
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring frustrated radical pairs through the persistent radical effect: methods of generation and recent applications
Darshika
Singh
 and
Robert E.
Maleczka
Jr
*
and
Robert E.
Maleczka
Jr
*
Department of Chemistry, Michigan State University, 578 S. Shaw Ln, East Lansing, Michigan 48824-1322, USA. E-mail: maleczka@chemistry.msu.edu
First published on 16th April 2025
Abstract
Radicals have long fascinated chemists owing to their structure, reactivity, and other features. The recent discovery of frustrated radical pairs (FRPs) has added a new dimension to this field. These unique radicals, which do not conform to traditional radical behavior, have opened a world of intriguing possibilities. FRPs have been categorized into neutral and ionic frustrated radical pairs and both are addressed as FRPs in this review. These pairs consist of two different (transient and persistent) radicals or radical ion pairs that do not react with each other. Such orthogonal reactivities and the resultant “persistent radical effect” enable chemical transformations that are difficult to achieve using traditional radical chemistry. This highlight uses recent examples to explore the different ways of generating these radical pairs and their working principle, highlighting the novelty and potential of this emerging field.
 Darshika Singh | Darshika Singh is originally from Uttar Pradesh, India. She completed her BSc and MSc from PPN Degree College (Kanpur, 2020) and Indian Institute of Technology Patna (2022). During her master's program, she earned a silver medal. Currently, Darshika is a PhD candidate in the Department of Chemistry at Michigan State University. As a third-year graduate student, her research project is on a Wittig rearrangement under the supervision of Prof. Robert Maleczka. Currently, she is working on promoting [1,2]- and [1,4]-N-Wittig rearrangements to afford novel silyl-containing products with potential applications in natural product synthesis. |
 Robert E. Maleczka | Robert E. Maleczka, Jr. is a Professor of Chemistry at Michigan State University (MSU). The Maleczka group's research interests include the invention of “green” reactions and strategies for organic synthesis. Honors bestowed on Dr Maleczka include being named a Fellow of the American Chemical Society (ACS) and the American Association for the Advancement of Science (AAAS). An active member of the profession, he currently Chairs the ACS Publications Committee and the C&EN Editorial Board and is a member of the Board of Directors of Organic Reactions, Inc. |
Introduction
Though the concept of chemical frustration has long been established,1 Frustrated Radical Pairs (FRPs) are relatively new to the field.2,3 FRPs bear similarities to Frustrated Lewis Pairs (FLPs). In their 2006 seminal report, Stephan and coauthors demonstrated that (C6H2Me3)2PH and B(C6F5)3 did not show traditional Lewis acid–base chemistry due to steric repulsion. Instead, what they termed as frustrated Lewis pairs was shown to activate dihydrogen (H2).4 After this breakthrough discovery, follow-up studies explored the different applications of FLP chemistry.5 Among the most important applications of FLPs were their use in activating small molecules, including H2, CO2, and CO (Fig. 1).6,7 Mechanistic investigations into such small molecule activations would ultimately lead to the discovery of frustrated radical pairs (FRPs).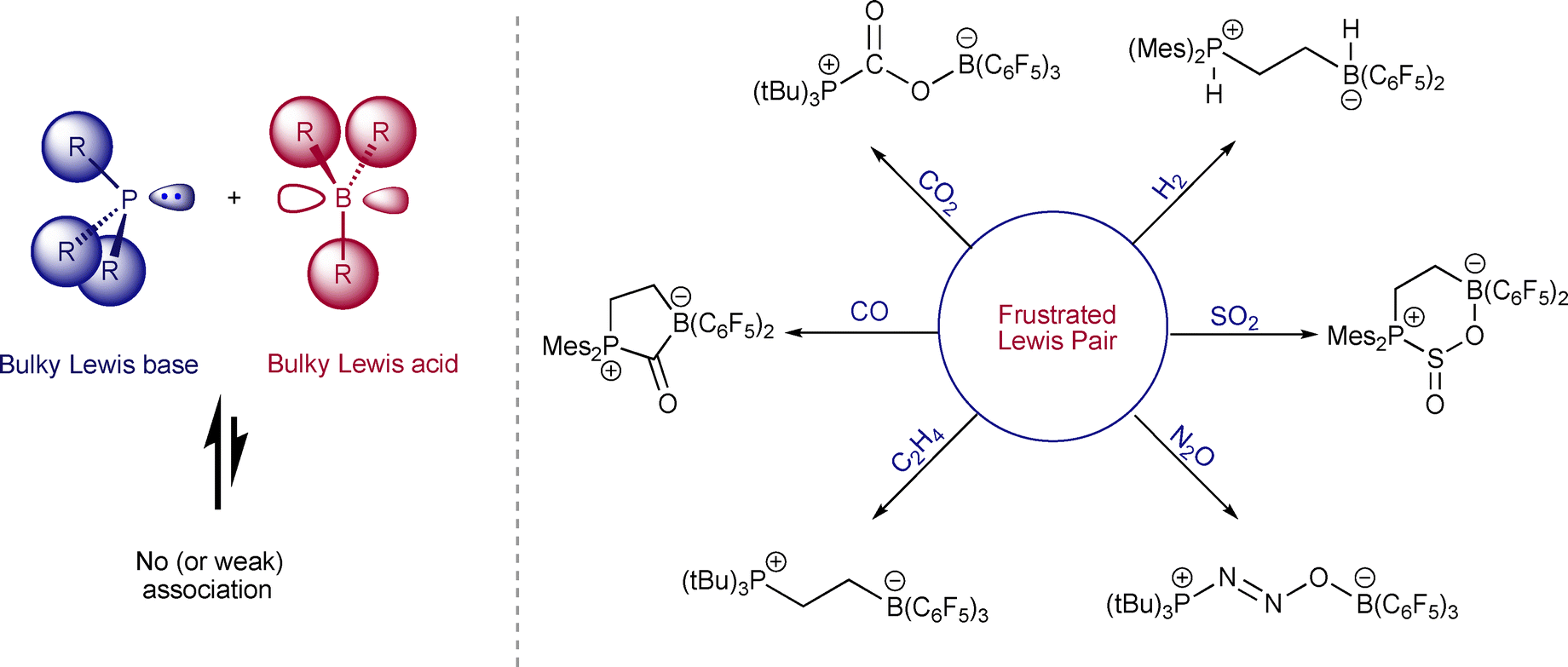 | ||
| Fig. 1 Frustrated Lewis pair and its applications for activating small molecules. | ||
