
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen hole states facilitated cleavage of Ni–O bonds in the rock-salt phase of a conversion-type anode†
Shengnan
Sun‡
 *a,
Jun
Zhou‡
ab,
Debbie Hwee Leng
Seng
a,
Hui Ru
Tan
a,
Shibo
Xi
c,
Xiping
Ni
a,
Fengxia
Wei
a,
Poh Chong
Lim
a,
Ming
Lin
a,
Yi
Ren
a,
Shijie
Wang
*ab and
Zhi Wei
Seh
*a
*a,
Jun
Zhou‡
ab,
Debbie Hwee Leng
Seng
a,
Hui Ru
Tan
a,
Shibo
Xi
c,
Xiping
Ni
a,
Fengxia
Wei
a,
Poh Chong
Lim
a,
Ming
Lin
a,
Yi
Ren
a,
Shijie
Wang
*ab and
Zhi Wei
Seh
*a
aInstitute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore. E-mail: sun_shengnan@imre.a-star.edu.sg; sj-wang@imre.a-star.edu.sg; sehzw@imre.a-star.edu.sg
bFuture Energy Acceleration & Translation (FEAT), Strategic Research & Translational Thrust (SRTT), Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, Innovis #08-03, Singapore 138634, Republic of Singapore
cInstitute of Sustainability for Chemicals, Energy and Environment (ISCE2), Agency for Science, Technology and Research (A*STAR), 1 Pesek Road, Jurong Island, Singapore 627833, Republic of Singapore
First published on 10th April 2025
Abstract
This work studies the influence of oxygen chemistry on the reduction potential in conversion-type NiO anodes in lithium-ion batteries and reveals that the hole states on oxygen are highly polarized and serve as a bridge that facilitates the reduction of Ni2+, representing a higher potential plateau associated with Ni–O bond cleavage.
The metal–oxygen interaction plays a crucial role in metal oxide-related electrochemistry. For example, the oxygen evolution reaction (OER) on oxides involves multiple steps: oxygen species adsorption,1 electron hopping from the adsorbate to the oxide surface,2 electron transport within the oxide,2,3 and oxygen desorption via bond cleavage.1 Additionally, the metal–lattice oxygen bond can break through lattice oxygen oxidation, contributing to dioxygen formation.4 Under OER conditions (applying oxidation potential), the cleavage of the metal–lattice oxygen bonds highly depends on the metal–oxygen covalency.5 For example, in Sr-doped LaCoO3, Sr-doping-induced hole doping enhances OER activity, where the valence state of the active metals increases or the oxygen vacancies are created as charge compensation.5 Shifting oxygen charge in spinel oxides also can be used to facilitate the OER.6 Density functional theory also unveiled that the oxygen vacancies can facilitate the OER in the metal–oxygen battery; for example, Nb2O5 for the sodium-oxygen battery.7
This insight motivated us to investigate the charge compensation effect in another electrochemical reaction involving metal–oxygen bond cleavage: the lithiation of metal oxide under a reduction potential in a conversion-type Li-ion battery anode. In this process, metal cations are reduced to metal while oxygen anions form Li2O.8 Each transition-metal oxide exhibits distinct reduction potentials and specific capacities.8,9 Li doping can induce charge compensation (hole doping) and improve anode performance. For example, Li0.10Ni0.90O demonstrates higher capacity and reduction plateau (1–0.9 V by Li et al.10) compared to pristine NiO (0.6 V by Poizot et al.8) during the first lithiation.11 Such improvements are also observed in medium/high-entropy oxides such as (LiMgCoNiZn)O and (LiMgCoNiCuZn)O, where Li doping increases oxygen vacancies and enhances lithiation performance.12 These results highlight the influence of oxygen chemistry on the cleavage of the metal–oxygen bond during lithiation. The difficulty of cleaving the metal–oxygen bonds can be reflected by the reduction potential in the lithiation process. Though oxygen vacancies have been reported to benefit the anode performance,12 to our knowledge, few works focused on the correlation between the reduction potential of conversion-type anodes and the oxygen chemistry from the aspect of metal–oxygen bond cleavage. In this work, we were the first to indicate the dependence of the reduction potential on oxygen holes in the metal–oxygen bonds, which is associated with the first lithiation process of conversion-type anodes.
