
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing sulfilimine as an oxidizing directing group in Cp*Co(III)-catalyzed [4+2] annulation with alkynes and 1,3-diynes†
Arijit
Ghosh
and
Amit B.
Pawar
 *
*
School of Chemical Sciences, Indian Institute of Technology Mandi, Mandi, Himachal Pradesh 175005, India. E-mail: amitpawar@iitmandi.ac.in
First published on 8th May 2025
Abstract
We have developed the first Cp*Co(III)-catalyzed [4+2] annulation utilizing sulfilimine as an oxidizing directing group in a redox-neutral fashion. The N–S bond of the sulfilimine serves as an internal oxidant, thereby eliminating the need for any internal oxidant. The reaction worked with various alkynes and also exhibited an excellent regioselectivity with 1,3-diynes furnishing the 3-alkynylated isoquinolones.
The use of oxidizing directing groups in transition metal-catalyzed C–H annulation has emerged as a powerful strategy for synthesizing a wide range of nitrogen heterocycles.1 Since the pioneering contributions of Fagnou, Glorius, and others, the application of oxidizing directing groups has grown significantly.2 This is due to the fact that in this particular strategy, the directing group plays the role of an external oxidant beside anchoring the metal to the specific site for performing C–H activation. Hence, the reaction can be performed under redox-neutral conditions without the requirement of any external oxidant. The oxidizing group strategy has been instrumental for C–H annulation reactions under rhodium and ruthenium catalytic systems. Apart from this, significant progress has been made in the area of Cp*Co(III)-catalysis using various oxidizing directing groups for the synthesis of important heterocycles such as isoquinolones, isoquinolines, and indoles (Scheme 1a).3 In the majority of these reports, N–O, N–N, and N–Cl bonds act as internal oxidants. To the best of our knowledge, the utilization of N–S bonds as an internal oxidant under cobalt-catalysis is scarce.4 Therefore, there is still significant scope for the development of novel Cp*Co(III)-catalyzed redox-neutral C–H annulation protocols utilizing novel oxidizing directing groups based on N–S, N–P and O–O bonds as an internal oxidant.
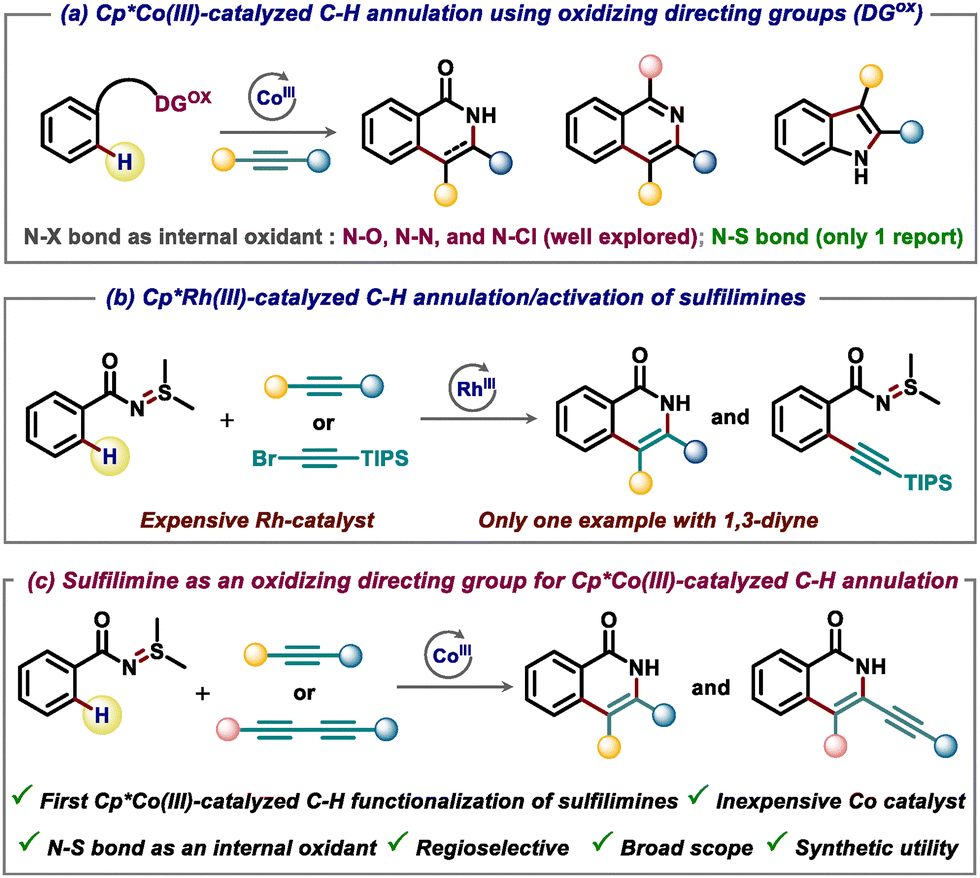 | ||
| Scheme 1 Oxidizing directing group strategy for C–H functionalization/annulation under Cp*Co(III) and Cp*Rh(III) catalysis. | ||
On the other hand, sulfilimines represent an important class of organic molecules widely utilized in amination reactions either as a nitrene precursor5 or as an N-nucleophile.6 Very recently, Huang and co-workers have successfully demonstrated the utility of sulfilimines in C–H activation and annulation reactions. However, the reaction was performed using precious Rh catalyst (Scheme 1b).7 Considering the high cost of the Rh-catalyst and need for the development of more sustainable C–H functionalization approaches, it is highly desirable to develop a method for the C–H functionalization of sulfilimines using first-row transition metal catalysts like Cp*Co(III) catalysts (Scheme 1c). Moreover, to the best of our knowledge there is no report on the redox-neutral synthesis of isoquinolones using N–S bonds as an internal oxidant under high-valent cobalt-catalysis.