The seeds of that discovery were planted in 2011 when Piers suggested four possible mechanisms for H2 activation using FLP chemistry.8 Other than the heterolytic cleavage of the H2 bond within the reactive pocket of the encounter complex (Fig. 2, pathway a),9 a homolytic pathway was also hypothesized (Fig. 2, pathway (b)). This latter mechanism postulates single electron transfer (SET) from a Lewis base t-Bu3P to Lewis acid B(C6F5)3 to form a radical anion/radical cation ion pair, which then homolytically cleaves a hydrogen bond. This radical ion pair was later recognized as the frustrated radical ion pair. Piers acknowledged that due to the mismatch of redox potential for B(C6F5)3 and t-Bu3P, any formation of this radical cation/anion ion pair would be limited to subnanomolar concentrations. Later, in 2013, Stephan proposed a similar mechanism for activating N2O using Al(C6F5)3 and R3P (R = t-Bu, mesityl, naphthyl), which involves the frustrated radical ion pairs formed via SET. The presence of radical ion pairs was observed by an EPR study, marking these as the first reported examples of frustrated radical ion pairs.10 This discovery highlighted the potential of FRPs in C–H activation, inspiring further exploration and application in chemistry.
 | ||
| Fig. 2 Heterolytic (a) and homolytic (b) pathways to activate hydrogen. Pathway (b) led to the origin of the frustrated radical pair. | ||
Working principle
The chemical frustration in most FRPs arises from steric hindrance.11 The chemical bond between two frustrated radicals would lie between states I and II on the potential energy vs internuclear distance curve (Fig. 3A). At state I, two atoms/molecules are far apart and experience no attractive force. As atoms/molecules begin to experience attractive forces, state II is reached, where the potential energy is lowest. This repulsive force then dominates III (Fig. 3A).12 Regarding FRPs, the two bulky radicals would approach each other (due to inherent radical properties), aiming to reach the lowest energy state by radical combination, but steric repulsion forbids such a process. Therefore, they lie at state IV (shaded area), which is between states I and II. At state IV, two bulky radicals experience attractive forces, but steric prohibit the coupling process, and thus, the radical pair is frustrated. | ||
| Fig. 3 (A) Potential energy vs. internuclear diagram representing chemical frustration. (B) The persistent radical effect in frustrated radical pair. | ||
This begs the question of why the two TEMPO radicals are not considered FRPs (Fig. 4)? Afterall, TEMPO is a bench-stable radical (lifetime ∼2 years) and is widely used in organic synthesis.13
 | ||
| Fig. 4 No radical recombination in TEMPO radical. | ||
The answer lies in the different reactivity of the two frustrated radicals, which is not applied in the case of the identical TEMPO radicals. The distinct reactivity of FRPs is best understood by consideration of the persistent radical effect (PRE). The PRE is a kinetic phenomenon that explains the high cross-selectivity of radical–radical couplings.14–16 If two radicals with different lifetimes are generated at equal rates, the longer-lived “persistent” radical will accumulate over time compared to the short-lived “transient” radical, resulting in high cross-coupling selectivity. Since FRPs cannot cross-couple due to steric hindrance, they follow another pathway leading to distinct reactivities.14–16 When two frustrated radicals, a transient radical T˙ and persistent radical P˙, are generated in solution at equal rates, the hindered transient radical T˙ can generate another unhindered transient radical T′˙via different chemical transformations like rearrangement, fragmentation, atom or group transfer and addition to an unsaturated molecule. This unhindered transient radical T′˙ later cross-couples with the hindered persistent radical P˙ (Fig. 3B).14–16 This unique reactivity has opened new avenues for this chemistry.
