
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium zincate-enabled divergent one-pot dual C–C bond formation in thiophenes†
Alexandre
Pierret
 a,
Kevin
Magra
a,
Hugo
Lopez
a,
Thomas
Kauffmann
a,
Clément
Denhez
b,
Ibrahim
Abdellah
c,
Christophe
Werlé
*ad and
Alexandre
Vasseur
*a
a,
Kevin
Magra
a,
Hugo
Lopez
a,
Thomas
Kauffmann
a,
Clément
Denhez
b,
Ibrahim
Abdellah
c,
Christophe
Werlé
*ad and
Alexandre
Vasseur
*a
aUniversité de Lorraine, CNRS, L2CM, F-54000 Nancy, France. E-mail: christophe.werle@univ-lorraine.fr; alexandre.vasseur@univ-lorraine.fr
bUniversité de Reims Champagne Ardenne, CNRS, ICMR UMR 7312, 51097 Reims, France
cUniversité de Lorraine, CNRS, L2CM, F-57000 Metz, France
dMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34 – 36, 45470 Mülheim an der Ruhr, Germany. E-mail: christophe.werle@cec.mpg.de
First published on 24th March 2025
Abstract
We present a lithium zincate-enabled, divergent one-pot synthesis for regioselective dual C–C bond formation in thiophenes. By modifying the zinc coordination environment, a single set of reagents (ZnCl2, R1Li, and diethyl (5-halo)thenylphosphate) was found to generate two distinct products. This approach extends the versatility of lithium organozincates to regioselective CAr(sp2)–Cthienyl(sp2) and Cthenyl(sp3)–CAr(sp2) couplings without requiring transition metals and/or arenes pre-activated with a boronic acid.
Recent outbreaks of epidemic and pandemic diseases, such as those caused by Ebola, SARS-CoV-2, and the Mpox virus, emphasise the urgency of rapid drug discovery. Each crisis spurs a swift search for treatments, driving efforts to develop new therapies or improve existing ones. In this context, synthetic chemists, alongside biologists, are equally mindful of the importance of efficient methods for accessing diverse molecular libraries.1 High-throughput screening of structurally related compounds often serves as a starting point for identifying potential treatments,2 requiring synthetic methods that are both rapid and scalable. Synthetic strategies that quickly produce diverse, yet structurally related molecules are critical in this process. “One-pot synthesis” is particularly advantageous for such demands as it enables multiple bond-forming reactions within a single reaction vessel.3 This strategy encompasses domino, consecutive, and independent reaction sequences, streamlining processes by eliminating the need to isolate intermediates.4 Similarly, ligand-enabled metal-promoted divergent synthesis offers flexibility5 by allowing different products to form from the same starting material by adjusting the ligand environment around the metal. However, these strategies are generally applied independently and have limited integration. A reagent capable of adapting to a wide range of reaction pathways could combine the principles of one-pot and divergent synthesis, offering increased versatility.
We hypothesised that lithium zincate (LiZn) of the PAIRiodic table,6—a synergistic complex combining group 1 and group 12 elements—could fulfil this role (Scheme 1a). When ZnCl2 reacts with three equivalents of an organolithium compound (R1Li), a 16-electron mono-anionic complex, (R1)3ZnLi, is formed. This complex features a dynamic Li–Zn bond (Scheme 1b), whose strength and adaptivity were recently demonstrated computationally for Et3ZnLi·2THF (Scheme 1c).7 Lithium triorganozincates demonstrate significant structural flexibility in THF, existing as a network of dynamic equilibria between three distinct forms (Scheme 1d).7 This flexibility underpins their broad reactivity profile,8 which includes metallation via I/Zn exchange,9 deprotonation,10 and homologation through 1,2-ligand migration.11 Additionally, they participate in 1,2- and 1,4-ligand additions to electrophiles,12 enabling both ligand-enabled divergent synthesis13 and sequential carbon–carbon bond formation in one-pot synthesis.14 In addition to these capabilities, reacting ZnCl2 with four equivalents of R1Li produces an 18-electron di-anionic complex, (R1)4ZnLi2 (Scheme 1b)15 which is thermodynamically more stable, kinetically more active and provides additional synthetic opportunities such as Br/Zn exchange metallations,15 cross-coupling reactions,16 conversions of carboxylic acid to ketones17 and transition metal catalyst-free conjugate addition to nitroolefins.18 With these characteristics established, we conceptualised two lithium zincate-mediated pathways as a framework for achieving regioselective functionalisation of thiophene derivatives. Using diethyl (5-halo)thenylphosphate 1 as a model substrate, we sought to explore their feasibility.