We commenced our investigation by using S,S-dimethyl-N-acylsulfilimine (1a) and diphenyl acetylene (2a) as model substrates where the N–S bond is expected to play the role of an internal oxidant. After performing a series of reactions, we found that the optimal reaction conditions of treatment of diphenyl acetylene (2a, 0.1 mmol) with S,S-dimethyl-N-acylsulfilimine (1a, 0.15 mmol) in the presence of [Cp*Co(CO)I2] (10 mol%), AgOAc (20 mol%), and CsOAc (20 mol%) at 100 °C for 24 h in HFIP furnished the desired isoquinolone derivative 3aa in 76% yield (Table 1, entry 1) (isolated yield 72%). When the reaction was performed using an alkyne having alkyl substituents (3-hexyne, 2b), it furnished the required product in 67% isolated yield. When the reaction was performed without AgOAc, it furnished only trace amount of the product (Table 1, entry 2), demonstrating that AgOAc is crucial for this transformation. Other acetate additives like KOAc, NaOAc, PivOH and AcOH were found to be less effective as compared to the CsOAc (Table 1, entries 3 and 4). Employing a solvent other than HFIP, such as 1,2-DCE, resulted in no detection of the product and a decrease in the yield for TFE (Table 1, entries 5 and 6). The reaction was conducted with other silver additives, such as AgSbF6 and AgNTf2, resulting in a lower yield in both instances (Table 1, entry 7). The reaction was tested by reducing the time to 12 hours and decreasing the temperature to 80 °C, but in both cases the yield dropped (Table 1, entries 8 and 9). Furthermore, when the reaction was performed in the absence of CsOAc, it resulted in a decrease in the product yield (Table 1, entry 10). The reaction yielded no product without a cobalt catalyst (Table 1, entry 11).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldc (%) |
| a Reaction conditions: 1a (0.15 mmol, 1.5 equiv.), 2a (0.10 mmol), [Cp*Co(CO)I2] (10 mol%), Ag(I) salt (20 mol%), and additive (20 mol%) and solvent (0.6 mL) at the indicated temperature and time. b Isolated yields are given under the standard conditions. c Yields are based on crude 1H NMR (internal standard: 1,1,2,2 tetrachloroethane). n.d. = not detected. | ||
| 1 | None | 76 |
| 2 | Without AgOAc | Trace |
| 3 | KOAc/NaOAc instead of CsOAc | 72/68 |
| 4 | PivOH/AcOH instead of CsOAc | 46/48 |
| 5 | 1,2-DCE as a solvent instead of HFIP | n.d. |
| 6 | TFE as a solvent instead of HFIP | 65 |
| 7 | AgSbF6/AgNTf2 instead of AgOAc | 40/50 |
| 8 | 12 h instead of 24 h | 60 |
| 9 | 80 °C instead of 100 °C | 58 |
| 10 | Without CsOAc | 65 |
| 11 | Without [Cp*Co(CO)I2] | n.d. |

Upon successfully optimizing the reaction conditions, we turned our attention to investigating the scope of various sulfilimine derivatives and alkynes (Scheme 2).8 The sulfilimines having electron-donating groups, such as Me and tBu, at the para-position furnished products in good yields with both diaryl and dialkyl alkynes, i.e., diphenyl acetylene and 3-hexyne (3ba–3ca; 3bb–3cb). The reaction was found to be compatible with sulfilimines having electron-withdrawing groups such as F and CF3 at the para-position, and annulated products in moderate yields (3da–3ea; 3db–3eb). The reaction was found to be highly regioselective for meta-substituted sulfilimines