A recent report by the Lin group exploited this phenomenon by first generating a hexamethyldisilazane and TEMPO radical FRP via in situ single electron transfer (SET). The hexamethyldisilazane radical (the hindered transient radical) was able to abstract a hydrogen atom from cyclohexane (N–H vs. C–H; 109 kcal mol−1, 98 kcal mol−1) to form cyclohexyl radical (an unhindered transient radical). Notably, the TEMPO radical does not abstract the H-atom as the BDE of the O–H bond is 70 kcal mol−1, which is 30% weaker than the typical O–H bond.13 The unhindered transient cyclohexyl radical is subsequently trapped by the persistent TEMPO radical to give a cross-coupled product 1 (Scheme 1).17
 | ||
| Scheme 1 (A) Reaction conditions for C–H bond activation using FRP. (B) Trapping of cyclohexyl radical with TEMPO governed by the persistent radical effect. | ||
In 2021, the Tang group reported on the sulfenylation of indoles under aerobic conditions by irradiation of B(C6F5)3 and indole-based electron donor–acceptor (EDA) complex (Scheme 2). Here again, the persistent radical effect can be invoked. Specifically, transient [B(C6F5)3]−˙, after reacting with oxygen, gives an intermediate that abstracts an H-atom from thiophenol to generate another unhindered transient radical (3a). Transient radical 3a couples with the persistent indole-based radical cation to provide product 4 upon proton removal (Scheme 2B).18 This report, along with other similar studies, has played a pivotal role in demonstrating the unique reactivity of frustrated radical pairs, enlightening the scientific community about the potential of this chemistry.19–22
 | ||
| Scheme 2 (A) Reaction scheme for sulfonation of indole. (B) The proposed reaction mechanism, governed by the persistent radical effect. | ||
Ways of generating FRPs and their applications
FRPs can be categorized as neutral or ionic depending on the reductant and oxidant. If the reductant and oxidant are ionic, single electron transfer (SET) from the reductant to the oxidant generates a neutral FRP.23 Conversely, a neutral reductant and oxidant pair generate an ionic FRP. Both are addressed as FRPs in this highlight. To date, three ways have been shown to generate FRPs from FLPs: (a) by thermal SET, (b) by photoinduced SET, and (c) by substrate-assisted generation of FRPs from FLPs.10,24 Each of these methods and applications of the so-generated FRPs are further described below.Thermal SET for the generation of FRP
In considering the generation of FRPs from FLP, it is helpful to examine the process through the lens of Mulliken theory. Donors and acceptors interact to form electron donor–acceptor (EDA) complexes [D–A], which generally have a new absorption band (typically lying in the 400–700 nm range) different from that of the individual donors and acceptors. The frontier orbitals of electron donor (D) and electron acceptor (A) interact to form new molecular orbitals that cause this new absorption band (Fig. 5A). EDA complexes are analogs to the weakly bound species known as an encounter complex (E) in FLP chemistry.22,25 Encounter complexes are held together by weak non-covalent interactions, not classical dative bonds.9,26 Upon thermal excitation of these EDA or encounter complex (E), SET from a donor (D) to an acceptor (A) forms a radical ion pair [D˙+, A˙−] (FRP) as long as the energy gap (ΔE) for SET is < 9 kcal mol−1.27,28 These radical ion pairs [D˙+, A˙−] often undergo back electron transfer to generate [D, A] complex. According to Boltzmann distribution, for a 0.06![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M solution, this corresponds to equilibrium with an energy gap (ΔG) of approximately 9 kcal mol−1 (0.4 eV) between the EDA complex (ground-state) and the radical pair. This leads to the formation of detectable radicals in concentrations as low as 10−8M via thermal SET (Fig. 5B). The Slootweg group has shown that this energy gap depends on the Lewis base's ionization energy and Lewis acid's electron affinity.29
