
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePCP and POCOP complexes of calcium†
Alessandro
Messori‡
a,
Marcel
Potocnik‡
a,
Sara
Belazregue
a,
Richard
Collins
 a,
Tobias
Krämer
b and
F. Mark
Chadwick
*a
a,
Tobias
Krämer
b and
F. Mark
Chadwick
*a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 82 Wood Lane, London, UK
bSchool of Chemistry, Trinity College Dublin, College Green, Dublin 2, Ireland
First published on 28th April 2025
Abstract
The first synthesis of PCP and POCOP complexes of calcium are described. Ca(N(SiMe3)2)2 is reacted with Li[tBuPCP] or Li[tBuPOCOP] to form (tBuPCP)Ca(N(SiMe3)2) (1a) and (tBuPOCOP)Ca(N(SiMe3)2) (1b). Reaction of 1b or Ca(N(SiMe3)2)2with excess Li[tBuPOCOP] leads to a homoleptic complex (tBuPOCOP)2Ca, 2. The (iPrPOCOP) ligand exclusively forms the homoleptic complex (iPrPOCOP)2Ca, 3.
In comparison to transition metals, the organometallic chemistry of the alkaline-earth metals has been severely neglected. Due to both their perceived lack of redox activity and their electropositive nature, their chemistry has often been relegated to use as synthons for the more popular transition metals, most notably as Grignard reagents.1,2 However, recent awareness of the sustainability issues surrounding metals commonly used in catalysis has brought the spotlight onto the more earth abundant metals, and particularly those of group 2.3
In the last couple of decades, the alkaline-earth metals’ chemistry has been further developed. Groups have developed polymerisation, hydroamination and hydrosilylation catalysis using these metals.3–7 Calcium has been deployed into the field of small molecule activation, including CO homologation, the reduction of benzene and N2 activation.8–10 Though group 2 pincer complexes have recently been used for hydrogenation chemistry, the chemistry is dominated by the use of the β-diketimidate ‘nacnac’ ligand.11 Since Chisholm's breakthrough paper in 2004, the simple (nacnac)Ca fragment has been the feature of over 100 papers.12–15 We recently developed a route to ‘RPCP’M complexes that precludes the traditional requirement of oxidative addition – this ligand shares some of the ‘nacnac’ ligand's primary features (monoanionic, ease of installation of bulk close to the metal centre) whilst retaining useful differences (e.g. tridentate vs. bidentate).16,17 We therefore wished to investigate whether it was possible to make RPCP/RPOCOP calcium complexes (RPCP = 2,6-(R2PCH2)2C6H3; RPOCOP = 2,6-(R2PO)2C6H3). To date there are only two reports of group 2 RPCP complexes (both reporting a (RPCP)2Mg fragment),18,19 and there are no reports of group 2 RPOCOP complexes. Previous attempts to make ‘(RPCP)Ca’ complexes resulted in a mixture of products which cleaved the P–C bonds.18 Very recently, Milstein has developed a route to anionic ‘PNP’ pincer complexes of calcium, which showed interesting small molecule reactivity with N2O.20 Herein, we report the formation of the first RPCP and RPOCOP Ca complexes.
Both (tBuPCP)Ca(HMDS) (tBuPCP = 2,6-(tBu2PCH2)2C6H3; HMDS = N(SiMe3)2, 1a) and (tBuPOCOP)Ca(HMDS) (tBuPOCOP = 2,6-(tBu2PO)2C6H3, 1b) can be made by analogous routes (Scheme 1). The appropriate lithium precursor is reacted with Ca(HMDS)2(THF)2 in pentane. The solution remains colourless throughout. Once reaction is complete (1a: 3 days, 1b 2 hours) the solvent is removed, and the product extracted in pentane. Pure 1a and 1b may be crystallised from cold pentane in good yields (1a: 62%; 1b: 50%).