 | ||
| Scheme 1 Development of a lithium-zincate system for divergent bond formation. (a) Conceptual representation of the “LiZn element” as a synergistic assembly. (b) Structures of mono- and di-anionic zincates. (c) Representation of Li–Zn bonding. (d) Structural equilibria in THF. (e) Conceptual strategy. | ||
In the first pathway (Scheme 1e), treating 1a (X = I) with two equivalents of lithium triorganozincate (R1)3ZnLi (2) initiates an I/Zn exchange to produce intermediate I. The coexistence of a formally negatively charged Zn centre and a potentially cationic centre at the thenyl position could drive a homologation reaction via a 1,2-migration of an R1 ligand, yielding intermediate II with temporary dearomatisation. A formally [1,5] metallotropic rearrangement would then restore aromaticity, producing the homologated thenylzinc intermediate III, which would react with an electrophile 3 to yield the final product (4).
In contrast, the second pathway (Scheme 1e) employs two equivalents of di-anionic tetraorganonzincate (5), where homologation proceeds via nucleophilic substitution on 1a or 1b (X = Br). This generates an isomeric product (6) through an X/Zn exchange followed by electrophilic trapping. This pathway emphasises controlled sequential reactivity, requiring homologation to occur faster than metallation to maintain pathway fidelity. Directing each pathway enables access to a diverse array of heterocycles, highlighting the versatility of the lithium zincate-mediated approach. However, several challenges need to be addressed to ensure successful implementation. These include maintaining the stability of the thiophene framework under the polar organometallic conditions required for pathway 1,19 managing the competition between halogen/metal exchange and nucleophilic substitution that determines entry into either pathway 1 or 2, and achieving selective reactivity of III and V with electrophile 3 (transfer of the thenyl or thienyl ligand rather than the alkyl R1 ligand).
To overcome these challenges, we developed a LiZn element-promoted divergent process that regioselectively forms two carbon–carbon bonds five atoms apart in a single reaction vessel. By modifying the Zn coordination environment, we achieved significant structural transformations through the distinct reaction pathways. Given the widespread applications of thiophenes in pharmaceuticals20 and materials science,21 this investigation focuses on leveraging diethyl (5-halo)thenyl-phosphate 1 to explore regioselective functionalisation strategies.
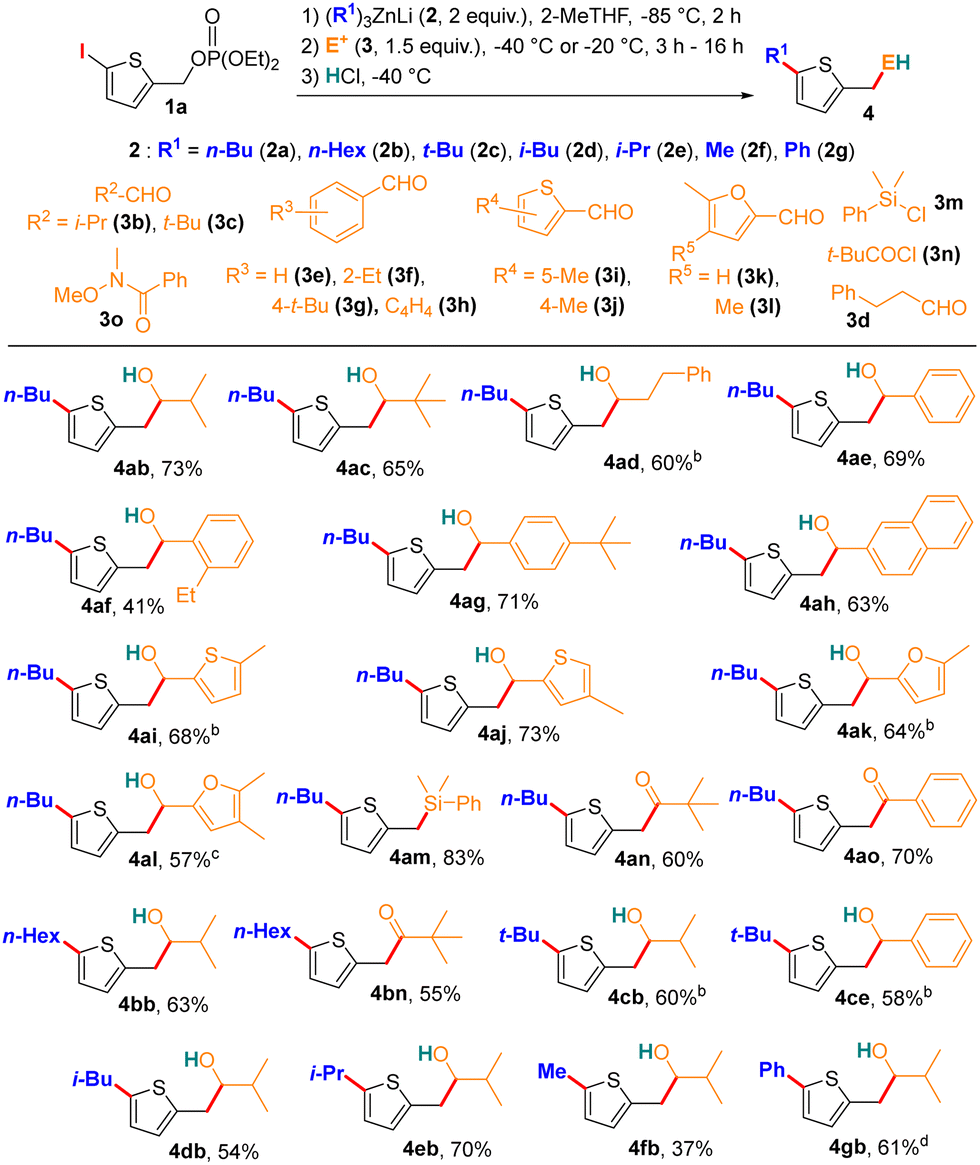