M solution, this corresponds to equilibrium with an energy gap (ΔG) of approximately 9 kcal mol−1 (0.4 eV) between the EDA complex (ground-state) and the radical pair. This leads to the formation of detectable radicals in concentrations as low as 10−8M via thermal SET (Fig. 5B). The Slootweg group has shown that this energy gap depends on the Lewis base's ionization energy and Lewis acid's electron affinity.29
 | ||
| Fig. 5 (A) General molecular orbital diagram for the formation of EDA complex and its excitation. (B) Schematic diagram representing the formation of FRPs through thermal single electron transfer. | ||
The Wang group provided evidence for the formation of methylene-bridged triarylamine radical cation 5a. When a 1:1 mixture of triphenylamine 5 and B(C6F5)3 in dichloromethane was stirred for 72 h at room temperature, SET from triarylamine to B(C6F5)3 was observed. EPR studies and UV-vis provided evidence for triphenylamine radical cation 5a formation (Scheme 3A).30 Recently, the Ooi group has shown the thermal SET between a mixture of B(C6F5)3 and N-methyl-N-((trimethylsilyl)methyl)aniline derivative 6 under similar conditions. N-Methyl-N-((trimethylsilyl)methyl)aniline derivative 6 was chosen due to its lower oxidation potential (0.23 V), which, upon SET, formed radical cation 6a (stability stemmed from the hyper-conjugation effect of the silicon–carbon bond), which was supported by ESR and UV-vis studies (Scheme 3B).31
 | ||
| Scheme 3 Generation of FRP via thermal single electron transfer by (A) Wang and coworkers (2013), (B) Ooi and coworkers (2020). | ||
Another landmark paper was recently published by the Lin group in which they used the hexamethyldisilazide anion (HMDS−) and the N-oxoammonium cation 2,2,6,6-tetramethyl-1-oxo-piperidinium (TEMPO+) 7. Single electron transfer from HMDS− to TEMPO+ resulted in the formation of neutral FRP that consists of a persistent radical TEMPO˙ 7a and transient radical HMDS˙ (Scheme 4A).32 The EPR signal was observed for stable TEMPO˙ (sterically encumbered). However, strong H-atom acceptor HMDS˙ could not be seen in EPR (N–H BDE ∼109 kcal mol−1). Together, these species could do regioselective activation of C–H bonds in small and complex molecules (reaction conditions mentioned in Scheme 1A), which could be controlled by tuning the structure of the donor molecule (Scheme 4B). After activation of the C–H bond, TEMPO trapped species 7b showed a reduction, halogenation, deuteration, and nucleophilic substitution, among many other things, showing the diversification of this product.14 Following their work, they have recently demonstrated that FRPs containing transient alkoxy radicals and persistent TEMPO radicals, by operating under PRE, undergo β-scission, radical cyclization, and remote C–H functionalization (Scheme 5).33
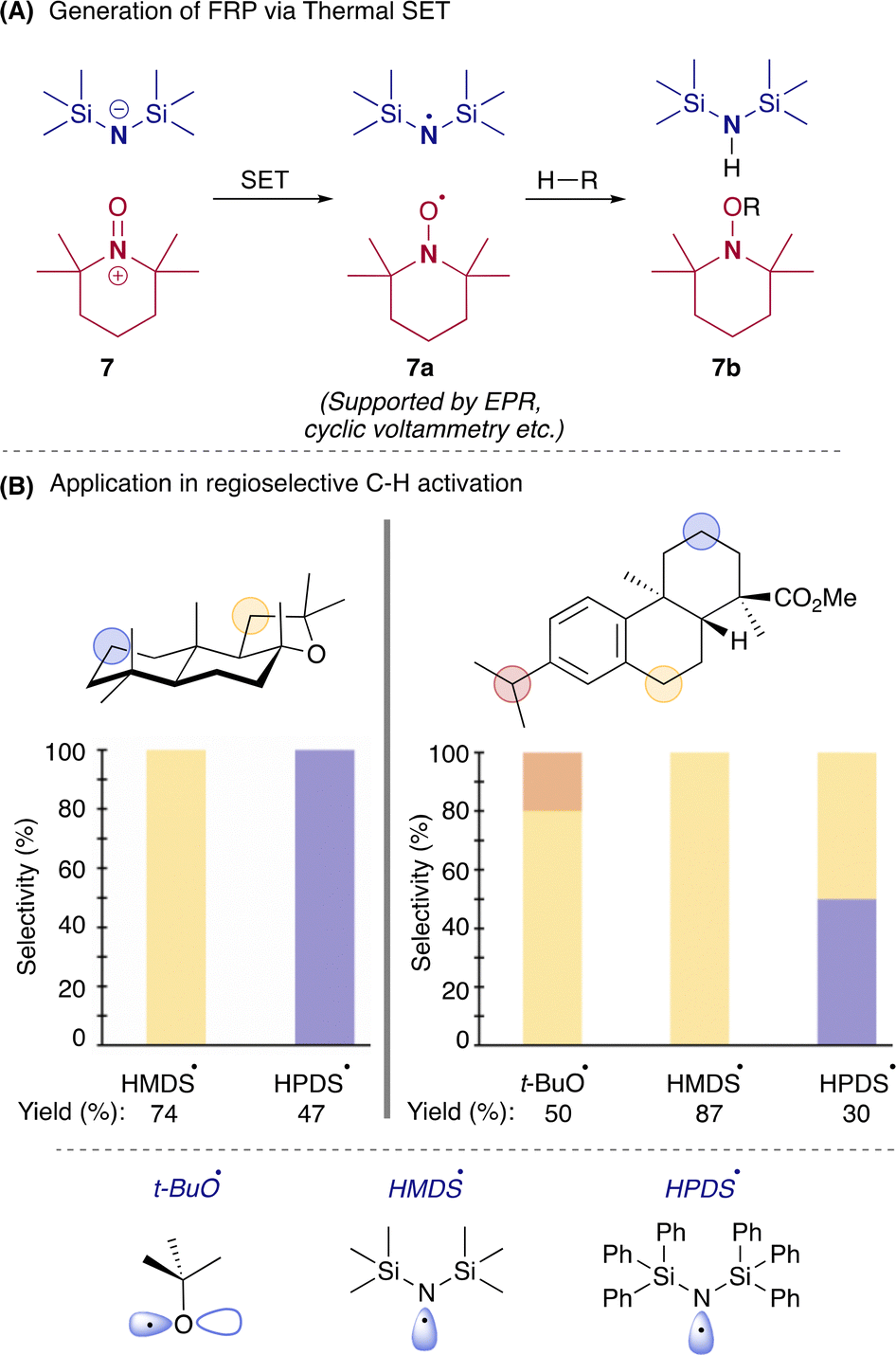 | ||
| Scheme 4 (A) Generation of FRP via Thermal SET. (B) Application in regioselective C–H bond activation. | ||
 | ||
| Scheme 5 Application of FRP in β-scissions, alkene addition and 1,5-HAT reactions. | ||
Photoinduced SET for the generation of FRP
The energy difference between the encounter complex (E) (or EDA complex) and the corresponding radical ion pair plays a vital role in determining the nature of single electron transfer. In this way, the generation of FRPs from FLPs is viable even when ΔE > 9 kcal mol−1. In 2020, Slootweg demonstrated that in the case of a significant energy gap (35.7–71.4 kcal mol−1, λ = 400–800 nm), upon irradiation of visible light, SET can occur in encounter complex to form radicals (Fig. 6A). Moreover, it is possible to tune this energy gap and predict the nature of SET based on the ionization energy (IE) of the donor and the electron affinity (EA) of the acceptor.29 | ||
| Fig. 6 (A) Schematic diagram representing generation of FRP by photoinduced SET (B). Ionization energy (IE) Lewis base and electron affinity of Lewis acid calculated by Slootweg group using the SCRF/ωB97X-D/6-311+G(d,p) level of theory (solvent = toluene). | ||