To investigate the role of oxygen chemistry in metal–oxygen bond cleavage in the lithiation, NiO and Li-doped NiO were selected as the study models for the following reasons. Firstly, Ni2+ reduces directly to Ni without the intermediate state in NiO.13 Secondly, charge compensation in Li-doped NiO occurs primarily on the O 2p orbital,14 avoiding interference from Ni3+ states. Beyond the effects of electronic states, the oxide surface area also impacts the observed reduction potential, with a large surface area for a higher reduction potential.15 In this work, we synthesized Li-doped NiO powders LixNi1−xO (x = 0, 0.05, 0.12, 0.17) using a similar sol–gel method16 to minimize differences in the surface area. Our results show that the Li doping induces extra charge (hole states) near the Fermi level on oxygen, and these hole states predominantly align along the nickel–oxygen bond direction. This chemically active oxygen correlates with the higher reduction potential observed during lithiation associated with the cleavage of the Ni–O bond, suggesting that oxygen serves as a bridge in facilitating the reduction of metal cations during the lithiation process. Besides, Li doping also lowers the formation energy of Li insertion into NiO. Our scenario can be extended to a counterexample: Mg0.05Ni0.95O, where oxygen holes are inhibited, and the reduction potential plateau is decreased.
The synthesized oxides are designated as NiO-syn, Li0.05Ni0.95O, Li0.12Ni0.88O and Li0.17Ni0.83O, based on the actual Li concentration in the oxides (Table S1 and Fig. S1, ESI†). Their surface areas are small and of negligible differences (Fig. 1a and Fig. S2, ESI†). No obvious impurity is observed from the X-ray diffraction (XRD) Rietveld refinements (Fig. S3–S6, ESI†). Li doping causes the diffraction peaks to shift to the high-angle direction, indicating the reduced lattice parameters. Note that Li-doped NiO has a pure rocksalt phase, while NiO-syn contains mixed rocksalt and monoclinic phases, which have similar XRD patterns.17 In Fig. 1a, we label these diffraction peaks according to the rocksalt phase. Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) results show that Li doping lowers the intensity of the Ni–O and Ni–Ni bonds (Fig. S7f, ESI†), and the fitted Ni–O bond lengths of the commercial NiO, the synthesized NiO and Li0.05Ni0.95O are close to 2.08 Å, and Li0.12Ni0.88O and Li0.17Ni0.83O have a relatively shorter Ni–O bond length of 2.07 Å (Fig. S8f, ESI†). In the first lithiation curve of NiO (Fig. 1b), the solid electrolyte interphase formation dominates the initial charge process,18 while the conversion reaction Ni2+ → Ni0 dominates the part exhibiting a potential plateau.11,13,18 At a reducing current density of 0.05 A g−1, the reduction potential plateau at 100 mA h g−1 increases dramatically from 0.67 V (NiO) to 0.92 V (Li0.05Ni0.95O), indicating that the Li doping facilitates Ni reduction. Higher Li concentration slightly increases the Ni reduction potential plateau further to 0.95 V (Li0.12Ni0.88O) and 0.99 V (Li0.17Ni0.83O). Below 25 mA h g−1, the dip in potential corresponds to the Ni nucleation barrier.13 During the first de-lithiation process, both NiO and Li-doped NiO exhibit a similar plateau, consistent with the previous report.11 Initial cyclic voltammetry (CV) curves (Fig. 1c) of the LixNi1−xO series show that increasing the Li concentration shifts the reduction peaks positively, from 0.30 V (NiO) to 0.57 V (Li0.05Ni0.95O) and 0.65 V (Li0.12Ni0.88O). Li0.17Ni0.83O exhibits two distinct reduction peaks at around 0.56 V and 0.20 V, respectively. Moreover, the onset potential for Li+ increases from 0.86 to 0.90, 0.97 and 1.05 V with Li doping, suggesting that more Li doping benefits the LixNi1−xO reduction. The LixNi1−xO series have a similar oxidation peak at around 2.17 V in the CV curves (Fig. 1c), which agrees with the similar potential plateau in the de-lithiation curves (Fig. 1b). Increasing the carbon black ratio in the electrode coating composite rules out the influence of the contact area between NiO and the conductive matrix on the reduction potential, and a comparison between NiO-syn and commercial NiO (NiO-comm) shows no significant differences in the reduction potential plateaus (Fig. S10, ESI†). Considering the kinetic effects in CV and galvanostatic tests, the galvanostatic intermittent titration technique (GITT) shows that the synthesized NiO, Li0.05Ni0.95O, Li0.12Ni0.88O and Li0.17Ni0.83O have open circuit potentials at 0.869, 1.122, 1.192, and 1.228 V at 100 mA h g−1, respectively, which agrees with the potential trend from CV and galvanostatic tests (Fig. S11, ESI†).