having Me and Br substituents (1f and 1g), furnishing a single isomer product in which the C–H activation occurs at the less hindered position in moderate yields (3fb, 3gb). However, in the case of 3-Cl substituted sulfilimine, we have observed the formation of a non-separable mixture of regioisomeric products, of which the product 3hb is the major isomer. The reaction was sluggish with an ortho-substituted sulfilimine derivative (1i) having a fluorine substituent, furnishing the required annulated product 3ia in 22% yield. The reaction was found to be compatible with sulfilimine derivatives having a naphthalene and thiophene moiety, resulting in the formation of the required products in low to moderate yields (3jb, 3kb).
 | ||
Scheme 2 Scope of sulfilimines and alkynes. Reaction conditions: 1 (1.5 equiv.), 2a (0.30 mmol.), [Cp*Co(CO)I2] (10 mol%), AgOAc (20 mol%), and CsOAc (20 mol%) in HFIP (1.8 mL) at 100 °C for 24 h. Isolated yields are given. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Inseparable regioisomers in 1.0:0.05 ratio. b2.0 equiv. of sulfilimine 1a was used. cThe other regio-isomer was formed in 24% yield.9dInseparable regioisomers in a 1.0:0.6 ratio. Inseparable regioisomers in 1.0:0.05 ratio. b2.0 equiv. of sulfilimine 1a was used. cThe other regio-isomer was formed in 24% yield.9dInseparable regioisomers in a 1.0:0.6 ratio. | ||
Next, the scope of the annulation reaction was further examined for various di-substituted acetylene derivatives (Scheme 2). The diaryl-acetylene derivatives having electron-donating as well as electron-withdrawing groups such as Me, OMe, CO2Et, and F at the para-position delivered the corresponding isoquinolone derivatives in moderate to good yields (3ac–3af). The disubstituted alkyne, 1,2-bis(3,4-dimethoxyphenyl)ethyne (2g) afforded the corresponding isoquinolone derivative in 58% yield (3ag). Similarly, diaryl-alkynes having different substituents like Me, OMe, and Cl at the meta-position yielded the annulated products in yields of 58%, 68%, and 68%, respectively (3ah–3aj). The reaction was found to be compatible with the heteroaryl alkyne having thiophene rings (2k), furnishing the desired product 3ak in 80% yield. The scope was further extended to dialkyl alkyne, i.e., 4-octyne, which furnished the product 3al in 60% yield. The structure of the product was confirmed by X-ray crystallographic analysis.9 Furthermore, reactions with unsymmetrical alkynes such as 1-phenyl-1-propyne and 1-phenyl-1-butyne resulted in the formation of a mixture of regioisomers (3am–3am′; 3an–3an′). However, out attempt to utilize a terminal alkyne (phenyl acetylene) as a coupling partner was futile.
After testing the scope of various alkyne derivatives towards the Cp*Co(III)-catalyzed [4+2] annulation reaction using sulfilimine as an oxidizing directing group, we were interested in utilizing 1,3-diynes as a coupling partner (Scheme 3).8 In recent years, 1,3-diynes have been employed in various C–H annulation reactions for the synthesis of a diverse array of heterocyclic compounds.10 However, due to the presence of the two alkyne units, utilization of 1,3-diynes as a coupling partner in C–H annulation becomes challenging due to chemo-selectivity and regio-selectivity issues.