In 2011, while suggesting a mechanism for generating frustrated radical pairs, Piers accounted for the mismatch of redox potential in (t-Bu)3P/B(C6F5)3 FLP. Therefore, he mentioned that this pair is not suitable for thermal SET.8 Later, Slootweg calculated the energy gap between an FLP and its corresponding radical ion pair based on IE and EA of the corresponding donor and acceptor (Fig. 6B). In this way, the calculated energy gaps (sum of IE and EA) between the Lewis acid/base pair and its corresponding radical ions were 67.4 kcal mol−1 for (t-Bu)3P/B(C6F5) and 57.8 kcal mol−1 for (Mes)3P/B(C6F5)3. The authors suggested that these FLPs will undergo a photoinduced SET process as the value lies in the visible light range (Fig. 7). Upon irradiation of visible light for 90 seconds on corresponding FLP, an EPR signal was observed that was attributed to the superposition of two radical species (Mes3P˙+/B(C6F5)3˙− and (t-Bu)3P˙+ and B(C6F5)3˙−). This work demonstrated that the energy required for single electron transfer could be decreased to fall under the thermal process by wisely choosing Lewis acid and Lewis base pair.29
 | ||
| Fig. 7 DFT- calculated energy required for forming an FRP using the Lewis base ionization energy (IE) and the Lewis acid's electron affinity (EA). Level of theory – SCRF/ωB97X-D/6-311+G(d,p) (solvent = toluene). | ||
As previously alluded to, the Tang group utilized this concept on the B(C6F5)3 and indole-based EDA complex. Upon irradiation of Blue LED on this EDA complex for 7–12 h, FRP 8a was generated, which was utilized in the sulfenylation of indoles under aerobic conditions. Upon irradiation by a 30 W blue LED lamp at 298 K for 10 min with blue LEDs under air, ethylene chloride solution containing 8 (0.1 M) and B(C6F5)3 (0.005 M) gave an intense EPR signal (g = 2.00296) (Scheme 6A).18 Ooi and co-workers observed that an equimolar mixture of 4-bromo-N,N-dimethylaniline 9 and B(C6F5)3 in dichloromethane did not give a signal. Interestingly, irradiation with a 405 nm LED light source led to the detection of FRP 9a (Scheme 6B). Compared to N-trimethylsilylmethyl 6 (Scheme 3B), this different behavior of 4-bromo-N,N-dimethylaniline 9 could be attributed to its lower reactivity and its higher oxidation potential (6: 0.23 V, 9: 0.50 V vs. Fc/Fc+). In addition, rates of different back electron transfers are also crucial. External energy from photoirradiation is required due to the higher energy barrier for SET and the faster back electron transfer of 9a. Gibbs free energy value calculated by DFT calculation for SET reactions also supported that energy was 4.2 kcal mol−1 higher for 9 and 9a (Scheme 6B) than between 6 and 6a (Scheme 3B).31 These FRPs are also applied in carbon–carbon bond formation reactions. This FRP in the presence of methyl vinyl ketone afforded compound 10a with 31% yield in 36 hours (Scheme 6C). Recently, the same group has also shown such a phenomenon on an intramolecular FLP p-diarylboryl halothiophenolates, which, upon absorbing visible light, showed an intramolecular charge transfer to form a radical pair consisting of a thiyl radical and boron radical anion.19 Another emerging subfield related to FRP chemistry is generation and use of poly(FRPs). Recent work utilized photoinduced SET to generate poly(FRPs), which were shown to catalyze hydrogenations, gas-gelations, and radical-mediated photocatalytic perfluoroalkylations.34
 | ||
| Scheme 6 (A), (B) examples of the generation of FRP by photoinduced thermal SET. (C) Application of Ooi's FRP in carbon–carbon bond formation. | ||
Substrate-assisted generation of FRPs from FLPs
Weaky-associated FLP encounter complexes (E) can also lead to the formation of a reactive pocket that helps in the activation of small molecules. Such FLP encounter complexes can thereby react with a small substrate (S), which in turn can generate a frustrated radical pair (Fig. 8). | ||
| Fig. 8 Generation of FRP through the substrate-assisted method. | ||
An example of substrate-assisted generation FRPs from FLPs can be found in Stephan's work on the capture of N2O by a boron and phosphorous-based FLP (B(C6F5)3/t-Bu3P). Building off the knowledge that the formation of Lewis acid–base adduct (t-Bu3PO)B(C6F5)3 is possible through ejection N2 under thermolysis or photolysis condition,35 Stephan showed that altering the Lewis acid affects the chemistry dramatically. Namely, the R3P/Al(C6F5)3 pair could not only capture N2O to form an R3P(N2O)Al(C6F5)3 (R = Mes, t-Bu) adduct, but upon reacting with another equivalent of Al(C6F5)3 liberated N2 and generated transient FRPs 11 (Scheme 7A).