 | ||
| Fig. 1 (a) XRD patterns, (b) the first lithiation–delithiation curves at 0.05 A g−1 and, (c) the first CV curves at the scan rate of 0.1 mV s−1 of the synthesized LixNi1−xO (x = 0, 0.05, 0.12, 0.17) from open circuit voltage after at least 24 hours of stabilization. | ||
The charge compensation on the oxygen orbital in Li-doped NiO is confirmed by the presence of a peak at ∼528 eV in the O K-edge electron energy loss spectroscopy (EELS, Fig. 2a). Peaks below 535 eV are attributed to transitions to the Ni 3d–O 2p covalent bond.19 For NiO, the peaks at ∼531 eV are ascribed to the transition from O 1s2–Ni (3d8L + 3d9L−1) to O 1s–Ni 3d9L.20 Here, L and L−1 denote the ligand and the ligand with a hole, respectively. With increasing Li doping amount, the peak at ∼531 eV weakens while an additional peak at ∼528 eV appears and intensifies, corresponding to the transition from O 1s2–Ni 3d8L−1 to O 1s–Ni 3d8L.20b,c The final state, 3d8L, suggests oxygen oxidization and hole formation in oxygen,21 driven by the strong Ni 3d–O 2p hybridization.22 Besides, the Ni 2p3/2 XPS results also support the oxygen charge deviation (Fig. 2b). For NiO, the Ni 2p3/2 XPS displays three distinct peaks at around 853.8 eV, 855.8 eV and 860.9 eV, corresponding to the final states associated with the local screening 3d9L−1, and nonlocal screening 3d9L, and 3d8, respectively.20c,23 Li doping weakens the peak related to the local screening 3d9L−1 final state and strengthens the peak related to the nonlocal screening 3d9L. This suggests that higher Li concentration promotes the oxygen charge dislocation, which interacts with Ni cations, facilitating hole transport and contributing to electrical conductivity.24 While oxygen orbitals primarily accommodate charge compensation after Li doping,14 some experimental25 and theoretical works20 suggest partial oxidation of Ni from 2+ to 3+. The X-ray absorption near edge structure (XANES) results (Fig. 2c) show a shift in the Ni K-edge to higher energy in Li0.05Ni0.95O, indicating a higher Ni oxidation state. However, for Li0.12Ni0.88O and Li0.17Ni0.83O, no further shift in the Ni K-edge is observed. Due to the lack of standard Ni3+ oxides with a similar crystal structure, the degree of Ni oxidation cannot be quantified by XANES results. The Ni-L3,2 edge EELS (Fig. 2d) corresponds to the transition from Ni 2p to Ni 3d.26 The broadened profiles of the Ni L3-edge peaks are observed for Li0.12Ni0.88O and Li0.17Ni0.83O. This additional broadening can be attributed to an increase in covalency and the presence of extra screening channels.27 A comparison of the reported Ni3+ EELS curves28 and Ni2+ EELS curves14 indicates that no significant Ni3+ exists in our samples.