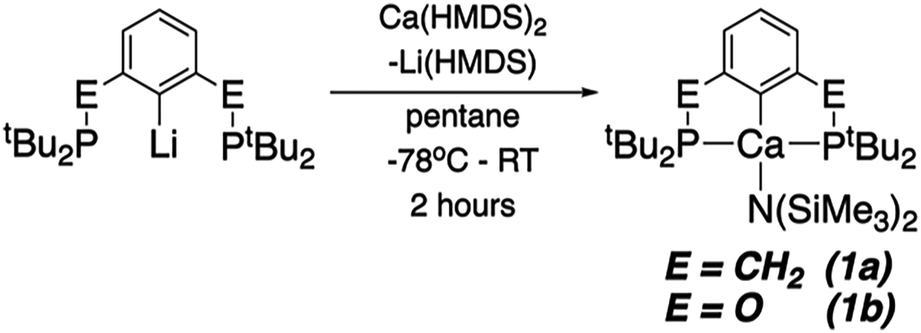 | ||
| Scheme 1 The synthetic route to 1a and 1b. | ||
1a and 1b represent the first examples of ‘PCP’ complexes of calcium. Indeed, pincer complexes of Ca are relatively rare.20–28 Both give the expected 31P{1H} NMR signals (at δ 24.7 and 122.1 ppm for 1a and 1b, respectively). Cyclic voltammetry studies were also undertaken however no electrochemical events were observed, even measuring to −3.0 V (vs. Fc/Fc+).
Fig. 1 shows the solid-state X-ray derived structure of 1a, 1b is isostructural. The calcium centre is tetracoordinate, and adopts an extremely distorted see-saw geometry; 1b is slightly closer to a square planar geometry than 1a (τ4 parameters 0.572 for 1a, 0.557 for 1b).29
 | ||
| Fig. 1 The single crystal X-ray diffraction structure of 1a. 1b is isostructural. Thermal probability ellipsoids at 50%. Hydrogen atoms and the methyl groups of the tert-butyl groups are omitted for clarity. Colours: black (carbon), purple (phosphorus), turquoise (calcium), blue (nitrogen), yellow (silicon). | ||
1a and 1b are air- and moisture-sensitive solids, which can be kept at room temperature in a N2 glovebox. We have not been successful in accessing Ca PCP complexes from CaI2, Ca(CH2Ph)2(THF), Ca(OTf)2 nor directly from Ca metal. 1a and 1b are quite reticent in terms of reactivity. Protonation reactions (with one equivalent of HOTf or Et3NHCl) resulted in pincer ligand protonation preferentially over amide protonation. Reactions with H2 and common hydride sources (NaH and NaBH4) resulted in no reaction, even upon heating.
Reaction of 1b did occur with NaBEt3H and similar hydride sources (KBEt3H, LiBEt3H, N-selectride). Upon initial addition, three distinct new 31P{1H} resonances appear, at δ 124 ppm, δ 126 ppm and δ 130 ppm. Upon addition of excess hydride source, the resonance at δ 130 ppm grows to be the sole product. It was posited that the resonances at δ 124 ppm and δ 126 ppm could be the targeted (tBuPOCOP)CaH species so attempts were made to isolate these. On one occasion crystallisation from pentane resulted in a crystal suitable for X-ray diffraction which elucidated one of these species to be not the targeted hydride, but instead (tBuPOCOP)2Ca (2).
2 can be intentionally made by reacting 2.5 equivalents of Li[tBuPOCOP] with Ca(HMDS)2 (Scheme 2). Analogous reactions with Li[tBuPCP] were not clean, yielding multiple, unidentified products. Recrystallisation from a pentane solution yields 2 in moderate yields (39%). Changing the alkyl group on the phosphine backbone from tert-butyl to iso-propyl resulted in the exclusive formation of the homoleptic product (iPrPOCOP)2Ca (3) even if a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Ca and pincer is used.
:1 mixture of Ca and pincer is used.
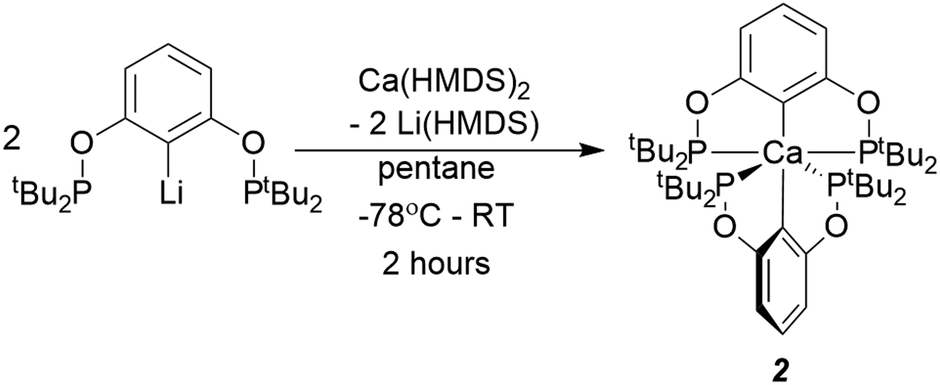 | ||
| Scheme 2 The synthesis of 2. | ||
For both 2 and 3, the two RPOCOP ligands are equivalent at room temperature in the solution phase, giving a single broad resonance in the 31P{1H} NMR (2: δ 126 ppm; 3: δ 113 ppm). The 1H NMR spectra give the expected resonances, with a single doublet corresponding to the tert-butyl protons for 2 (δ 1.21 ppm), and the septet and two apparent doublets of doublets corresponding to the iso-propyl protons in 3 (δ 1.93 ppm, δ 1.14 ppm, δ 1.04 ppm respectively). The single crystal X-ray derived structure of 2 is shown in Fig. 2. 3 is isostructural.