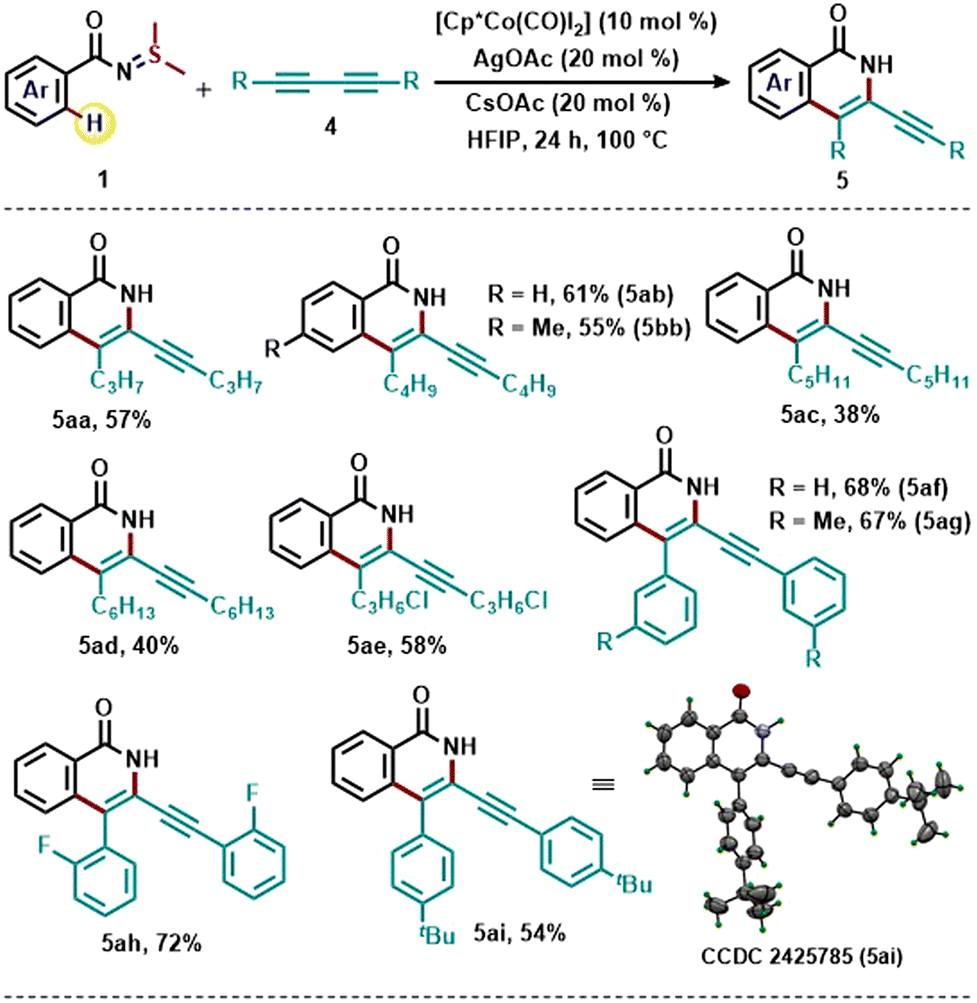 | ||
| Scheme 3 Scope of 1,3-diynes for the regioselective synthesis of 3-alkynylated isoquinolones. Reaction conditions: 1 (1.5 equiv.), 4 (0.30 mmol), [Cp*Co(CO)I2] (10 mol%), AgOAc (20 mol%), and CsOAc (20 mol%) in HFIP (1.8 mL) at 100 °C for 24 h. Isolated yields are given. | ||
We were pleased to see that reaction of S,S-dimethyl-N-acylsulfilimine (1a) with various dialkyl 1,3-diynes under the optimized conditions resulted in the formation of 3-alkynylated isoquinolones in moderate to good yields. The reaction also tolerated the primary chloride functionality present in the 1,3-diyne moiety. The protocol also works well with 1,3-diynes having aryl groups.11 The regioselectivity of alkyne insertion leading to the formation of 3-alkynylated isoquinolones was unambiguously confirmed by the X-ray analysis of the product 5ai.