As an initial step, we identified optimal conditions to carry out the consecutive reaction sequence in pathway 1 (Scheme 1e). Treating 1a with (n-Bu)3ZnLi at −85 °C in 2-MeTHF for two hours, followed by the addition of 1 M HCl (3a), afforded the desired product 4aa along with three minor Wurtz-type homocoupling by-products (7, 8, and 9), in a favourable 4aa/(7+8+9) ratio of 90/10 (see table in Section 3a, S7 in the ESI† for optimisation details). These results align with prior lithium zincate-assisted functionalisation studies on 4-iodobenzyl derivatives, where 2-MeTHF served as a bio-solvent that promoted sequential steps and minimised Wurtz-type by-products.22 Notably, using THF instead of 2-MeTHF reversed the product ratio, giving a 20/80 distribution of 4aa to by-products, underscoring the role of the solvent in selectively driving the transformation sequence. A key difference in this reaction sequence compared to previous reports is the temperature requirement for the 1,2-migration homologation step. With 1a, the homologation was completed at −85 °C, whereas a warmer environment (−40 °C) was necessary for similar transformations with 4-iodobenzyl mesylate substrates. The synthetic scope of the reaction was next examined under the optimised conditions (Table 1).
| a Conditions: (1) 1a (0.6 mmol), 2 (1.2 mmol), 2-MeTHF (10 mL), −85 °C, 2 h; (2) 3 (0.9 mmol), −40 °C, 3 h; (3) HCl 1 M (8 mL), −40 °C; isolated yield. b 3 added at −40 °C and mixture stirred at −20 °C for 3 h. c 3 added at −40 °C and mixture stirred at −20 °C for 16 h. d (1) 1a (0.6 mmol), 2 (1.2 mmol), 2-MeTHF (10 mL), −85 °C, 30 min; (2) −40 °C, 1 h 30; (3) PhLi (0.6 mmol), −40 °C, 20 min; (4) 3 (0.9 mmol), −40 °C and then −20 °C for 16 h; (5) HCl 1 M (8 mL), −20 °C. |
|---|
|
Various electrophiles were tested to achieve a second C–C bond formation five atoms away from the homologation site in a single operation. We found that the intermediate III (Scheme 1e), formed after the metallotropic rearrangement, was generally less reactive than its counterpart derived from 4-iodobenzyl mesylate.22 Nevertheless, it reacted readily with secondary aldehyde 3b at −40 °C over three hours to afford homothenyl alcohol 4ab in an isolated yield of 73%. Notably, this transformation proceeded with complete selectivity, as no transfer of the butyl ligand to 3b was observed. Several homothenyl alcohols were synthesised by reacting intermediate III with a range of aldehydes, including tertiary (4ac), enolisable primary (4ad), aromatic (4ae–4ah), and heteroaromatic (4ai–4al) aldehydes, with isolated yields generally exceeding 60%, except for 4af (41%). In some cases, warming the reaction mixture from −40 °C to −20 °C after adding the electrophile improved yields. We also observed efficient C–Si bond formation with electrophile 3m, yielding 4am in 83% yield. Our approach was further validated for synthesising sterically hindered (4an, 60%) and less hindered (4ao, 70%) homothenyl carbonyl compounds. Sterically hindered products were selectively synthesised using the corresponding acid chlorides (e.g., 3n for 4an), while