) for their fellowship. KBG acknowledges the Start-up Research Grant (SRG/2022/000737), ANRF-India and CSIR-Aspire (01/WS(023)/2023-24/EMR-II/ASPIRE) for the financial support.
References
- E. Fabbri and T. J. Schmidt, ACS Catal., 2018, 8, 9765–9774 CrossRef CAS.
- N. Sato and G. Okamoto, Electrochim. Acta, 1965, 10, 495–502 CrossRef CAS.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque, R. Kötz and T. J. Schmidt, Sci. Rep., 2015, 5, 12167 CrossRef CAS PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- X. Ren, T. Wu, Y. Sun, Y. Li, G. Xian, X. Liu, C. Shen, J. Gracia, H.-J. Gao, H. Yang and Z. J. Xu, Nat. Commun., 2021, 12, 2608 CrossRef CAS PubMed.
- S. Ghosh, B. P. Bloom, Y. Lu, D. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2020, 124, 22610–22618 CrossRef CAS.
- W. Mtangi, V. Kiran, C. Fontanesi and R. Naaman, J. Phys. Chem. Lett., 2015, 6, 4916–4922 CrossRef CAS PubMed.
- W. Mtangi, F. Tassinari, K. Vankayala, A. Vargas Jentzsch, B. Adelizzi, A. R. A. Palmans, C. Fontanesi, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2017, 139, 2794–2798 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS.
- K. Chae, N. A. R. C. Mohamad, J. Kim, D.-I. Won, Z. Lin, J. Kim and D. H. Kim, Chem. Soc. Rev., 2024, 53, 9029–9058 RSC.
- Y. Liang, M. Lihter and M. Lingenfelder, Isr. J. Chem., 2022, 62, e202200052 CrossRef CAS.
- S. Sanvito, Nat. Mater., 2011, 10, 484–485 CrossRef CAS PubMed.
- R. A. Rosenberg, D. Mishra and R. Naaman, Angew. Chem., Int. Ed., 2015, 54, 7295–7298 CrossRef CAS PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- F. Tassinari, K. Banerjee-Ghosh, F. Parenti, V. Kiran, A. Mucci and R. Naaman, J. Phys. Chem. C, 2017, 121, 15777–15783 CrossRef CAS.
- P. K. Bhartiya, M. Srivastava and D. Mishra, Int. J. Hydrogen Energy, 2022, 47, 42160–42170 CrossRef CAS.
- U. Utkarsh, S. R. Pramatha, A. Balo, U. K. Gosh, K. V. Rao and K. B. Ghosh, J. Mater. Chem. A, 2024, 12, 20354–20363 RSC.
- Y. Liang, K. Banjac, K. Martin, N. Zigon, S. Lee, N. Vanthuyne, F. A. Garcés-Pineda, J. R. Galán-Mascarós, X. Hu, N. Avarvari and M. Lingenfelder, Nat. Commun., 2022, 13, 3356 CrossRef CAS PubMed.
- P. Vensaus, Y. Liang, N. Zigon, N. Avarvari, V. Mujica, G. J. A. A. Soler-Illia and M. Lingenfelder, J. Chem. Phys., 2024, 160, 111103 CrossRef CAS PubMed.
- K. B. Ghosh, W. Zhang, F. Tassinari, Y. Mastai, O. Lidor-Shalev, R. Naaman, P. Möllers, D. Nürenberg, H. Zacharias, J. Wei, E. Wierzbinski and D. H. Waldeck, J. Phys. Chem. C, 2019, 123, 3024–3031 CrossRef CAS.
- M. Gazzotti, A. Stefani, M. Bonechi, W. Giurlani, M. Innocenti and C. Fontanesi, Molecules, 2020, 25, 3988 CrossRef CAS PubMed.
- T. Feng, W. Chen, J. Xue, F. Cao, Z. Chen, J. Ye, C. Xiao and H. Lu, Adv. Funct. Mater., 2023, 33, 2215051 CrossRef CAS.