 | ||
| Scheme 7 (A) Generation FRP using N2O. (B) Application in C–H activation. | ||
For ([Mes3P˙]+)/[(μ-O˙)(Al(C6F5)3)2]− FRP 11a, EPR studies and its UV-vis spectrum indicated the presence of the phosphoniumyl radical cation, but an EPR signal for the Al2 oxyl radical anion [(μ-O˙)(Al(C6F5)3)2]− was not observed. This failure was attributed to that species shorter lifetime. Nonetheless, it was found that [(μ-O˙)(Al(C6F5)3)2]− capable of undergoing hydrogen atom transfer(HAT) with the toluene solvent as [(μ-HO)(Al(C6F5)3)2]− was observed (Scheme 7B). Additionally, FRP ([t-Bu3P˙]+)/[(μ-O˙)(Al(C6F5)3)2]−11b, where the ligand on phosphorous switched from Mes to t-Bu, underwent C–H bond activation of the t-butyl group when the solvent was fluorobenzene (Scheme 7B).10 These reactions of 11a and 11b are the first examples of C–H bond activation via FRP chemistry. It is worth noting that, unlike with 11a, an EPR signal was not observed for [t-Bu3P˙]+.10 This difference was explained by the fact that, as Slootweg and co-workers determined by transient absorption spectroscopy,29 [t-Bu3P˙]+ has a shorter lifetime than [Mes3P˙]+ (6 vs. 273 ps respectively).
FRPs can also be generated from FLPs in which SET cannot occur spontaneously. In such cases, adding a substrate that will accept an electron from the Lewis base and donate it to the Lewis acid facilitates the generation of the FRP.
Tetrachloro-p-benzoquinone (TCQ) is among the substrates that can facilitate the generation of an FRP from an FLP by a concerted Lewis acid/base action.26,27 This was demonstrated by Stephan and co-workers, who detected a visible absorption maximum at 573 nm when half an equivalent of TCQ 14 was added to Mes3P/B(C6F5)3 in toluene at −78 °C. This measurement is consistent for [Mes3P]˙+, though only a weak EPR signal was observed.36 A Mes3P/B(C6F5)3 FLP in the presence of TCQ gave [Mes3P˙]+2[(C6F5)3BOC6Cl4OB(C6F5)3]2−14a (Scheme 8A). The identification of intermediate radical salts supported that the mechanism followed SET from the Lewis base to the Lewis acid.37 Furthermore, Slootweg24 found that this reaction also proceeds in the dark. B(C6F5)3 and TCQ 14 are not strong enough electron acceptors (electron affinity (EA) = −3.31 and −4.45 eV, respectively38) to facilitate a thermal SET. It was suggested that when B(C6F5)3 coordinates to TCQ 14 to form TCQ-B(C6F5)3 adduct 14b, the electron affinity (EA = −5.57 eV) increases. As a result, rapid SET from Mes3P to 14b forms the radical ion pair [Mes3P]˙+[C6Cl4O2B(C6F5)3]˙−14c (Scheme 8B).24,39,40 In short, adding TCQ facilitated the SET process in Mes3P/B(C6F5)3 to generate FRP.41
 | ||
| Scheme 8 (A) FRP Generation upon adding 1/2 equivalent of Tetrachloro-p-benzoquinone (TCQ) (B) Proposed mechanism for the generation of FRP 8c where TCQ EA increases by coordination with Lewis acid B(C6F5)3. | ||
Conclusion
Compared to the frustrated Lewis pair, frustrated radical pair chemistry is relatively new. Nonetheless, frustrated radical pairs have shown applications in weak and strong bond activation, sulfenylation, small molecule activation, dehydrohalogenation reactions, radical cyclization and many more. Some examples have shown that selectivity in this chemistry can be easily tuned and used in diverse chemical modifications, making it even more exciting and showing that it could be an essential synthesis toolkit. As this field continues to evolve, it enhances understanding of fundamental principles like FLP, PRE, and radical chemistry. It also holds promises of advancing its application to different fields of chemistry.Author contributions
The authors contributed equally to the writing of this Highlight.Data availability
All data are stored in the originally, already published papers cited in this Highlight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of Michigan State University.References
- G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalog Company, Inc., 1923 Search PubMed.