 | ||
| Fig. 2 Electronic state characterization of EELS of the synthesized LixNi1−xO. (a) EELS of O K-edge, (b) XPS of Ni 2p, (c) XANES of Ni K-edge, and (d) EELS of Ni L3,2-edge. The commercial NiO data are included. The spectra in (a), (b) and (d) are normalized to the peak heights. | ||
The charge compensation (hole states) by Li doping is further confirmed by density functional theory (DFT) calculations. The density of states (DOS) of pristine NiO (Fig. 3a) shows that it is insulating with the Ni 3d–O 2p hybridized orbitals dominating the valence band near the Fermi level, while the lowest unoccupied states above the Fermi level are primarily derived from Ni 3d orbitals, consistent with the report by Wrobel.29 In contrast, Li-doped NiO (Fig. 3b) features Ni 3d–O 2p hybridized states spanning the Fermi level, leading to hole states above the Fermi level. These states around the Fermi level contribute dominantly to the charge transport30 and the conduction behavior at moderate temperatures, as argued by Feinleib and Adler31 and effectively pin the Fermi energy.32 For the specific atom O1 and Ni2 along the Li–O bond direction, the projected DOS shows that states spanning the Fermi level are primarily derived from O 2pz and Ni 3dz2, which are spin-polarized (Fig. 3c and d). The corresponding real-space distribution of these hole states is shown in Fig. 3e–g. Fig. 3f and g show that Li doping induces significant extra charge on oxygen (O1) nearest to the doped Li, and these extra charges are predominantly polarized along the Li–O bond directions. The influence of doped Li extends to the Ni atom (Ni2) along the Li–O–Ni bond but does not affect the charge of the nearest Ni atom (Ni1), despite Ni1 being bonded to O1. This can be attributed to the absence of direct interaction between Li and Ni1, with the extra charge on O1 not polarized to Ni1. Interestingly, although the polarized extra charge on O1 has minimal impact on the Ni1–O1 bond, a slight extra charge is observed on the O3 atom, which is bonded to the Ni2 (the Ni atom carrying the polarized extra charge). In contrast, no extra charge can be observed in the second nearest oxygen atom (O2), located at a corner of the Li–Ni–O cubane body diagonal and not bonded to any Ni along the Li–O bond direction (Fig. 3c). These results suggest that the induced extra charge predominantly aligns along the Li–O bond direction, and the extra charge on Ni can, in turn, induce extra charge on oxygen in the Ni–O bond, even when that oxygen is not in the Li–O bond direction.
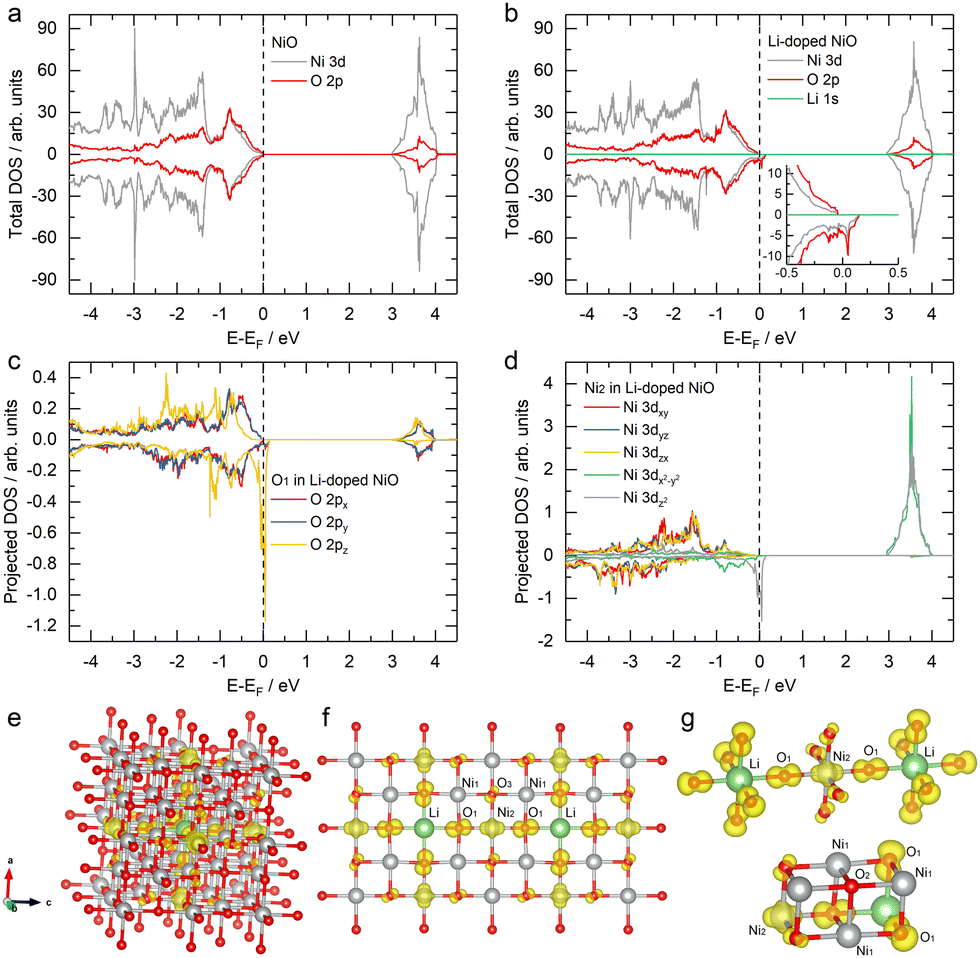 | ||
| Fig. 3 The DOS of (a) NiO and (b) Li-doped NiO, projected DOS on (c) O 2p orbitals and (d) Ni 3d orbitals for specific atoms in Li-doped NiO. The structure of Li-doped NiO with decomposed charge densities for the hole states (the isosurface value of 3 × 10−3 e− Å−3) from (e) bulk view, (f) cross-section view, and (g) specific atom view. The green, grey and red balls represent the lithium, nickel and oxygen atoms, respectively. The yellow contour represents the electron density in (e)–(g). | ||
For the process of Li insertion into the oxide prior to Ni reduction, formation energy calculations (Fig. S12 and S13, ESI†) reveal that Li insertion into undoped NiO is energetically unfavourable (positive formation energy), while it becomes favourable (negative formation energy) in Li-doped NiO. For NiO, the inserted Li breaks the original Ni–O bond and forms bonds with lattice oxygen. In Li-doped NiO, the most stable configuration involves two Li trapped in a Ni–O cage, each bonded to three neighbouring oxygens but not to each other. For surface Li absorption, the most stable configuration involves added Li bonding to two surface oxygens near a doped Li site. Similar bonds of the surface Li can also be found in the subsurface configurations.