Weinreb amides proved essential for less hindered compounds, as PhCOCl mainly led to double addition by-products (see Section 3c, S8 for details, ESI†). Beyond (n-Bu)3ZnLi, other lithium trialkylzincates with more nucleophilic (4bb, 4bn) or bulkier ligands (4cb, 4ce, 4db, 4eb) were also compatible, demonstrating the versatility of this methodology. Additionally, methyl ligands, traditionally considered less nucleophilic, could participate in the 1,2-migration, as seen with Me3ZnLi (2f), yielding 4fb in fair yield (37%). This contrasts with earlier findings where only the hybrid zincate Me2PhZnLi facilitated similar migrations.22 A major challenge in expanding the substrate scope involved achieving C(sp2)–C(sp2) bond formation via a 1,2-migration of an aryl ligand. Notably, reacting 1a with Ph3ZnLi (2g) at −85 °C followed by −40 °C produced the intermediate III, which gave 4gb in a 61% yield when activated by one equivalent of PhLi before adding 3b. Notably, 4gb exemplifies a rare aryl-heteroaryl coupling via 1,2-migration of a Ph ligand without a transition metal23 and highlights the potential of metallotropic rearrangement to facilitate one-pot remote formation of a second C–C bond.
The next objective was to develop a ligand-enabled divergent pathway relying on two independent reactions in a precise order: homologation via nucleophilic substitution of the phosphate by the zinc R1 ligand, followed by Zn/X exchange metallation. Treating 1a with 2 equivalents of the di-anionic reagent (n-Bu)4ZnLi2 (5a) in 2-MeTHF at −85 °C yielded a mixture of 6aa and 10 in a 39/61 ratio after hydrolysis (Scheme 2), with pathway 1 favoured. The product 6aa arises from hydrolysis of Va (pathway 2), while 10 results from the reaction of IIIa with n-BuI released during the I/Zn exchange (pathway 1). This result indicates that metallation proceeds faster than nucleophilic substitution, a finding further supported by solvent effects: replacing 2-MeTHF with THF reversed the 6aa/10 ratio, while using substrate 1b (X = Br) delayed metallation, providing only 6aa (see table in Section 3b, S7, for further details, ESI†).
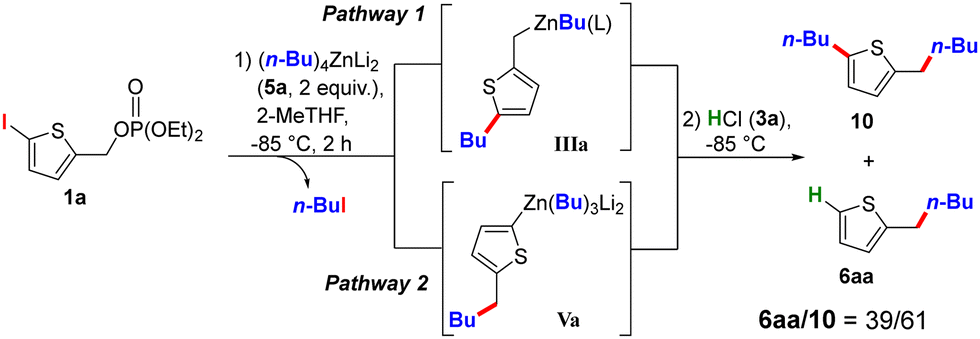 | ||
| Scheme 2 Preliminary results supporting pathway 2. | ||
Given the limited data on the reactivity of Va,24 we investigated its reactions with aldehydes (Table 2). Va reacted successfully with secondary and tertiary aliphatic acyclic aldehydes (6ab, 6ac, 6ap) and cyclic aliphatic aldehydes (6aq, 6ar, 6as), yielding thienyl alcohols in 62% to 87%. This approach also enabled the synthesis of thienyl aldehyde 6at (61%) and several ketones (6an, 6ao, 6au, 6av), with Weinreb amides (3o, 3u, 3v) delivering higher yields than acid chlorides (3n). Va could also be successfully converted into ester compounds, as shown by the isolation of 6aw and 6ax in 65% and 76%, respectively.