 | ||
| Fig. 2 The single crystal X-ray derived structure of 2. Thermal ellipsoids at 50% probability, hydrogen atoms and tert-butyl methyl groups omitted for clarity. Colours: Red (oxygen) otherwise same as Fig. 1. 3 is isostructural. | ||
For 2, the calcium centre sits upon a special position (C2 axis), so that the two POCOP ligands are symmetry related. The binding of the two ligands almost fully encapsulates the calcium centre.
The geometry of the calcium centre is intriguing. Though a six coordinate complex, it does not adopt the expected octahedral coordination. On inspection, it seems that the geometry is entirely driven by the steric encumbrance of the phosphine groups on the ligands. If one only considers these four ligating groups (P(1), P(2), P(1′), P(2′)), and ignores the carbon atoms from the arene ring (C(1), C(1′)), the structure reveals itself to be approximately tetrahedral. Indeed, using the same treatment on 1a and 1b reveals the Ca centre to be adopting a trigonal planar geometry with P(1), P(2) and N(1) (Σ angles around Ca (ignoring C) 1a: 359.0°; 1b: 359.5°).
Some key structural metrics from 1a, 1b, 2 and 3 are summarised in Table 1. The Ca–C distances are approximately the same throughout and lie in the range of other structurally characterised Ca–Caryl complexes.30 The bond lengths are significantly longer than the previously characterised Mg PCP complexes, reflecting the increased ionic radius (Mg–P distances (Å) 2.770(1)/2.761(1) for Mg(MePCP)2 and 2.692(1)/2.961(1)/2.863(1)2.689(1) for Mg(PhPCP)2).18,19 The slight increase in Ca–P bond lengths in 2 compared to 1b is presumeably due to increased steric crowding. Though there are structurally characterised homoleptic RPCP complexes, 2 and 3 mark the first homoleptic ‘RPOCOP’ complexes.
| 1a | 1b | 2 | 3 | |
|---|---|---|---|---|
| Ca(1)–P(1) | 2.9796(6) | 2.9439(8) | 3.1753(5) | 2.966(1) |
| Ca(1)–P(2) | 2.9859(6) | 2.9587(7) | 3.0653(5) | 3.013(1) |
| Ca(1)–C(1) | 2.513(2) | 2.516(2) | 2.528(2) | 2.526(3) |
| Ca(1)–N(1) | 2.285(2) | 2.279(2) | n/a | n/a |
| Ca to C(1)–P(1)–P(2) plane | 1.174(2) | 0.799(2) | 1.420(2) | 1.202(3) |
| P(1)–Ca(1)–P(2) | 130.43(1) | 130.07(2) | 120.00(2) | 122.8(1) |
In most RPCP/RPOCOP pincer complexes the metal centre is ligated by the PCP in an almost perfect meridional fashion. By contrast in our complexes the metal is significantly out of the C(1)–P(1)–P(2) plane, particularly for 1a and 2.
In the reaction of 1b with PhSiH3, rather than forming the expected Ca hydride, we found that the silane attached to the ligand instead (presumably with concomitant formation of ‘CaH(N(HMDS)2)), producing (tBuPOCOP)SiH2Ph, 4 (Scheme 3).
 | ||
| Scheme 3 The synthesis of (tBuPOCOP)SiH2Ph. | ||
4 can be isolated and crystallised from a pentane solution cooled to −40 °C. Its structure is shown in Fig. 3 and it is clear there is no interaction between the silane and the pendant phosphine arms.