Next, we focused on the synthetic utility of the synthesized isoquinolone derivative 3aa (Scheme 4).9 Various annulation reactions were performed using benzoquinone, ethyl acrylate, and diphenyl acetylene under Ir, Rh, and Ru catalytic systems to furnish 6, 7, and 8 in moderate to good yields.12 Later, the N-benzylation of the isoquinolones followed by Cp*Co(III)-catalyzed C–H olefination with naphthoquinone13 resulted in the formation of 9. Finally, we carried out preliminary mechanistic studies to gain some information about the reaction mechanism (Scheme 5). In an intermolecular competitive experiment (Scheme 5a), it was found that sulfilimine having an electron-donating Me substituent (1b) at the para-position reacts preferably over the sulfilimine having an electron-withdrawing F substituent (1d). This observation indicates that the C–H activation occurs through an electrophilic activation mechanism.
 | ||
| Scheme 4 Synthetic utilities.9 | ||
 | ||
| Scheme 5 Preliminary mechanistic studies. | ||
Later, the kinetic isotope effect (KIE) was determined by performing parallel reactions in separate vessels as well as a competitive reaction in the same vessel using 1a and [D5]-1a (Scheme 5b). The KIE values from these experiments were found to be 2.2 and 3.3, respectively, indicating that the C–H activation step is likely to be the rate-determining step of the reaction.
A plausible mechanism for the cobalt-catalyzed annulation of sulfilimine is illustrated in Scheme 6. Initially, the catalytically active species A is generated by treating [Cp*Co(CO)I2] with AgOAc and CsOAc. This species then undergoes a rate-limiting cyclometallation with sulfilimine 1, leading to the formation of cobaltacycle B. Subsequently, the insertion of an alkyne or 1,3-diyne into the carbon–cobalt bond results in the formation of a seven-membered metallacycle C. The intermediate C undergoes reductive elimination with subsequent loss of Me2S, and ligand exchange yielding species D. Finally, protodemetalation of D regenerates the catalytically active Cp*Co(III) species A, while affording the desired annulated product (3 or 5).
 | ||
| Scheme 6 Plausible mechanism. | ||
In conclusion, we have developed an efficient Cp*Co(III)-catalyzed [4+2] C–H annulation strategy for synthesizing isoquinolone derivatives, utilizing sulfilimine as an oxidizing directing group. Notably, this work represents the first example of Cp*Co(III)-catalyzed C–H functionalization of sulfilimines. The reaction works under redox-neutral conditions and employs the N–S bond of sulfilimines as an internal oxidant. The reaction exhibits high regioselectivity with 1,3-diynes and proceeds smoothly with a broad range of substitutions on both the sulfilimines and alkynes. Additionally, post-synthetic modifications of the synthesized isoquinolones were carried out successfully.
A. G. thanks the Ministry of Human Resources Development (MHRD), India, for the research fellowship. A. B. P. thanks the Science and Engineering Research Board (SERB), New Delhi, India for financial support in the form of a Core Research Grant (CRG/2023/002075).
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 3al and 5ai have been deposited at the CCDC under 2425784 and 2425785.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977–1979 CrossRef CAS PubMed; (b) Z. Hu, X. Tong and G. Liu, Chin. J. Org. Chem., 2015, 35, 539 CrossRef CAS; (c) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC; (d) H. Wang and H. Huang, Chem. Rec., 2016, 16, 1807–1818 CrossRef CAS PubMed; (e) Z. Wang, P. Xie and Y. Xia, Chin. Chem. Lett., 2018, 29, 47–53 CrossRef CAS; (f) Y. Luo and Y. Xia, Handbook of CH-Functionalization, John Wiley & Sons, Ltd, 2022, pp. 1–28 Search PubMed.
- (a) J. Wu, X. Cui, L. Chen, G. Jiang and Y. Wu, J. Am. Chem. Soc., 2009, 131, 13888–13889 CrossRef CAS PubMed; (b) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908–6909 CrossRef CAS PubMed; (c) K.-H. Ng, A. S. C. Chan and W.-Y. Yu, J. Am. Chem. Soc., 2010, 132, 12862–12864 CrossRef CAS PubMed; (d) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS PubMed; (e) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed.