Our method's versatility extends to zinc ligands beyond n-Bu. For instance, reagent 5b, with n-hexyl ligands, yielded products 6bb and 6bc in over 59% yield. Even bulkier ligands, such as those in 5c (t-Bu) and 5d (i-Bu), proved effective, as shown by products 6cb, 6dt, and 6db, with yields between 54% and 72%. Notably, 5e, containing less transferable ligands than n-Bu, afforded products 6eb and 6ee in fair yields (49–53%).
Finally, the presented one-pot strategy also supports C(sp3)–C(sp2) cross-coupling. This was demonstrated by the synthesis of 6fu and 6fb, with yields of 58% and 50%, respectively. To our knowledge, this achievement represents a rare C(sp3)–C(sp2) cross-coupling in the absence of a boronic acid and/or transition metal catalyst.25 In total, nine constitutional isomers of the products shown in Table 2 were successfully synthesised, underscoring the broad applicability of this approach.
In summary, we developed a divergent, one-pot synthetic method for regioselective formation of two carbon–carbon bonds five atoms apart, leveraging the reactivity of lithium zincate (LiZn) element. By adjusting zinc's coordination environment, a single set of starting materials (ZnCl2, organolithium R1Li, and diethyl (5-halo)thenylphosphate) yielded distinct products: pathway 1 forms product 4via a consecutive reaction sequence, while pathway 2 produces its constitutional isomer 6 through independent sequential reactions. This outcome underscores the versatility of lithium organozincates, with pathway 1 adapting our remote functionalisation approach22 to heterocycles for the first time and pathway 2 representing a new methodology. A critical factor was the solvent choice: bio-solvent 2-MeTHF promoted pathway 1, while THF favoured pathway 2. Across 44 examples, this method enabled heteroaryl-aryl couplings without transition metal and/or boronic acid activation, highlighting its synthetic versatility. Other heterocycles are currently under study, and the role of the bio-solvent is also being examined. The results will be disclosed in due course.
The authors acknowledge financial support from the French Agence Nationale de la Recherche (ANR-19-CE07-0032), the French Ministry of Higher Education and Research (MESR), and the Max Planck Society. They also thank F. Dupire (MASSLOR), P. Lemière (SynBioN), D. Dodin for designing custom devices to enhance chiller functionality & the “Plateforme de RMN” (Pôle CPM/UL).
Open Access funding provided by the Max Planck Society.
Data availability
The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Hayashi, J. Org. Chem., 2021, 86, 1–23 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. S. Sittampalam, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- Y. Hayashi, Acc. Chem. Res., 2021, 54, 1385–1398 CrossRef CAS PubMed.
- N. J. Green and M. S. Sherburn, Aust. J. Chem., 2013, 66, 267–283 CAS.
- Y. Wang, J. Feng, E.-Q. Li, Z. Jia and T.-P. Loh, Org. Biomol. Chem., 2024, 22, 37–54 CAS.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- A. Pierret, C. Lefebvre, P. C. Gros, C. Denhez and A. Vasseur, Eur. J. Org. Chem., 2023, e202300954 CAS.
- M. Uchiyama and C. Wang, Top. Organomet. Chem., 2014, 47, 159–202 CAS.
- M. Balkenhohl and P. Knochel, Chem. – Eur. J., 2020, 26, 3688–3697 CAS.
- (a) D. K. Wanic, R. Melvin and G. Barker, Synthesis, 2023, 3487–3501 CAS; (b) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802–3824 CAS.
- I. Marek, Tetrahedron, 2002, 58, 9463–9475 CAS.
- Selected papers on reactivity of zincate reagents with electrophiles: (a) S. Cho, E. J. McLaren and Q. Wang, Angew. Chem., Int. Ed., 2021, 60, 26332–26336 CAS; (b) S. Cho and Q. Wang, Org. Lett., 2020, 22, 1670–1674 CAS.
- (a) S. E. Baillie, V. L. Blair, D. C. Blakemore, D. Hay, A. R. Kennedy, D. C. Prydeb and E. Hevia, Chem. Commun., 2012, 48, 1985–1987 CAS; (b) T. Imahori, M. Uchiyama, T. Sakamoto and Y. Kondo, Chem. Commun., 2001, 2450–2451 CAS.