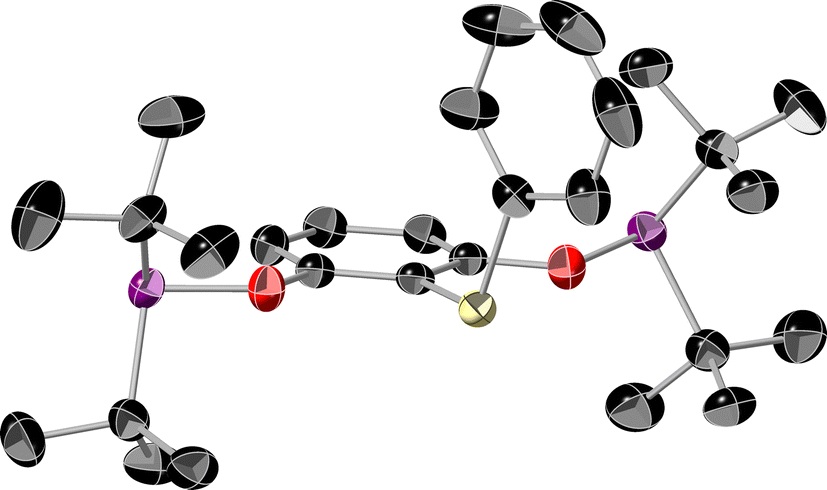 | ||
| Fig. 3 The single crystal X-ray diffraction structure of 4. Thermal ellipsoids set at 50% and hydrogen atoms omitted for clarity. Colours as in Fig. 1. | ||
A series of density functional theory (DFT) calculations have been carried out to interrogate the nature of bonding in complexes 1a, 1b, 2 and 3. Geometries of all complexes were optimised using the M06L functional in conjunction with the def2-TZVP (Ca, P, N, O) and def2-SVP (C, H) basis sets, giving excellent agreement with their crystallographic counterparts (Table S1, ESI†). Detailed analysis of the corresponding wavefunctions obtained from subsequent single point calculations (M06-D3/def2-TZVPP) reveals that for all complexes the HOMO is dominantly localised on the aryl ipso-carbon. The significant lone pair character of this centre explains why the observed reaction of 1b with phenylsilane does not yield the targeted calcium hydride complex (Fig. 4). The LUMO resides on P centres with admixture of 4s and 3d orbital character from Ca2+.
 | ||
| Fig. 4 Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of complexes 1a and 3 (isovalue 0.03 a.u). Hydrogens cloaked for clarity. | ||
The distorted see-saw geometry around the tetracoordinate calcium centre in both 1a and 1b is closely reproduced in the optimised geometries (calculated τ4 values of 0.55 and 0.60, respectively), although the PES is relatively flat. Structural isomers of both complexes with enforced square-planar geometries and (near) linear Cipso–Ca–N angles are destabilised by ∼3 kcal mol−1 relative to the global minimum, both of which relax back to the distorted geometry upon release of the constraints. Similar observations are made for complexes 2 and 3, for which pseudo-octahedral guess structures reverse back to the experimentally observed distorted geometries upon geometry optimisation.
The presence of non-covalent closed-shell interactions between the calcium centres and the ligands in all complexes was confirmed by analysis of the topology of their total electron densities using quantum theory of atoms in molecules (QTAIM). Linear bond paths between Ca2+ and the corresponding donor atoms (P, N and C) are observed for all complexes, approximately coinciding with their respective internuclear bond axes. Bond critical and ring critical points relating to these interactions were readily identified in the molecular graph of each species. The QTAIM descriptors associated with each BCP signal small electron densities, small positive Laplacians and small negative potential energy densities, the latter being approximately matched by the kinetic energy density, i.e., |G(r)| ≈ |V(r)|. Taken together with small covalent bond orders, these data are characteristic of dative interactions in all species.20,31
The absence of covalent bonding between Ca2+ and donor atoms in all complexes is consolidated by intrinsic bond orbitals (IBO) and natural bond orbitals (NBO), underpinning that these interactions are largely due to electron donation from the P, N and C centres with minor contribution from the Ca2+ orbital manifold (Fig. S14, ESI†). A quantitative measure of the donor–acceptor interactions is obtained from second order perturbation theory of the Fock matrix in the NBO basis, with the vacant 4s orbital of Ca2+ serving as the main acceptor orbital. Donations from the P and C lone pairs in complexes 1a (E(2) = 12.1–15.8 kcal mol−1) and 1b (E(2) = 14.2–14.4 kcal mol−1) govern the interactions, while donation from the HMDS ligand are somewhat weaker in both cases (E(2) = 4.0–5.0 kcal mol−1). Likewise, in 2 and 3, the estimates of E(2) for LP(P) → Ca2+ and LP(C) → Ca2+ donations are in the range 9.9–18.2 kcal mol−1. Atomic charges on the calcium ion obtained from both NPA (1.60–1.73) and QTAIM (1.50–1.55) schemes indicate that this centre adopts an oxidation states close to +2 in all complexes.