Based on our proposed scenario, we continue to examine the reduction potential of a counterexample rocksalt Mg0.05Ni0.95O (Fig. S14, ESI†), in which the oxygen hole formation is inhibited. The reasons are as follows. (1) The charge compensation is not expected due to isovalent Mg2+ and Ni2+. (2) During the lithiation process, Mg2+ is not reduced to metallic Mg,33 minimizing the influence of a second metal. (3) It is established that the non-local screening from oxygen is inhibited after Mg substitution,34 meaning that oxygen hole formation is suppressed and electron delocalization from oxygen is restricted. In the first lithiation process (Fig. S15, ESI†), compared with NiO-syn, Mg0.05Ni0.95O has a lower reduction potential plateau in the first lithiation curve and a lower onset potential in the negative scan of the first CV curve. This indicates that the cleavage of the Ni–O bond becomes more difficult by Mg substitution. It fits our scenario on the relation between the cleavage of the Ni–O bond and the oxygen hole formation. We also examine the lithiation and CV curves of Mg0.50Ni0.50O (Fig. S15, ESI†), which does not exhibit an obvious lithiation plateau and CV response with very low specific capacity. It could be explained that excess Mg substitution obstructs the charge transport pathway caused by the Ni 3d–O 2p interaction and lowers the intrinsic conductivity, which makes the oxide more difficult to be reduced.
In summary, we utilized NiO and Li-doped NiO as models to investigate the correlation between the hole states on oxygen and the higher reduction potential in the first lithiation. Our findings suggest that the oxygen acts as a bridge that facilitates metal reduction. We found that the significant hole states on oxygen induced by Li doping are highly polarized along the Li–O bond direction, but negligible holes emerge in the diagonal direction of Li–Ni–O cubane. In addition, Li doping is helpful to lower the formation energy of additional Li into the bulk oxides. The case of Mg0.05Ni0.95O, where the hole states are inhibited and the reduction potential is lower, further supports this correlation. This work serves as a guide to enable orbital physics for optimizing the reduction potential in the metal–oxygen cleavage process in the energy field. More importantly, we noticed that such reaction potential adjustments by doping are also applicable to the intercalation-type anodes, for example, the constructed molecule-layered MnO2-pillared structures.35
Z. W. S. was supported by the Singapore National Research Foundation (NRF Investigatorship NRF-NRFI09-0002) and the Agency for Science, Technology and Research (MTC Programmatic Fund M23L9b0052). We acknowledge the computational resources supported by the National Supercomputing Centre (NSCC) Singapore and NUS IT's Research Computing group. This research used the TEM resources and facilities at the A*STAR Institute of Materials Research and Engineering (A*STAR IMRE). This work is supported by the Agency for Science, Technology and Research (A*STAR) through the Low Carbon Energy Research Finding Initiative (LCERFI01-0033|U2102d2006).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Suntivich, et al. , Science, 2011, 334, 1383–1385 CrossRef PubMed.