- L. J. C. van der Zee, J. Hofman, J. M. van Gaalen and J. C. Slootweg, Mechanistic Studies on Single-Electron Transfer in Frustrated Lewis Pairs and its Application to Main-Group Chemistry, Chem. Soc. Rev., 2024, 53, 4862–4876 RSC.
- A. Dasgupta, E. Richards and R. L. Melen, Frustrated Radical Pairs: Insights from EPR Spectroscopy, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Reversible, Metal-Free Hydrogen Activation, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan, The Broadening Reach of Frustrated Lewis Pair Chemistry, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- R. Pal, M. Ghara and P. K. Chattaraj, Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs, Catalysts, 2022, 12, 201 CrossRef CAS.
- W. E. Piers, A. J. V. Marwitz and L. G. Mercier, Mechanistic Aspects of Bond Activation with Perfluoroarylboranes, Inorg. Chem., 2011, 50, 12252–12262 CrossRef CAS PubMed.
- A. T. Littlewood, T. Liu, L. E. English, L. Chen, T. A. Barendt and A. R Jupp, Quantifying Interactions in the Active Encounter Complex of Frustrated Lewis Pairs, Nat. Commun., 2025, 16, 3666 CrossRef PubMed.
- G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, C–H Bond Activation by Radical Ion Pairs Derived from R3P/Al(C6F5)3 Frustrated Lewis Pairs and N2O, J. Am. Chem. Soc., 2013, 135, 6446–6449 CrossRef PubMed.
- F.-G. Fontaine and D. W. Stephan, On the Concept of Frustrated Lewis Pairs, Phil. Trans. R. Soc. A, 2017, 375, 20170004 CrossRef PubMed.
- P. L. A. Popelier and R. J. Gillespie, Chemical Bonding and Molecular Geometry from Lewis to Electron Densities, Oxford University Press, 2001 Search PubMed.
- F. Montanari, S. Quici, H. Henry-Riyad and T. T. Tidwell, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2006 Search PubMed.
- H. Fischer, The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed.
- K.-S. Focsaneanu and J. C. Scaiano, The Persistent Radical Effect: From Mechanistic Curiosity to Synthetic Tool, Helv. Chim. Acta, 2006, 89, 2473–2482 CrossRef CAS.
- D. Leifert and A. Studer, The Persistent Radical Effect in Organic Synthesis, Angew. Chem., Int. Ed., 2020, 59, 74–108 CrossRef CAS PubMed.
- Z. Lu, M. Ju, Y. Wang, J. M. Meinhardt, J. I. Martinez Alvarado, E. Villemure, J. A. Terrett and S. Lin, Regioselective Aliphatic C–H Functionalization Using Frustrated Radical Pairs, Nature, 2023, 619, 514–520 CrossRef CAS PubMed.
- W. Yuan, J. Huang, X. Xu, L. Wang and X.-Y. Tang, B(C6F5)3-Catalyzed Electron Donor–Acceptor Complex-Mediated Aerobic Sulfenylation of Indoles under Visible-Light Conditions, Org. Lett., 2021, 23, 7139–7143 CrossRef CAS PubMed.
- T. Kikura, Y. Taura, Y. Aramaki and T. Ooi, p-Diarylboryl Halothiophenols as Multifunctional Catalysts via Photoactive Intramolecular Frustrated Lewis Pairs, J. Am. Chem. Soc., 2024, 146, 20425–20431 CrossRef CAS PubMed.
- F. Xie, J. He and Y. Zhang, Frustrated Radical Pair-Initiated Hydrodehalogenation of Alkyl and Aryl Halides Under Visible Light, ChemistrySelect, 2024, 9, e202400477 CrossRef CAS.
- L. L. Liu, L. L. Cao, D. Zhu, J. Zhou and D. W. Stephan, Homolytic Cleavage of Peroxide Bonds via a Single Electron Transfer of a Frustrated Lewis Pair, Chem. Commun., 2018, 54, 7431–7434 RSC.