A series of PCP and POCOP calcium complexes have been made, characterised and their structures investigated by computational calculations. The complexes’ geometries are derived predominantly from the steric effects of the phosphine side arms of the pincer ligands. The HOMO of the complexes is the interaction between the aryl-carbon and the calcium centre, which results in the onward reactivity being focused on these atoms, rather than the HMDS ligand.
Data availability
Crystallographic information files (.cif) for all complexes have been deposited in the CCDC (2423347 (1a), 2423349 (1b), 2423348 (2), 2423351 (3) and 2423350 (4)). Other experimental data in support of this paper are presented in the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- V. Grignard, C. R. Hebd. Seances Acad. Sci., 1900, 130, 1322–1324 CAS.
- M. Westerhausen, Coord. Chem. Rev., 2008, 252, 1516–1531 CrossRef CAS.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- E. Fazekas, P. A. Lowy, M. Abdul Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- C. Brinkmann, A. G. M. Barrett, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2012, 134, 2193–2207 CrossRef CAS PubMed.
- J. Escorihuela, A. Lledós and G. Ujaque, Chem. Rev., 2023, 123, 9139–9203 CrossRef CAS PubMed.
- M. Magre, M. Szewczyk and M. Rueping, Curr. Opin. Green Sustainable Chem., 2021, 32, 100526 CrossRef CAS.
- M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed.
- S. Brand, H. Elsen, J. Langer, W. A. Donaubauer, F. Hampel and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 14169–14173 CrossRef CAS PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- Y. Liang, U. K. Das, J. Luo, Y. Diskin-Posner, L. Avram and D. Milstein, J. Am. Chem. Soc., 2022, 144, 19115–19126 CrossRef CAS PubMed.
- M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed.
- S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- B. Rösch and S. Harder, Chem. Commun., 2021, 57, 9354–9365 RSC.
- Y. C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS.
- B. Stadler, H. H. Y. Meng, S. Belazregue, L. Webster, A. Collauto, K. M. Byrne, T. Krämer and F. M. Chadwick, Organometallics, 2023, 42, 1278–1285 CrossRef CAS PubMed.
- L. Webster, T. Krämer and F. M. Chadwick, Dalton Trans., 2022, 51, 16714–16722 RSC.
- A. Koch, S. Krieck, H. Görls and M. Westerhausen, Inorganics, 2016, 4, 39 CrossRef.
- A. Pape, M. Lutz and G. Müller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2281–2284 CrossRef.
- Y. Liang, I. Efremenko, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2024, 63, e202401702 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2010, 29, 4203–4206 CrossRef CAS.
- A. Koch, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2017, 36, 994–1000 CrossRef CAS.
- J. Jenter, R. Köppe and P. W. Roesky, Organometallics, 2011, 30, 1404–1413 CrossRef CAS.
- M. J. C. Dawkins, A. N. Simonov and C. Jones, Dalton Trans., 2020, 49, 6627–6634 RSC.
- P. S. Kubiak, A. L. Johnson, P. J. Cameron and G. Kociok-Köhn, Eur. J. Inorg. Chem., 2019, 3962–3969 CrossRef CAS.
- J. Langer, M. Köhler, H. Görls and M. Westerhausen, Chem. – Eur. J., 2014, 20, 3154–3161 CrossRef CAS PubMed.
- A. Koch, Q. Dufrois, M. Wirgenings, H. Görls, S. Krieck, M. Etienne, G. Pohnert and M. Westerhausen, Chem. – Eur. J., 2018, 24, 16840–16850 CrossRef CAS PubMed.
- J. A. Darr, S. R. Drake, D. J. Otway, S. A. S. Miller, D. M. P. Mingos, I. Baxter, M. B. Hursthouse and K. M. A. Malik, Polyhedron, 1997, 16, 2581–2588 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Koch, S. Krieck, H. Görls and M. Westerhausen, Organometallics, 2017, 36, 2811–2817 CrossRef CAS.
- C. Lepetit, P. Fau, K. Fajerwerg, M. L. Kahn and B. Silvi, Coord. Chem. Rev., 2017, 345, 150–181 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2423347–2423351. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00872g |
| ‡ Both these authors contributed equally and have permission to represent themselves as first author. |
| This journal is © The Royal Society of Chemistry 2025 |