- U. Utkarsh, S. Sahu, A. Balo, D. Barik and K. Banerjee Ghosh, ACS Appl. Energy Mater., 2025, 8, 1722–1730 CrossRef CAS.
- W. Zhang, K. Banerjee-Ghosh, F. Tassinari and R. Naaman, ACS Energy Lett., 2018, 3, 2308–2313 CrossRef CAS.
- W. Zhang, W. Wang, Y. Hu, H. Guan and L. Hao, Int. J. Hydrogen Energy, 2021, 46, 3504–3509 CrossRef CAS.
- A. Vadakkayil, C. Clever, K. N. Kunzler, S. Tan, B. P. Bloom and D. H. Waldeck, Nat. Commun., 2023, 14, 1067 CrossRef CAS PubMed.
- C. J. Mingoes, B. C. Schroeder and A. B. Jorge Sobrido, ACS Mater. Au, 2024, 4, 204–213 CrossRef CAS PubMed.
- Y. Jin, W. Fu, Z. Wen, L. Tan, Z. Chen, H. Wu and P. Wang, J. Am. Chem. Soc., 2024, 146, 2798–2804 CrossRef CAS PubMed.
- J. Ran, M. Si and D. Gao, Chem. Eng. J., 2024, 493, 152545 CrossRef CAS.
- Y. Li, L. Qiu, R. Tian, Z. Liu, L. Yao, L. Huang, W. Li, Y. Wang, T. Wang and B. Zhou, Small, 2024, 20, 2408248 CrossRef CAS PubMed.
- A. Vadakkayil, W. A. Dunlap-Shohl, M. Joy, B. P. Bloom and D. H. Waldeck, ACS Catal., 2024, 14, 17303–17309 CrossRef CAS PubMed.
- H. Lee, C. U. Lee, J. Yun, W. Jeong, C.-S. Jong, J. Son, Y. S. Park, G. Jang, S. Moon and J. Moon, ECS Meeting Abstracts, 2023, 2318 CrossRef.
- J. Son, G. Jang, S. Ma, H. Lee, C. U. Lee, S. Yang, J. Lee, S. Moon, W. Jeong, J. H. Park, C. Jung, J. Kim, J. Park and J. Moon, Adv. Sci., 2024, 11, 2403326 CrossRef CAS PubMed.
- H. Lee, S. Ma, S. Oh, J. Tan, C. U. Lee, J. Son, Y. S. Park, J. Yun, G. Jang and J. Moon, Small, 2023, 19, 2304166 CrossRef CAS PubMed.
- M. Zhang, Q. Dai, H. Zheng, M. Chen and L. Dai, Adv. Mater., 2018, 30, 1705431 CrossRef PubMed.
- Z. Li, Y. Xiao, C. Jiang, B. Hou, Y. Liu and Y. Cui, Natl. Sci. Rev., 2024, 11, nwae332 CrossRef PubMed.
- X. Wang, Q. Yang, S. Singh, H. Borrmann, V. Hasse, C. Yi, Y. Li, M. Schmidt, X. Li, G. H. Fecher, D. Zhou, B. Yan and C. Felser, Nat. Energy, 2024, 10, 101–109 CrossRef.
- Z. Feng, C. Dai, H. Wang, R. Guo, J. You and X. Liu, ChemCatChem, 2023, 15, e202300688 CrossRef CAS.
- U. Lüders, M. Bibes, K. Bouzehouane, E. Jacquet, J.-P. Contour, S. Fusil, J.-F. Bobo, J. Fontcuberta, A. Barthélémy and A. Fert, Appl. Phys. Lett., 2006, 88, 082505 CrossRef.
- H. Wang, Y. Dong, J. Ying, Z. Zhu, Y. Feng, Y.-X. Xiao, G. Tian, L. Shen, W. Geng, Y. Lu, S. Wu and X.-Y. Yang, Chem. Commun., 2025, 61, 4343–4346 RSC.
- F. A. Garcés-Pineda, M. Blasco-Ahicart, D. Nieto-Castro, N. López and J. R. Galán-Mascarós, Nat. Energy, 2019, 4, 519–525 CrossRef.