- (a) T. Harada, T. Katsuhira, A. Osada, K. Iwazaki, K. Maejima and A. Oku, J. Am. Chem. Soc., 1996, 118, 11377–11390 CrossRef CAS; (b) T. Harada, T. Kaneko, T. Fujiwara and A. Oku, J. Org. Chem., 1997, 62, 8966–8967 CrossRef CAS.
- M. Uchiyama and Y. Kondo, J. Synth. Org. Chem., Jpn., 2006, 64, 1180–1190 CrossRef CAS.
- C. Wang, T. Ozaki, R. Takita and M. Uchiyama, Chem. – Eur. J., 2012, 18, 3482–3485 CAS.
- R. Murata, K. Hirano and M. Uchiyama, Chem. – Asian J., 2015, 10, 1286–1290 CAS.
- (a) M. Dell’Aera, F. M. Perna, P. Vitale, A. Altomare, E. Hevia and V. Capriati, Eur. J. Inorg. Chem., 2024, e202400505 Search PubMed; (b) M. Dell’Aera, F. M. Perna, P. Vitale, A. Altomare, A. Palmieri, L. C. H. Maddock, L. J. Bole, A. R. Kennedy, E. Hevia and V. Capriati, Chem. – Eur. J., 2020, 26, 8742–8748 CrossRef PubMed.
- Dearomatisation/rearomatisation processes involving thenyl substrates are reported only under Pd-catalysed conditions, see: S. Zhang, X. Yu, X. Feng, Y. Yamamoto and M. Bao, Chem. Commun., 2015, 51, 3842–3845 CAS.
- (a) R. M. D. da Cruz, F. J. B. Mendonça-Junior, N. B. de Mélo, L. Scotti, R. S. A. de Araújo, R. N. de Almeida and R. O. de Moura, Pharmaceuticals, 2021, 14, 692 Search PubMed; (b) Archna, S. Pathania and P. A. Chawla, Bioorg. Chem., 2020, 101, 104026 CAS.
- L. Li, J. Li, L. Guo, Y. Xu, Y. Bi, Y. Pu, P. Zheng, X.-K. Chen, Y. Wang and C. Li, Chem. Sci., 2024, 15, 11435–11443 CAS.
- A. Pierret, C. Denhez, P. C. Gros and A. Vasseur, Adv. Synth. Catal., 2022, 364, 3805–3816 CAS.
- Two other examples without transition metal: (a) H. J. Jeong, S. Chae, K. Jeong and S. K. Namgoong, Eur. J. Org. Chem., 2018, 6343–6349 CAS; (b) A. Hernán-Gómez, E. Herd, M. Uzelac, T. Cadenbach, A. R. Kennedy, I. Borilovic, G. Aromı and E. Hevia, Organometallics, 2015, 34, 2614–2623 Search PubMed.
- In contrast, the hybrid zincate Bu3PhZnLi2 is well known for reacting non-selectively with aldehydes, favouring the 1,2-addition of the butyl ligand to the electrophile over the aryl ligand, see: Y. Kondo, M. Fujinami, M. Uchiyama and T. Sakamoto, J. Chem. Soc., Perkin Trans. 1, 1997, 799–800 CAS.
- For rare examples of cross coupling involving arylboronic acids or arylboronic acid pinacol esters in the absence of transition metal, see: (a) J. Procter, J. J. Dunsford, P. J. Rushworth, D. G. Hulcoop, R. A. Layfield and M. J. Ingleson, Chem. – Eur. J., 2017, 23, 15889–15893 Search PubMed; (b) R. B. Bedford, N. J. Gower, M. F. Haddow, J. N. Harvey, J. Nunn, R. A. Okopie and R. F. Sankey, Angew. Chem., Int. Ed., 2012, 51, 5435–5438 CrossRef CAS PubMed . For examples in the presence of a transition metal, see: ; (c) K. A. C. Bastick and A. J. B. Watson, Synlett, 2023, 2097–2102 CAS; (d) F. Trauner, B. Boutet, F. Rambaud, V. N. Ngo and D. Didier, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-52hq3.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation for new compounds including NMR spectra. See DOI: https://doi.org/10.1039/d5cc01079a |
| This journal is © The Royal Society of Chemistry 2025 |