- Selected examples: (a) H. Wang, J. Koeller, W. Liu and L. Ackermann, Chem. – Eur. J., 2015, 21, 15525–15528 CrossRef CAS PubMed; (b) B. Sun, T. Yoshino, M. Kanai and S. Matsunaga, Angew. Chem., Int. Ed., 2015, 54, 12968–12972 CrossRef CAS PubMed; (c) A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef CAS PubMed; (d) G. Sivakumar, A. Vijeta and M. Jeganmohan, Chem. – Eur. J., 2016, 22, 5899–5903 CrossRef CAS PubMed; (e) Y. Liang and N. Jiao, Angew. Chem., Int. Ed., 2016, 128, 4103–4107 CrossRef; (f) X. Yu, K. Chen, S. Guo, P. Shi, C. Song and J. Zhu, Org. Lett., 2017, 19, 5348–5351 CrossRef CAS PubMed; (g) A. Lerchen, T. Knecht, M. Koy, C. G. Daniliuc and F. Glorius, Chem. – Eur. J., 2017, 23, 12149–12152 CrossRef CAS PubMed; (h) N. Muniraj and K. R. Prabhu, Org. Lett., 2019, 21, 1068–1072 CrossRef CAS PubMed; (i) A. Ghosh, T. Rana, N. Bhaduri and A. B. Pawar, Org. Lett., 2023, 25, 7878–7883 CrossRef CAS PubMed.
- F. Wang, Q. Wang, M. Bao and X. Li, Chin. J. Catal., 2016, 37, 1423–1430 CrossRef CAS.
- (a) X. Tian, L. Song, C. Han, C. Zhang, Y. Wu, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 2937–2940 CrossRef CAS PubMed; (b) X. Tian, L. Song, M. Rudolph, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 4327–4330 CrossRef CAS PubMed; (c) X. Tian, L. Song, M. Rudolph, F. Rominger, T. Oeser and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2019, 58, 3589–3593 CrossRef CAS PubMed; (d) X. Tian, L. Song, M. Rudolph, Q. Wang, X. Song, F. Rominger and A. S. K. Hashmi, Org. Lett., 2019, 21, 1598–1601 CrossRef CAS PubMed; (e) P. W. Antoni, A. V. Mackenroth, F. F. Mulks, M. Rudolph, G. Helmchen and A. S. K. Hashmi, Chem. – Eur. J., 2020, 26, 8235–8238 CrossRef CAS PubMed.
- (a) R. L. Grange, E. A. Clizbe, E. J. Counsell and P. A. Evans, Chem. Sci., 2015, 6, 777–781 RSC; (b) K. O. Marichev, K. Wang, K. Dong, N. Greco, L. A. Massey, Y. Deng, H. Arman and M. P. Doyle, Angew. Chem., Int. Ed., 2019, 58, 16188–16192 CrossRef CAS PubMed; (c) K. O. Marichev, K. Dong, L. A. Massey, Y. Deng, L. De Angelis, K. Wang, H. Arman and M. P. Doyle, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- J. Liu, X. Jia and L. Huang, Org. Lett., 2022, 24, 6772–6776 CrossRef CAS PubMed.
- We have observed the formation of around 30–35% of the corresponding benzamide derivatives.
- See ESI† for more details.
- (a) D. G. Yu, F. De Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650–9654 CrossRef CAS PubMed; (b) B. V. Pati, N. N. Puthalath, S. K. Banjare, T. Nanda and P. C. Ravikumar, Org. Biomol. Chem., 2023, 21, 2842–2869 RSC.
- In the case of reactions with diaryl 1,3-diynes, we have observed traces of unidentified impurities along with the required 3-alkynylated isoquinolones (5af–5ai).
- (a) F. Wang, G. Song, Z. Du and X. Li, J. Org. Chem., 2011, 76, 2926–2932 CrossRef CAS PubMed; (b) B. Li, H. Feng, N. Wang, J. Ma, H. Song, S. Xu and B. Wang, Chem. – Eur. J., 2012, 18, 12873–12879 CrossRef CAS PubMed; (c) T. Zhou, L. Li, B. Li, H. Song and B. Wang, Org. Lett., 2015, 17, 4204–4207 CrossRef CAS PubMed.
- T. Sharma, N. Sumit, N. Sachin, D. Chandra, S. S. Gupta and U. Sharma, Org. Lett., 2024, 26, 5027–5031 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2425784 and 2425785. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01230a |
| This journal is © The Royal Society of Chemistry 2025 |