- M. Ju, Z. Lu, L. F. T. Novaes, J. I. Martinez Alvarado and S. Lin, Frustrated Radical Pairs in Organic Synthesis, J. Am. Chem. Soc., 2023, 145, 19478–19489 CrossRef CAS PubMed.
- Frustrated Lewis Pairs, ed. J. Chris Slootweg and A. R. Jupp, Springer International Publishing, Cham, 2021, vol. 2 Search PubMed.
- F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin and J. C. Slootweg, Single-Electron Transfer in Frustrated Lewis Pair Chemistry, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS PubMed.
- L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, Probing the Association of Frustrated Phosphine–Borane Lewis Pairs in Solution by NMR Spectroscopy, J. Am. Chem. Soc., 2014, 136, 112–115 CrossRef CAS PubMed.
- A. R. Jupp, Evidence for the Encounter Complex in Frustrated Lewis Pair Chemistry, Dalton Trans., 2022, 51, 10681–10689 RSC.
- R. S. Mulliken, Molecular Compounds and their Spectra. II, J. Am. Chem. Soc., 1952, 74, 811–824 CrossRef CAS.
- R. S. Mulliken, Molecular Compounds and their Spectra. III. The Interaction of Electron Donors and Acceptors, J. Phys. Chem., 1952, 56, 801–822 CrossRef CAS.
- F. Holtrop, A. R. Jupp, N. P. van Leest, M. Paradiz Dominguez, R. M. Williams, A. M. Brouwer, B. de Bruin, A. W. Ehlers and J. C. Slootweg, Photoinduced and Thermal Single-Electron Transfer to Generate Radicals from Frustrated Lewis Pairs, Chem. – Eur. J., 2020, 26, 9005–9011 CrossRef CAS PubMed.
- X. Zheng, X. Wang, Y. Qiu, Y. Li, C. Zhou, Y. Sui, Y. Li, J. Ma and X. Wang, One-Electron Oxidation of an Organic Molecule by B(C6F5)3; Isolation and Structures of Stable Non-para-substituted Triarylamine Cation Radical and Bis(triarylamine) Dication Diradicaloid, J. Am. Chem. Soc., 2013, 135, 14912–14915 CrossRef CAS PubMed.
- Y. Aramaki, N. Imaizumi, M. Hotta, J. Kumagai and T. Ooi, Exploiting Single-Electron Transfer in Lewis Pairs for Catalytic Bond-Forming reactions, Chem. Sci., 2020, 11, 4305–4311 RSC.
- J. C. Siu, G. S. Sauer, A. Saha, R. L. Macey, N. Fu, T. Chauviré, K. M. Lancaster and S. Lin, Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO–N3 Charge-Transfer Complex, J. Am. Chem. Soc., 2018, 140, 12511–12520 CrossRef CAS PubMed.
- M. Ju, S. Lee, H. M. Marvich and S. Lin, Accessing Alkoxy Radicals via Frustrated Radical Pairs: Diverse Oxidative Functionalizations of Tertiary Alcohols, J. Am. Chem. Soc., 2024, 146, 19696–19703 CrossRef CAS PubMed.
- M. Wang, M. Shanmugam, E. J. L. McInnes and M. P. Shaver, Light-Induced Polymeric Frustrated Radical Pairs as Building Blocks for Materials and Photocatalysts, J. Am. Chem. Soc., 2023, 145, 24294–24301 CrossRef CAS PubMed.
- E. Otten, R. C. Neu and D. W. Stephan, Complexation of Nitrous Oxide by Frustrated Lewis Pairs, J. Am. Chem. Soc., 2009, 131, 9918–9919 CrossRef CAS PubMed.
- X. Pan, X. Chen, T. Li, Y. Li and X. Wang, Isolation and X-ray Crystal Structures of Triarylphosphine Radical Cations, J. Am. Chem. Soc., 2013, 135, 3414–3417 CrossRef CAS PubMed.
- L. Liu, L. L. Cao, Y. Shao, G. Ménard and D. W. Stephan, A Radical Mechanism for Frustrated Lewis Pair Reactivity, Chemistry, 2017, 3, 259–267 CrossRef CAS.
- Note: The electron affinity (EA) value mentioned for B(C6F5)3 –3.31 is different from what was mentioned in Fig. 6B –3.03. Both values were calculated using ωB97X-D/6-311+G(d,p), but with different solvents (C6H5Cl and toluene, respectively).
- J. M. Blackwell, W. E. Piers and M. Parvez, Mechanistic Studies on Selectivity in the B(C6F5)3-Catalyzed Allylstannation of Aldehydes: Is Hypercoordination at Boron Responsible?, Org.
Lett., 2000, 2, 695–698 CrossRef CAS PubMed.
- R. C. West and A. F. Hill, Advances in Organometallic Chemistry, Elsevier, 2005 Search PubMed.
- B. L. Thompson and Z. M. Heiden, Tuning the Reduction Potentials of Benzoquinone Through the Coordination to Lewis Acids, Phys. Chem. Chem. Phys., 2021, 23, 9822–9831 RSC.
| This journal is © The Royal Society of Chemistry 2025 |