- J. Yan, Y. Wang, Y. Zhang, S. Xia, J. Yu and B. Ding, Adv. Mater., 2021, 33, 2007525 CrossRef CAS PubMed.
- H. Zheng, Y. Wang, P. Zhang, F. Ma, P. Gao, W. Guo, H. Qin, X. Liu and H. Xiao, Chem. Eng. J., 2021, 426, 130785 CrossRef CAS.
- H. Zheng, Y. Wang, J. Xie, P. Gao, D. Li, E. V. Rebrov, H. Qin, X. Liu and H. Xiao, ACS Appl. Mater. Interfaces, 2022, 14, 34627–34636 CrossRef CAS PubMed.
- X. Qin, J. Teng, W. Guo, L. Wang, S. Xiao, Q. Xu, Y. Min and J. Fan, Catal. Lett., 2023, 153, 673–681 CrossRef CAS.
- D. Peng, C. Hu, X. Luo, J. Huang, Y. Ding, W. Zhou, H. Zhou, Y. Yang, T. Yu, W. Lei and C. Yuan, Small, 2023, 19, 2205665 CrossRef CAS PubMed.
- X. Li, C. Hao, Y. Du, Y. Lu, Y. Fan, M. Wang, N. Wang, R. Meng, X. Wang, Z. J. Xu and Z. Cheng, Chin. J. Catal., 2023, 55, 191–199 CrossRef CAS.
- P. Vensaus, Y. Liang, J.-P. Ansermet, G. J. A. A. Soler-Illia and M. Lingenfelder, Nat. Commun., 2024, 15, 2867 CrossRef CAS PubMed.
- L. Lin, R. Xin, M. Yuan, T. Wang, J. Li, Y. Xu, X. Xu, M. Li, Y. Du, J. Wang, S. Wang, F. Jiang, W. Wu, C. Lu, B. Huang, Z. Sun, J. Liu, J. He and G. Sun, ACS Catal., 2023, 13, 1431–1440 CrossRef CAS.
- T. Sun, Z. Tang, W. Zang, Z. Li, J. Li, Z. Li, L. Cao, J. S. Dominic Rodriguez, C. O. M. Mariano, H. Xu, P. Lyu, X. Hai, H. Lin, X. Sheng, J. Shi, Y. Zheng, Y.-R. Lu, Q. He, J. Chen, K. S. Novoselov, C.-H. Chuang, S. Xi, X. Luo and J. Lu, Nat. Nanotechnol., 2023, 18, 763–771 CrossRef CAS PubMed.
- Z. Zhang, J. Li, J. Qian, Z. Li, L. Jia, D. Gao and D. Xue, Small, 2022, 18, 2104248 CrossRef CAS PubMed.
- X. Lyu, Y. Zhang, Z. Du, H. Chen, S. Li, A. I. Rykov, C. Cheng, W. Zhang, L. Chang, W. Kai, J. Wang, L. Zhang, Q. Wang, C. Huang and E. Kan, Small, 2022, 18, 2204143 CrossRef PubMed.
- Y. Wang, T. Yang, S. Yue, H. Zheng, X. Liu, P. Gao, H. Qin and H. Xiao, ACS Appl. Mater. Interfaces, 2023, 15, 11631–11641 CrossRef CAS PubMed.
- Y. Zhang, P. Guo, S. Niu, J. Wu, W. Wang, B. Song, X. Wang, Z. Jiang and P. Xu, Small Methods, 2022, 6, 2200084 CrossRef CAS PubMed.
- Z. Li, Z. Lv, X. Liu, G. Wang, Y. Lin, G. Xie and L. Jiang, Renewable Energy, 2021, 165, 612–618 CrossRef CAS.
- A. N. Nair, S. Fernandez, M. Marcos-Hernández, D. R. Romo, S. R. Singamaneni, D. Villagran and S. T. Sreenivasan, Nano Lett., 2023, 23, 9042–9049 CrossRef CAS PubMed.
- H. Gajapathy, S. Bandaranayake, E. Hruska, A. Vadakkayil, B. P. Bloom, S. Londo, J. McClellan, J. Guo, D. Russell, F. M. F. De Groot, F. Yang, D. H. Waldeck, M. Schultze and L. R. Baker, Chem. Sci., 2024, 15, 3300–3310 RSC.
- Q. Li, C. Zeng, X. Chen, M. Zhang, Y. Ding, Y. Xian, X. Huang, P. Liu and C. Han, Int. J. Hydrogen Energy, 2024, 80, 435–439 CrossRef CAS.
- M. Müllner, M. Riva, F. Kraushofer, M. Schmid, G. S. Parkinson, S. F. L. Mertens and U. Diebold, J. Phys. Chem. C, 2019, 123, 8304–8311 CrossRef.
- R. Poulain, A. Klein and J. Proost, J. Phys. Chem. C, 2018, 122, 22252–22263 CrossRef CAS.
- Z. Liu, H. M. A. Amin, Y. Peng, M. Corva, R. Pentcheva and K. Tschulik, Adv. Funct. Mater., 2023, 33, 2210945 CrossRef CAS.
- E. M. Davis, A. Bergmann, H. Kuhlenbeck and B. Roldan Cuenya, J. Am. Chem. Soc., 2024, 146, 13770–13782 CrossRef CAS PubMed.
- W. Zhang and K. Zhou, Small, 2017, 13, 1700806 CrossRef PubMed.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- G. Abellán, J. A. Carrasco, E. Coronado, J. Romero and M. Varela, J. Mater. Chem. C, 2014, 2, 3723–3731 RSC.
- T. S. Munonde, H. Zheng and P. N. Nomngongo, Ultrason. Sonochem., 2019, 59, 104716 CAS.
- W. Han, J. Wei, K. Xiao, T. Ouyang, X. Peng, S. Zhao and Z. Liu, Angew. Chem., 2022, 134, e202206050 Search PubMed.
- H. Zhang, Z. Chen, X. Ye, K. Xiao and Z. Liu, Angew. Chem., Int. Ed., 2025, 64, e202421027 CrossRef CAS PubMed.
- F. A. Garcés-Pineda, J. Yu, C. A. Mesa, S. Plana-Ruiz, D. Ruano, Y. Liang, M. Lingenfelder, S. Giménez and J. R. Galán-Mascarós, Chem. Sci., 2025, 16, 5475–5482 RSC.
- J. Gracia, R. Sharpe and J. Munarriz, J. Catal., 2018, 361, 331–338 CrossRef CAS.
- J. Gracia, Phys. Chem. Chem. Phys., 2017, 19, 20451–20456 RSC.
- J. Gracia, J. Phys. Chem. C, 2019, 123, 9967–9972 CrossRef CAS.
- B. P. Bloom, Z. Chen, H. Lu and D. H. Waldeck, Natl. Sci. Rev., 2024, 11, nwae212 CrossRef PubMed.
- J. Cai, L. Wang and P. Wu, Phys. Lett. A, 2007, 362, 105–108 CrossRef CAS.
- L. Pan, M. Ai, C. Huang, L. Yin, X. Liu, R. Zhang, S. Wang, Z. Jiang, X. Zhang, J.-J. Zou and W. Mi, Nat. Commun., 2020, 11, 418 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, D. Ni, Z. Chen, X. Wang, Y. Bu and J. Ao, Adv. Funct. Mater., 2019, 29, 1902101 CrossRef.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. (David) Lou, Adv. Mater., 2020, 32, 2002235 CrossRef CAS PubMed.
- A. Zhang, Y. Liang, H. Zhang, Z. Geng and J. Zeng, Chem. Soc. Rev., 2021, 50, 9817–9844 RSC.
- Y. Qin, T. Yu, S. Deng, X.-Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y.-B. He, H.-J. Qiu, L. He, F. Kang, K. Li and T.-Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
Footnote |
| † DB and UU contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*