- J. Gracia, et al. , J. Catal., 2018, 361, 331–338 CrossRef.
- W. T. Hong, et al. , Energy Environ. Sci., 2017, 10, 2190–2200 RSC.
- (a) X. Rong, et al. , ACS Catal., 2016, 6, 1153–1158 CrossRef; (b) A. Grimaud, et al. , Nat. Chem., 2017, 9, 457–465 CrossRef PubMed.
- J. T. Mefford, et al. , Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- S. Sun, et al. , Angew. Chem., Int. Ed., 2019, 58, 6042–6047 CrossRef CAS PubMed.
- J.-H. Li and Y.-X. Yu, ChemSusChem, 2021, 14, 5488–5498 CrossRef CAS PubMed.
- P. Poizot, et al. , Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- J. Cabana, et al. , Adv. Mater., 2010, 22, E170–E192 CrossRef CAS.
- Z. Li, et al. , Mater. Chem. Phys., 2005, 91, 36–39 CrossRef CAS.
- Y. Li, et al. , Ceram. Int., 2016, 42, 14565–14572 CrossRef CAS.
- (a) E. Lökçü, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 23860–23866 CrossRef; (b) X. Liu, et al. , Small, 2022, 18, e2200524 CrossRef PubMed.
- A. Ponrouch, et al. , RSC Adv., 2014, 4, 35988–35996 RSC.
- J. van Elp, et al. , Solid State Commun., 1991, 80, 67–71 CrossRef CAS.
- M.-Y. Cheng, et al. , J. Power Sources, 2014, 253, 27–34 CrossRef CAS.
- S. Sun, et al. , J. Mater. Chem. A, 2024, 12, 20064–20076 RSC.
- W. Hong, et al. , Environ. Sci. Technol., 2023, 57, 20053–20063 CrossRef CAS.
- K. He, et al. , Nano Lett., 2015, 15, 1437–1444 CrossRef CAS.
- Y. Koyama, et al. , J. Phys. Chem. B, 2005, 109, 10749–10755 CrossRef CAS PubMed.
- (a) S. Hüfner, et al. , Z. Phys. B: Condens. Matter, 1992, 88, 247–248 CrossRef; (b) P. Kuiper, et al. , Phys. Rev. Lett., 1989, 62, 221–224 CrossRef CAS PubMed; (c) F. Reinert, et al. , Z. Phys. B: Condens. Matter, 1995, 97, 83–93 CrossRef CAS; (d) I. Davoli, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 2979–2982 CrossRef CAS PubMed.
- (a) S. Sun, et al. , Nat. Commun., 2024, 15, 260 CrossRef CAS PubMed; (b) B. H. Goodge, et al. , Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2007683118 CrossRef CAS PubMed.
- H. Chen and J. H. Harding, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 115127 CrossRef.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- J. Yamashita and T. Kurosawa, J. Phys. Chem. Solids, 1958, 5, 34–43 CrossRef CAS.
- (a) E. Antolini, Mater. Chem. Phys., 2003, 82, 937–948 CrossRef CAS; (b) I. J. Pickering, et al. , J. Am. Chem. Soc., 1993, 115, 4137–4144 CrossRef CAS.
- M. A. van Veenendaal and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 11326–11331 CrossRef CAS PubMed.
- M. Abbate, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 5419–5422 CrossRef CAS PubMed.
- F. M. de Groot, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459–5468 CrossRef CAS PubMed.
- F. Wrobel, et al. , Phys. Rev. B, 2020, 101, 195128 CrossRef CAS.
- V. Zayets, J. Magn. Magn. Mater., 2018, 445, 53–65 CrossRef CAS.
- (a) J. Feinleib and D. Adler, Phys. Rev. Lett., 1968, 21, 1010–1013 CrossRef CAS; (b) D. Adler and J. Feinleib, Phys. Rev. B: Solid State, 1970, 2, 3112–3134 CrossRef.
- S. Hüfner, Adv. Phys., 1994, 43, 183–356 CrossRef.
- K. Wang, et al. , Nat. Commun., 2023, 14, 1487 CrossRef CAS PubMed.
- (a) D. Alders, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 7716–7719 CrossRef PubMed; (b) S. Altieri, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 13403–13409 CrossRef CAS.
- J.-H. Li, et al. , J. Phys. Chem. C, 2021, 125, 3725–3732 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00987a |
| ‡ S. S. and J. Z. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |