
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling mechanistic insights and applications of aggregation-enhanced emission (AEE)-active polynuclear transition metal complexes†
Bishnu
Das
 *
*
Department of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, 741246, India. E-mail: bd18rs073@iiserkol.ac.in
First published on 21st March 2025
Abstract
Aggregation-enhanced emission (AEE) in polynuclear transition metal complexes (PTMCs) represents a major advancement in luminescent materials, overcoming the limitations of aggregation-caused quenching (ACQ) in traditional systems. Unlike conventional materials that suffer from quenching, AEE-active PTMCs exhibit enhanced luminescence in the aggregated state, driven by mechanisms such as restricted molecular motion, π–π stacking, and metal–metal interactions. These properties make PTMCs highly versatile for applications including chemical sensing, bioimaging, photodynamic therapy (PDT), optoelectronics (e.g., OLEDs, WOLEDs, and LEDs), and security technologies (e.g., anti-counterfeiting inks). They enable the sensitive detection of pollutants, facilitate high-performance bioimaging, and enhance the efficiency of energy devices. However, PTMCs face several challenges, including complex synthesis, limited thermal and photostability, solubility issues, and environmental and toxicity concerns. Additionally, high production costs, instability in different media, and the need for optimized energy transfer efficiency must be addressed to enhance their practical performance. This review explores the mechanisms behind AEE in PTMCs and discusses strategies for overcoming these challenges, including ligand engineering, hybrid material development, and sustainable synthesis methods. It also highlights their potential in advancing energy-efficient technologies, precision therapeutics, and secure communication systems, contributing to a more sustainable and innovative future.
1. Introduction
Luminescent metal complexes are essential for technological advancements, serving key roles in applications such as bioimaging, chemical sensing, energy-efficient lighting, and sophisticated security systems.1 However, a significant limitation of traditional luminescent systems is aggregation-caused quenching (ACQ), where their emission diminishes upon aggregation in the solid state.2 This poses a serious challenge for applications requiring solid-state performance, such as organic light-emitting diodes (OLEDs), biological imaging, and sensors. The discovery of aggregation-induced emission (AIE) provided a solution to this problem by enabling non-emissive materials to become luminescent upon aggregation.3 Building on this concept, aggregation-enhanced emission (AEE) refers to systems that are already emissive in solution but exhibit significantly stronger luminescence upon aggregation, making it a breakthrough in luminescent material design.4Polynuclear transition metal complexes (PTMCs), indispensable for their versatile applications in science, medicine, and device technologies, have attracted considerable interest among AEE-active materials because of their distinctive photophysical characteristics.5 These complexes harness the versatile characteristics of transition metals, such as strong spin–orbit coupling, tunable oxidation states, and intricate molecular geometry. Mechanistically, AEE in PTMCs is driven by phenomena like the restriction of inter- and intramolecular motions, π–π stacking interactions, and metal–metal interactions, which amplify luminescence and suppress non-radiative decay pathways. These properties enable PTMCs to address diverse needs across various fields. In chemical sensing, they offer high sensitivity for detecting pollutants, hazardous compounds, and volatile organic compounds. In healthcare, they facilitate advanced bioimaging with near-infrared (NIR) emission and exceptional photostability, while in photodynamic therapy (PDT), they act as photosensitizers, generating reactive oxygen species (ROS) to selectively target and destroy cancer cells or pathogens. In optoelectronics, PTMCs contribute to the development of efficient OLEDs, near-infrared LEDs, and white-light-emitting devices. Moreover, their ability to change emission properties in response to external stimuli makes them valuable in security applications like re-writable luminescent inks and anti-counterfeiting technologies.
The transformative potential of AEE-active PTMCs lies in their ability to combine enhanced emission efficiency with structural adaptability. This combination allows for superior performance in solid-state applications, such as higher sensitivity in sensing, improved penetration and clarity in bioimaging, and greater energy efficiency and durability in optoelectronic devices. By overcoming ACQ-related limitations, AEE-active materials redefine the capabilities of luminescent systems, fostering advancements in energy-efficient technologies, precision medicine, and secure communication systems.
Despite their promising potential, AEE-active PTMCs face several challenges, including concerns about their thermal and photostability, the high cost of synthesis due to reliance on rare and expensive metals, and environmental considerations related to sustainability. Additionally, a deeper understanding of the interplay between metal–metal interactions, aggregation dynamics, and ligand behavior is necessary to optimize their performance. Addressing these challenges requires innovative strategies, including advanced ligand engineering, which involves designing and modifying ligands to enhance the stability, efficiency, and functionality of AEE-active PTMCs, computational modeling to predict and optimize material properties, design new materials, and improve performance, as well as hybrid material integration, which combines organic and inorganic materials to improve stability and overall functionality, along with the adoption of green synthesis techniques.
In this review, we have presented an inclusive overview of the mechanistic insights into AEE in PTMCs, highlighting their diverse applications and addressing the existing challenges. By bridging fundamental research with application-driven innovations, AEE-active PTMCs have the potential to revolutionize multiple domains, including sustainable technologies, precision medicine, and next-generation photonic devices, contributing to a brighter and more advanced future.
2. Mechanisms of AEE in PTMCs
Aggregation-enhanced emission in polynuclear transition metal complexes arises from the interplay of several critical mechanisms, as summarized in Table 1. These include π–π stacking interactions, metal–metal interactions, restriction of inter- and intramolecular motion, excited-state stabilization, and molecular packing within crystal lattices. These processes enhance luminescence by minimizing non-radiative decay pathways and stabilizing the emissive states. Among these, metal–metal interactions stand out as particularly influential, serving as conduits for energy transfer, stabilization of excited states, and facilitation of aggregate formation. Below, we delve into how these mechanisms work in harmony to drive AEE.| Complex | Mechanism | Key features | Ref. |
|---|---|---|---|
| 1 | π–π stacking | Interacts with pyrene, naphthalene, and biphenyl; exhibits intense room-temperature phosphorescence due to the heavy-atom effect of mercury. | 8 |
| 2–4 | π–π stacking | Coumarin ring interactions lead to emissive aggregates with enhanced fluorescence quantum yields in solid-state and acetonitrile/water mixtures. | 9 |
| 5–9 | Metal–metal (Pt–Pt) | Strong Pt⋯Pt and π–π interactions lead to red-shifted emissions (∼830 nm), reversible aggregation behavior based on temperature. | 12 |
| 10–16 | Metal–metal (Pt–Pt) | Water-induced aggregation results in red-shifted emissions, hydrophobic folding modulates luminescence. | 13 |
| 17–18 | Metal–metal (Pt–Pt) | Temperature-responsive micellization; aggregation enhances red-NIR emission (19-fold for 17). | 14 |
| 19 | Metal–metal (Pt–Pt) | Hierarchical assembly into columnar structures with enhanced emission at 703 nm; serves as a luminescence turn-on switch. | 15 |
| 20–21 | Metal–metal (Pt–Pt) | Stereochemistry influences aggregation and self-assembly; trans isomers form crystalline fiber-like structures, cis isomers yield amorphous aggregates. | 16 |
| 22 | Metal–metal (Pt–Pt) | Ionochromic response to Na+ complexation, demonstrating reversible aggregation-enhanced emission for stimuli-responsive applications. | 17 |
| 23–28 | Metal–metal (Pt–Pt) | Phosphorescent complexes with optimized Pt⋯Pt distances, quantum yields up to 100%, enhanced diastereoselectivity. | 18 |
| 29 | Metal–metal (Au–Au) | Tunable monomer-excimer equilibrium; white-light emission in solution; solid-state emission varies with stacking and Au⋯Au interactions. | 20 |
| 30 | Metal–metal (Au–Au) | Aggregation-enhanced emission through self-assembly; thermoreversible gel-to-sol transition; luminescence color switching (red-green-blue). | 21 |
| 31–34 | Metal–metal (Au–Au) | Short Au⋯Au distances influence emission colors; structural control fine-tunes luminescence properties. | 22 |
| 35–37 | Metal–metal (Au–Au) | Dinuclear gold(I) units exhibit strong AEE; mechanical force enhances luminescence. | 23 |
| 38–45 | Metal–metal (Au–Au) | Crown ether-based complexes; cation (K+) binding and solvent composition modulate emission. | 24 |
| 46 | Metal–metal (Au–Au) | Reversible phosphorescent mechanochromism; aggregation enhances luminescence quantum yield. | 25 |
| 47 | Metal–metal (Au–Au) | Fluorene-based gold(I) complex with intense white-light emission in solid state. | 26 |
| 48–49 | Restriction of intermolecular motion | π–π interactions stabilize structure; aggregation enhances luminescence. | 28 |
| 50–51 | Restriction of intermolecular motion | Reversible piezochromic phosphorescence; metal–ligand charge transfer enhances emission. | 29 |
| 52 | Restriction of intermolecular motion | Tetraphenylethene-based metallacage; self-assembly enhances fluorescence through restricted rotation. | 30 |
| 53–55 | Restriction of intermolecular motion | Gold(I) complexes exhibit AEE and reversible mechanochromic luminescence; intermolecular forces enhance clustering. | 31 |
| 56 | Restriction of intermolecular motion | Mechanochromic fluorescence via crystalline-to-amorphous transitions; aggregation-induced green emission. | 32 |
| 57–58 | Restriction of intramolecular motion | Ionic dinuclear iridium(III) Schiff base complexes; aggregation enhances photoluminescence quantum yield. | 34 |
| 59–62 | Restriction of intramolecular motion | Hydrazide-bridged diiridium(III) complexes; π–π interactions restrict motion, enhancing emission. | 35 |
| 63–65 | Excited-state stabilization | Rhenium(I)-based molecular rectangles; aggregation minimizes solvent exposure, increasing emission intensity. | 37 |
| 66 | Crystal lattice/molecular packing | Dinuclear rhenium(I) complex with polymorph-dependent luminescence; molecular packing controls emission properties. | 39 |
2.1. π–π stacking interactions
π–π stacking interactions, the attractive forces between aromatic rings via their π-electrons, are fundamental to many molecular assemblies.6 These interactions result in the formation of ordered, rigid aggregates that are crucial for aggregation-enhanced emission.7 AEE describes the phenomenon where molecules that are weakly emissive in solution become highly luminescent upon aggregation. This transformation occurs as aggregation restricts intramolecular motions, such as vibrations and rotations, which would otherwise quench excited-state energy through non-radiative pathways. π–π stacking plays a pivotal role in this process by promoting tight molecular packing and stabilizing aggregate structures.A striking example of π–π stacking in AEE is observed in trinuclear mercury(II) complex 1 (Fig. 1), which interacts with polycyclic aromatic hydrocarbons, such as pyrene, naphthalene, and biphenyl. The resulting supramolecular stacks exhibit intense room-temperature phosphorescence with emissions in red, green, and blue wavelengths. These emissions, attributed to the heavy-atom effect of mercury, are enhanced by the tightly packed structures formed through π–π stacking. This strategy not only mitigates excimer emissions but also offers a promising pathway for the development of electroluminescent devices.8 Similarly, the AEE behavior observed in dinuclear gold(I) complexes 2–4 (Fig. 1) is driven by π–π stacking between coumarin rings, which leads to the formation of emissive aggregates. These aggregates show significantly enhanced fluorescence quantum yields in both solid-state and acetonitrile/water mixtures. TDDFT calculations further elucidate the influence of different syn and anti-conformations on the AEE behavior, highlighting how molecular design can be optimized to enhance the fluorescence properties for applications in optoelectronics and bioimaging.9
 | ||
| Fig. 1 Structures of pyrene, naphthalene, biphenyl and complexes 1–4. | ||
Both examples demonstrate how π–π stacking interactions can be leveraged to manipulate and enhance the optical properties of metal-based systems, advancing their potential in cutting-edge applications, such as energy-efficient lighting, advanced displays, and biomedical imaging.
2.2. Metal–metal interactions
Metal–metal interactions play a crucial role in driving aggregation-enhanced emission in polynuclear transition metal complexes.10 These interactions significantly influence the photophysical properties of PTMCs by enabling the formation of unique excimeric states, facilitating metal–metal-to-ligand charge transfer (MMLCT) in platinum systems, and ligand-to-metal–metal charge transfer (LMMCT) in gold systems. Both types of charge transfer stabilize excited states in aggregated systems. By bridging electronic communication between metal centers, these interactions suppress non-radiative decay pathways and amplify luminescence. Below, we delve into the influence of metal–metal interactions with a focus on systems involving platinum (Pt⋯Pt) and gold (Au⋯Au) interactions, illustrating their pivotal role in enhancing AEE.For example, dinuclear alkynyl platinum(II) terpyridyl complexes 5–9 (Fig. 2) exemplify the influence of strong Pt⋯Pt and π–π interactions on AEE behavior. Complexes with longer, flexible bridges (7–9) exhibited significant self-association at room temperature, evidenced by 1H NMR, UV-visible absorption, and emission spectroscopy, while shorter-bridge complexes (5, 6) did not aggregate. This intramolecular aggregation resulted in red-shifted emissions attributed to 3MMLCT transitions around 830 nm, with enhanced intensity at lower temperatures and reversible disaggregation upon heating. These findings highlight the importance of temperature-controlled self-association in platinum(II) systems and set the stage for further exploration of metal–metal interactions.12 Building on this, binuclear organoplatinum(II) complexes 10–16 (Fig. 2), featuring foldable oligo(ortho-phenyleneethynylene) linkers, displayed water-induced emission properties. Upon forming nanosized aggregates in water-acetonitrile mixtures with >40% water, these complexes showed significant red-shifts and enhanced emissions due to hydrophobic folding and inter/intramolecular interactions. Complexes with longer linkers (12–16) demonstrated pronounced effects, while shorter-linker complexes (10, 11) showed weaker responses. TEM and DLS confirmed aggregate formation and spectroscopic analyses revealed π–π and Pt⋯Pt interactions as critical factors driving emission enhancement. Notably, complex 14 exhibited increased emission intensity and conformational changes modulated by water content and temperature, further highlighting the intricate interplay of metal–metal interactions and aggregation.13 Further studies on platinum(II) terpyridyl-based metallo-supramolecular triblock copolymers 17 and 18 (Fig. 2) illustrated temperature-responsive micellization. The aggregation of platinum(II) terpyridyl moieties, driven by the phase transition of PEO-PPO-PEO[poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide)] triblock copolymers, resulted in significant spectral changes. Upon heating, complex 17 exhibited a remarkable 19-fold enhancement in red-NIR emission due to polymeric micelle formation and aggregation through Pt⋯Pt and π–π interactions. Complex 18 showed similar trends, although the emission enhancement was less pronounced. These findings highlight the potential of AEE for designing sensitive luminescent sensors capable of probing temperature-induced conformational and environmental changes.14 In a different context, a dinuclear alkynylplatinum(II) terpyridine complex 19 (Fig. 2), bridged by an amphiphilic binaphthol derivative, demonstrated AEE in aqueous acetonitrile solutions. The complex exhibited hierarchical assembly into cylindrical columnar structures driven by inter- and intramolecular Pt⋯Pt and π–π stacking interactions, resulting in significant emission enhancement at 703 nm, characteristic of 3MMLCT transitions. This effect served as a luminescence turn-on switch for monitoring the assembly process. UV-visible, CD, and electron microscopy analyses confirmed the formation of fibrillar networks indicative of supramolecular self-assembly, emphasizing the critical role of metallophilic interactions in functional luminescent material design.15 Stereoisomerism further modulated metal–metal interactions, as demonstrated in complexes 20 and 21 (Fig. 2). The presence or absence of bulky substituents on terpyridine ligands was found to significantly impact the extent of Pt⋯Pt and π–π stacking interactions, thus modulating the self-assembly behaviors in different solvent environments. Notably, solvent-dependent UV-visible absorption and fluorescence studies revealed that the aggregation of these complexes leads to red-shifted MMLCT transitions. The trans isomers, which exhibit a linear stacking arrangement, demonstrated increased crystallinity and distinct fiber-like self-assembled morphologies, whereas the cis isomers favored twisted conformations, leading to amorphous aggregates. The absence of bulky tert-butyl substituents resulted in stronger Pt⋯Pt interactions, enabling hierarchical supramolecular architectures such as helical ribbons and nanotubes. Electron microscopy (TEM and SEM) images further confirmed the formation of left-handed (M) and right-handed (P) helical ribbons, indicating the role of non-covalent interactions in supramolecular chirality. These findings highlight the importance of precise stereochemical control in tuning metal–metal interactions for achieving aggregation-enhanced emission properties.16 Another notable example is complex 22 (Fig. 2), a dinuclear organoplatinum(II) complex that exhibited AEE behavior along with ionochromism in response to Na+ cation complexation. Exposure to Na+ ions caused a shift in the emission spectrum, making this complex a promising candidate for ion-responsive applications. The behavior of this complex highlights the utility of Pt⋯Pt interactions and π–π stacking in designing materials sensitive to external stimuli such as solvent polarity or ion presence. Furthermore, the reversible nature of its aggregation-enhanced emission suggests potential for smart materials capable of dynamically switching their optical properties in response to environmental changes. These tunable luminescence and reversible emission characteristics make dinuclear platinum(II) complexes highly attractive for applications in bioimaging, molecular switches, and stimuli-responsive optoelectronic devices.17 Lastly, the development of phosphorescent platinum(II) complexes has been advanced through the use of soft-bridged binuclear platinum(II) complexes, such as (Race)23, (Race)24, (Race)25/26, and (L,S,L)27/28 (Fig. 2), which addressed challenges such as aggregation-caused quenching and the lack of metal-induced chirality. These complexes utilized smaller conjugated bridging ligands, improving solubility, enabling enantioseparation, and introducing point chirality for enhanced diastereoselectivity. The phosphorescence quantum yields of these complexes reached up to 100%, achieved through optimized intramolecular Pt⋯Pt distances. Remarkably, these complexes exhibited significant AEE properties, with emission quantum yields dramatically increasing in aggregated states compared to solutions, pushing forward the development of platinum(II) complexes with advanced photonic and sensing capabilities.18
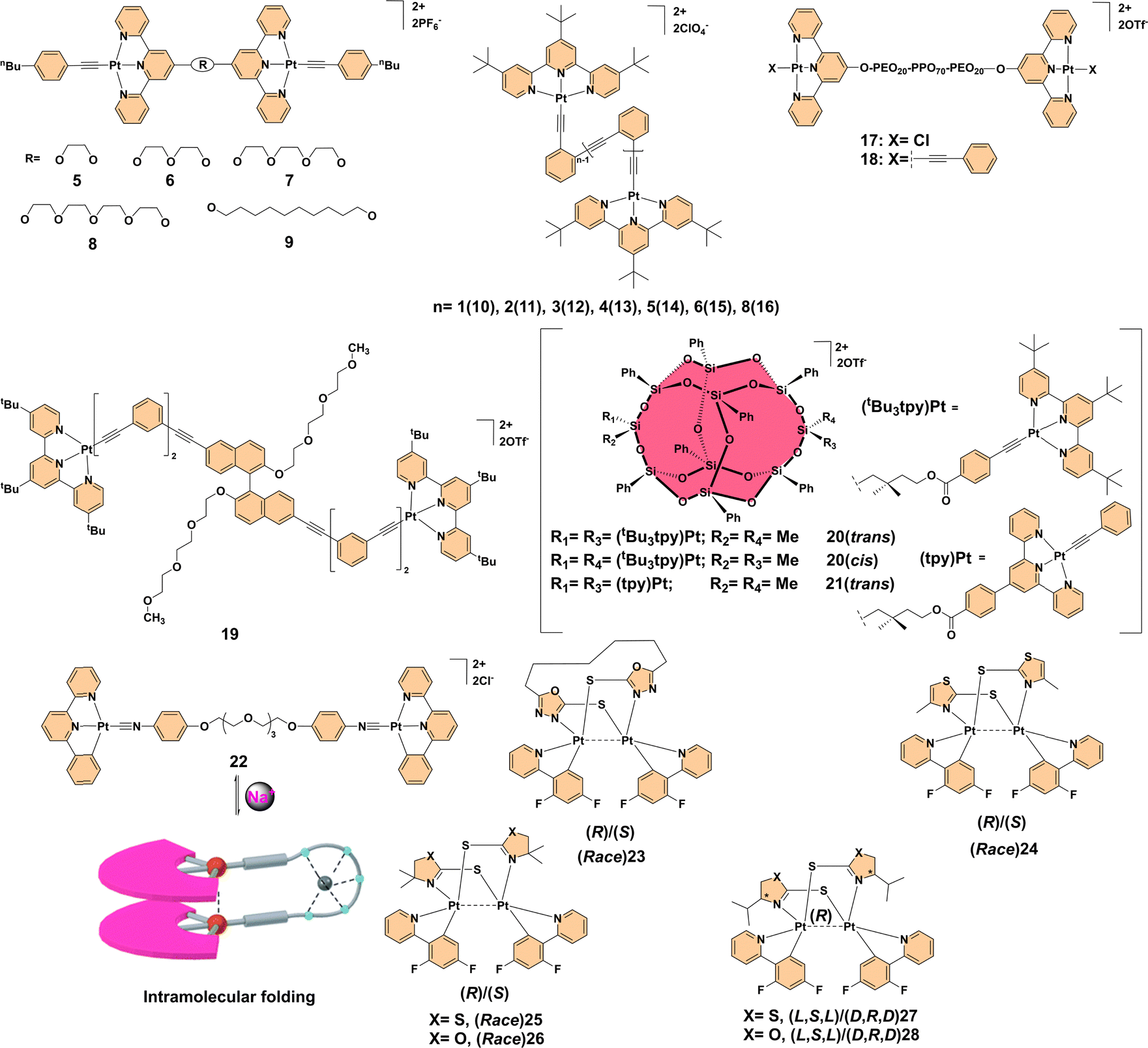 | ||
| Fig. 2 Structures of complexes 5–28. | ||
Collectively, these findings highlight the transformative role of Pt⋯Pt interactions in tuning luminescent properties, offering a versatile platform for developing advanced photonic, sensing, and environmental monitoring materials with unprecedented precision and functionality.
Building on the insights from platinum(II)-based systems, the trinuclear gold(I) pyrazolate cluster 29 (Fig. 3) exemplifies the role of aurophilic interactions in achieving dual emission properties. This cluster demonstrates a tunable monomer-excimer equilibrium, with emission profiles sensitive to factors like concentration, excitation energy, and temperature. Such dynamic behavior enables adjustable white-light emission in solution, while in the solid-state, distinct emission patterns emerge due to variations in stacking and Au⋯Au interactions. These findings align with the excimeric states and LMMCT-driven luminescence enhancements, highlighting a shared foundation across both systems for achieving AEE and tunable photophysical properties.20 Extending this concept further, the trinuclear gold(I) pyrazolate complex 30 (Fig. 3) demonstrates aggregation-enhanced emission behavior through self-assembly in hexane. Upon heating, the complex formed a gel that exhibited red luminescence (λem = 640 nm) through Au⋯Au metallophilic interactions. Notably, when doped with Ag+, the gel emitted a blue luminescence (λem = 458 nm), and upon removal of Ag+ with cetyltrimethylammonium chloride, the original red luminescence was restored. This thermoreversible gel-to-sol transition, accompanied by luminescence color switching (red-green-blue), highlighted the AEE properties of the complex, with phosphorescence lifetimes ranging from 3 to 6 μs. Scanning electron microscopy and X-ray diffraction revealed the formation of highly entangled fiber structures with a rectangular columnar packing, further confirming the association between self-assembly and the observed luminescence properties.21 Beyond these examples, additional gold(I) complexes 31–34 (Fig. 3) further demonstrate the fundamental role of Au⋯Au interactions in tuning photoluminescence behavior. Complex 31, with a short Au⋯Au distance of 3.38 Å, exhibits strong aurophilic interactions leading to purple emission, while complex 32 shows similar aggregation behavior but emits at different wavelengths. These findings highlight the importance of structural control in molecular aggregates, as luminescence properties can be fine-tuned by modifying aggregation patterns.22
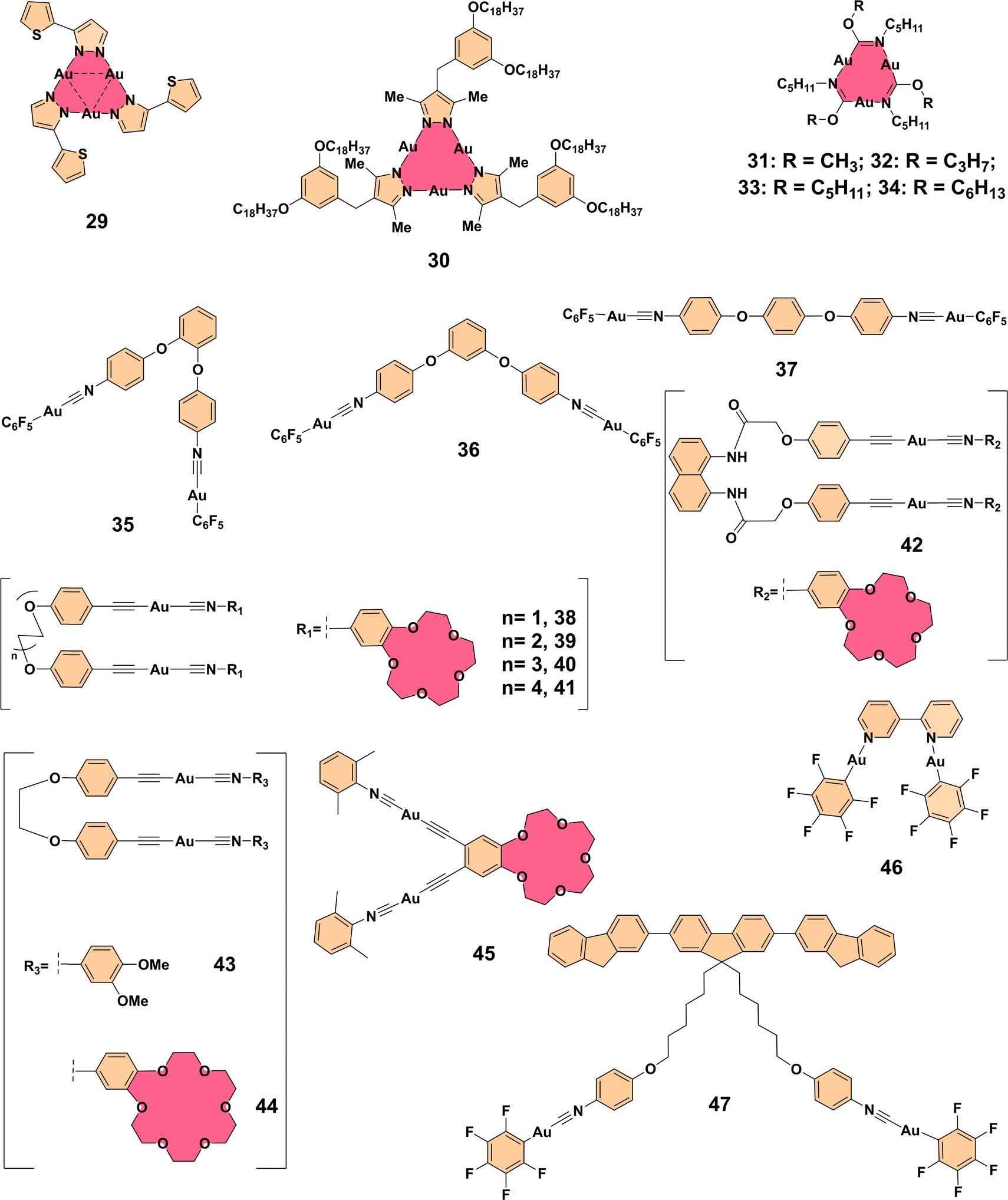 | ||
| Fig. 3 Structures of complexes 29–47. | ||
Expanding the scope to gold(I) systems, constitutional isomers containing dinuclear gold(I) units (35–37) (Fig. 3) exhibited notable AEE behavior, where aggregation led to enhanced fluorescence through aurophilic interactions between gold(I) centers. Complex 35 showed a strong AEE response with green emission arising from gold(I) unit aggregation, while complex 36 demonstrated luminescence enhancement under mechanical force. These studies highlight how metal–ligand interactions, particularly aurophilic bonding, can be harnessed to develop stimuli-responsive materials with advanced luminescent properties, providing a broader context for understanding metal–metal interaction dynamics.23 Further extending these principles, crown ether-based bis(alkynyl)-bridged gold(I) isocyanide complexes 38–45 (Fig. 3) displayed intriguing photophysical behaviors modulated by Au⋯Au interactions. Cation (K+) binding and solvent composition significantly influenced these interactions, leading to marked changes in emission spectra. For instance, complex 45 exhibited enhanced low-energy emission (∼640 nm) as water content increased, attributed to the emergence of LMMCT emission due to intermolecular aggregation. This illustrates how solvent and structural modulation enhance the understanding of aurophilic bonding and its role in facilitating AEE, linking back to other gold(I) complexes discussed earlier.24 Beyond conventional AEE systems, a bipyridine-based gold(I) complex 46 (Fig. 3) demonstrated remarkable aggregation-enhanced emission and reversible phosphorescent mechanochromism. Upon aggregation in a dimethylformamide–water mixture, the luminescence quantum yield increased significantly due to weak intermolecular aurophilic Au⋯Au interactions. These interactions were integral to the complex's aggregation behavior and mechanochromic properties, highlighting the pivotal role of metal–metal interactions in enabling the unique luminescent and self-assembly characteristics of AEE-active materials. Together with earlier examples, this study emphasizes the versatility and critical role of metal–metal interactions in shaping AEE behaviors across various systems.25 Expanding on these photophysical properties, a novel fluorene-based gold(I) complex 47 (Fig. 3) was reported, exhibiting intriguing aggregation-enhanced emission behavior. This complex demonstrated the unusual property of emitting intense white light in its aggregated state, making it a promising candidate for white organic light-emitting diodes (WOLEDs). Complex 47 does not require complex doping or mixing procedures to achieve white light emission in the solid state, distinguishing it from traditional AEE molecules. The fluorescence behavior of the complex in dimethylformamide–water mixtures showed a shift from blue-green emission to broad white light emission as the water content increased, primarily due to aggregation. Additionally, complex 47 exhibited excellent solid-state fluorescence with a high quantum yield, further enhancing its potential in optoelectronic applications. This study expands the possibilities for designing new AEE compounds and advanced white light-emitting systems.26
Overall, these studies highlight the versatile and critical role of aurophilic interactions in shaping the luminescent and self-assembly properties of gold(I) complexes, facilitating the development of advanced materials with tunable photophysical behaviors and practical applications in optoelectronics and sensing.
2.3. Restriction of intermolecular motion
The restriction of intermolecular motion plays a pivotal role in the manifestation of aggregation-enhanced emission in various complex systems.27 This phenomenon occurs when molecules transition from a disordered state, where their rotational and vibrational motions are relatively free, to an ordered state that limits such movements. In the ordered state, these restrictions reduce non-radiative decay channels, allowing photoluminescence to dominate. The key factor facilitating the restriction of molecular motion is the presence of strong intermolecular interactions such as π–π stacking, hydrogen bonding, and metal-based interactions. Such interactions contribute to the stabilization of particular structures, improving the emission characteristics of the materials in their solid state.One example of how these interactions enhance luminescence is seen in the dinuclear complexes 48 and 49 (Fig. 4), where strong intermolecular π–π interactions in the solid state induce aggregation-enhanced emission. In solution, the photoluminescence of these complexes is weak due to triplet ligand-centered (3LC) states, but in the solid state, the complexes exhibit intense orange-red emission (λem = 597 nm), arising from metal-to-ligand–ligand charge-transfer (3M(LL)CT) states. The aggregation in complex 49, aided by the π–π interactions between adjacent pyridine rings in the iridium(III)-centered moieties and the extended stacking of the platinum(II) moieties, demonstrates how restricted molecular motion can significantly enhance luminescence and provide insights for designing AEE materials for optoelectronic applications.28 A similar phenomenon is observed in the dinuclear cationic iridium(III) complexes 50 and 51 (Fig. 4), which exhibit both aggregation-enhanced emission and highly reversible piezochromic phosphorescence. In solution, the Schiff base bridging ligands induce intra-ligand charge transfer (ILCT) excited states, suppressing luminescence due to non-radiative decay. However, in the solid state, the complexes transition to metal-to-ligand charge transfer (MLCT) and ligand-to-ligand charge transfer (LLCT) excited states, significantly enhancing phosphorescence. This transformation is attributed to intermolecular π–π interactions between the pyrazole rings, which induce face-to-face aggregation. The reversibility of these interactions, observed through mechanical grinding and recrystallization, further highlights the dynamic nature of molecular packing and its impact on luminescent behavior.29 The importance of intermolecular interactions is further exemplified in the tetraphenylethene (TPE)-based metallacage 52 (Fig. 4), where self-assembly of TPE-pyridine ligands and palladium(II) ions results in aggregation-enhanced emission. In solution, the fluorescence of this complex is suppressed due to the free rotation of peripheral benzene rings, but upon aggregation in poor solvents like water, restricted rotation enhances fluorescence intensity. X-ray crystallography reveals a hydrophobic cavity stabilized by hydrogen bonds and electrostatic interactions, which encapsulates nitrate ions and facilitates aggregation, leading to the formation of well-dispersed nanospheres. The reversible stimuli-responsive behavior of cage 52, triggered by chloride and silver ions, highlights the role of dynamic intermolecular interactions in achieving AEE and responsiveness, showing its potential as a versatile fluorescent host in supramolecular chemistry.30 Similarly, the diisocyano-based dinuclear gold(I) complexes 53–55 (Fig. 4) exhibit remarkable aggregation-enhanced emission and reversible mechanochromic luminescence behaviors. These complexes transition from weakly emissive states in pure solvents to highly luminescent nano-aggregates in water-rich mixtures due to changes in molecular packing and the enhancement of gold–gold (aurophilic) interactions. Intermolecular forces such as CH⋯F, C⋯F, and weak π–π interactions promote the clustering of molecules in less polar environments, leading to enhanced emission intensity and blue-shifts in luminescence. The mechanochromic behavior of these complexes, which arises from reversible phase transitions between crystalline and metastable amorphous states, further emphasizes how the restriction of molecular motion under different external stimuli (such as grinding or solvent exposure) can activate luminescence.31 Finally, the fluorene-based dinuclear gold(I) complex 56 (Fig. 4) exemplifies how intermolecular interactions govern aggregation-enhanced emission and crystallization-induced emission enhancement (CIEE). The complex shows a transition from faint fluorescence in pure dimethylformamide to robust yellow emission in dimethylformamide–water mixtures with high water fractions, driven by intermolecular aurophilic interactions. The mechanochromic fluorescence of complex 56 stems from morphological transformations between crystalline and amorphous states, further confirming that the interplay between aggregation, molecular packing, and mechanical stimuli is crucial for achieving reversible fluorescence changes. The strong green emission observed in the crystalline form of complex 56 highlights the enhanced luminescence upon aggregation and reinforces the potential of such complexes for developing advanced, multi-stimuli responsive luminescent materials.32
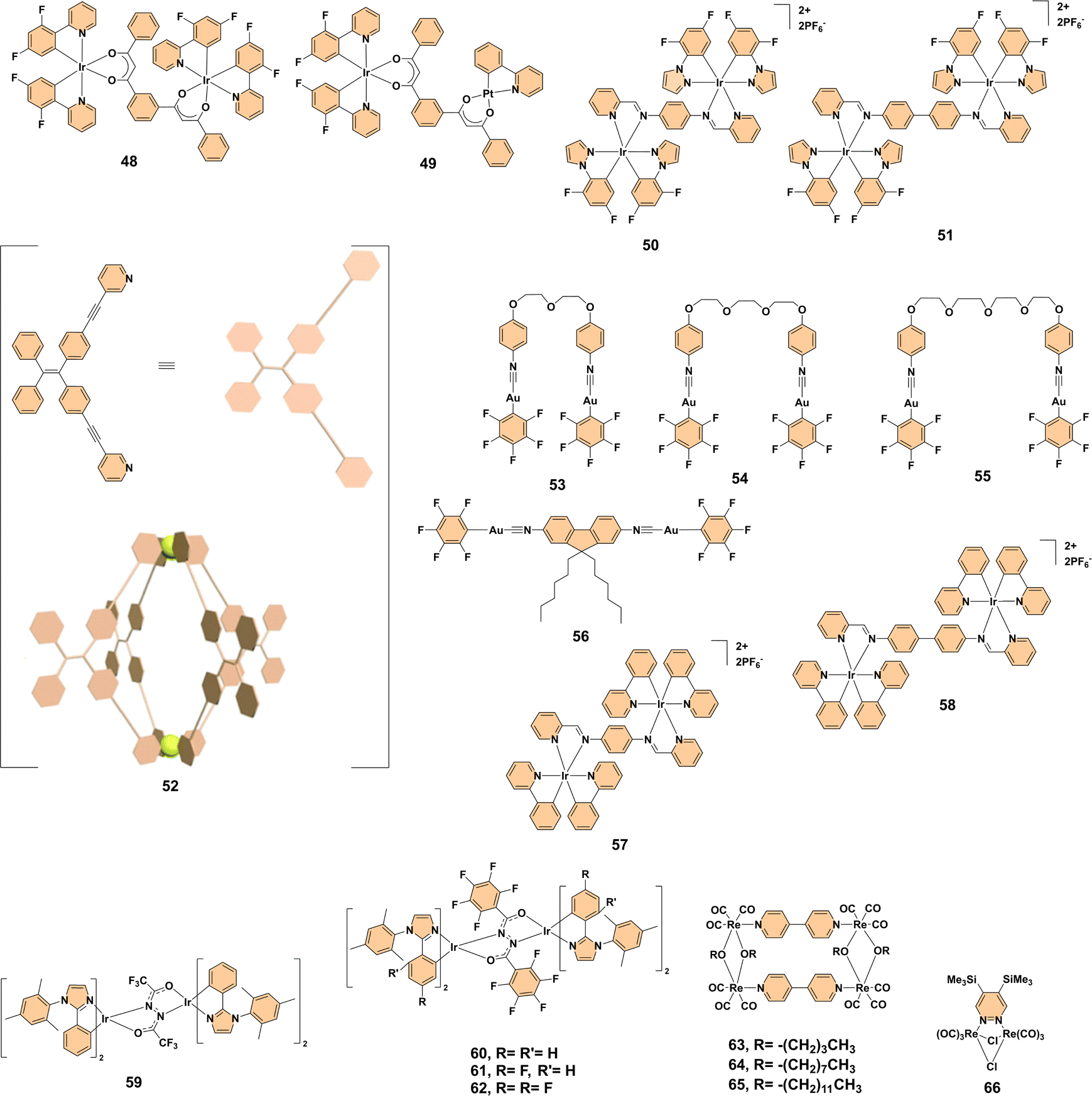 | ||
| Fig. 4 Structures of complexes 48–66. | ||
Taken together, these examples clearly illustrate the pivotal role of intermolecular interactions and the restriction of molecular motion in enhancing aggregation-enhanced emission and mechanochromic behaviors. Whether through π–π stacking, aurophilic interactions, or hydrogen bonding, these interactions drive the aggregation process, leading to the formation of ordered structures that promote efficient luminescence. The ability to reversibly transition between ordered and disordered states opens new possibilities for innovative applications, including optoelectronics, pressure sensors, and re-writable phosphorescent data storage devices.
2.4. Restriction of intramolecular motion
Like the restriction of intermolecular motion, the restriction of intramolecular motion also plays a crucial role in enhancing the photoluminescence properties of many luminescent metal complexes.33 This restriction typically occurs when molecular aggregates transition from a disordered state in solution to a more ordered state in the solid state, which restricts the motion of the complex's molecular components. In particular, when intramolecular rotation or relaxation is suppressed, the emission intensity is often significantly improved due to the reduced non-radiative decay pathways. This process is often linked to the formation of planar or rigidified geometries that facilitate more efficient radiative transitions, as seen in aggregation-enhanced emission behavior.For instance, the ionic dinuclear iridium(III) Schiff base complexes 57 and 58 (Fig. 4) exhibit notable AEE with photoluminescence quantum yields (PLQYs) of 37.3% and 26.4%, respectively, in neat films. In solution, these complexes show weak emission due to unrestricted intramolecular motion, which is primarily a result of the lack of π–π interactions in the excited states. However, upon aggregation in solid films or mixtures with high water content, intermolecular interactions such as π–π stacking and CH⋯π interactions come into play. These interactions lead to the planarization of the molecular geometry, which restricts intramolecular rotation and promotes radiative transitions, thus enhancing the photoluminescent properties of the complexes.34 Similarly, the hydrazide-bridged diiridium complexes 59–62 (Fig. 4), featuring bulky 1,2-diarylimidazole cyclometalating ligands, demonstrate enhanced emissivity due to the presence of rigidifying intramolecular π–π interactions. These interactions restrict the motion of the bridging ligands and suppress non-radiative decay pathways, leading to significantly higher PLQYs of 47–55 ± 10%. Complex 59, lacking these rigidifying interactions, exhibits lower emissivity due to increased non-radiative decay. In contrast, complexes 60–62, with their intramolecular π–π interactions, exhibit high photoluminescence quantum yields and show aggregation-enhanced emission behavior, where emission intensity is greatly enhanced in the solid state.35
Thus, the presence of rigidifying interactions that restrict intramolecular motion is essential for improving the luminescent properties of both dinuclear iridium(III) Schiff base complexes and hydrazide-bridged diiridium(III) complexes. This highlights the importance of controlling molecular geometry and aggregation state in the design of high-performance luminescent materials with tunable emission characteristics.
2.5. Excited-state stabilization
Excited-state stabilization upon aggregation is another key mechanism that enhances luminescence in aggregation-enhanced emission systems.36 When molecules aggregate, their exposure to solvents is reduced, limiting vibrational relaxation and minimizing the distortion of the excited-state environment. This stabilization of the excited state leads to higher quantum yields and improved luminescence properties. The following example illustrates the crucial role of excited-state stabilization in AEE.The self-assembly of rhenium(I)-based molecular rectangles 63–65 (Fig. 4) demonstrated significant AEE, particularly when the solvent was changed from organic to aqueous and long alkyl chains were introduced. The aggregation of complex 65 in an aqueous medium led to a dramatic increase in emission intensity, quantum yield, and lifetime. This enhancement was attributed to the stabilization of the excited state, where aggregation reduced vibrational motion and minimized solvent exposure, leading to a less distorted excited-state environment. Specifically, the emission quantum yield of complex 65 increased from 0.39 × 10−3 in acetonitrile to 6.5 × 10−3 in a 10% acetonitrile–90% water mixture, while the lifetime rose from 11 ns to 212 ns.37
So, the results highlight the importance of excited-state stabilization in enhancing fluorescence in aggregated states, highlighting the potential for improved luminescence properties in supramolecular systems, which could be leveraged for applications in sensing, imaging, and advanced materials development.
2.6. Crystal lattice and molecular packing
The molecular packing and organization in the crystal lattice during aggregation have a significant impact on the photophysical properties of aggregation-enhanced emission materials.38 The arrangement of molecules in the solid-state influences intermolecular interactions, which can alter emission characteristics. The following example demonstrates how crystal lattice and molecular packing contribute to enhanced luminescence in AEE systems.The aggregation-enhanced emission properties of the dinuclear rhenium(I) complex 66 (Fig. 4) were investigated. It exists in two polymorphs: yellow (66Y) and orange (66O). Both polymorphs exhibited intense photoluminescence, with quantum yields of 0.56 for 66Y and 0.52 for 66O, significantly higher than the 0.06 observed in solution. The enhanced emission in the solid state was attributed to the restricted rotation of the Me3Si groups within the crystal lattice. Despite the absence of strong intermolecular interactions, the distinct molecular packing and local dipole arrangements in the two polymorphs led to differences in absorption and emission maxima. Specifically, 66O exhibited red-shifted spectral features compared to 66Y. This study highlighted how even subtle differences in crystal packing and molecular organization can modulate the photophysical properties of the complex, emphasizing the importance of lattice arrangement in controlling solid-state emission behavior.39
This demonstrates the crucial role of crystal lattice and molecular packing in tuning the emission properties of AEE systems, which could be leveraged for the design of materials with tailored photophysical behaviors for applications in sensors, light-emitting devices, and advanced materials.
3. Applications of PTMCs
Polynuclear transition metal complexes are widely used in chemical sensing, detecting hazardous substances with high sensitivity; in biological imaging and theranostics, enabling targeted visualization and therapy; and in optoelectronics, powering efficient OLEDs, NIR LEDs, and advanced security technologies, showing their versatility and innovation potential. We have summarized all these examples in Table 2 for a clearer overview of their diverse applications.| Complex | Application type | Key features | Ref. |
|---|---|---|---|
| 67, 68 | Chemical sensing | 500-fold luminescence enhancement upon aggregation; selective fluorescence quenching for picric acid detection. | 42 |
| 69 | Anion detection | “Turn-on” phosphorescence upon ClO4− ion exchange; 20-fold increase in photoluminescence in aqueous media; potential for biological imaging. | 43 |
| 70 | Environmental monitoring | AEE, piezochromic luminescence, and vapochromism; reversible color change in phosphorescence with VOCs; stable in air. | 45 |
| 71 | Explosives detection | Fluorescence quenching for picric acid and TNT detection via photo-induced electron transfer; suitable for on-site detection (paper strips, soil). | 46 |
| 72–74 | Environmental monitoring | Morphology-driven emission enhancement; selective detection of nitroaromatic explosives and nitro-containing antibiotics. | 47 |
| 75 | Biomolecular sensing | Strong aggregation and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Pt-5′-GMP binding mode; enhanced hydrolytic stability (t1/2 = 55.5 min); Pt⋯Pt stacking interactions. :2 Pt-5′-GMP binding mode; enhanced hydrolytic stability (t1/2 = 55.5 min); Pt⋯Pt stacking interactions. |
50 |
| 76 | Biomolecular sensing | Limited aggregation; 1:1 Pt-5′-GMP binding mode; lower hydrolytic stability (t1/2 = 16.9 min). |
50 |
| 77–80 | DNA sensing | Strong aggregation-enhanced emission upon DNA binding; complex 79 showed ∼40-fold fluorescence enhancement; high hydrolytic stability. | 51 |
| 81 | Enzymatic assays | Near-infrared AEE at 785 nm; luminescence quenched by G-quadruplex DNA and restored upon DNA degradation; enzymatic activity monitoring. | 52 |
| 82, 83 | Bacterial imaging | Zinc(II)-dipicolylamine core; selectively binds bacterial membranes; high specificity for Gram-negative and Gram-positive bacteria. | 55 |
| 84–87 | Cell imaging | Binuclear copper(I) complexes; aggregation-enhanced emission; phosphorescent properties; stable in air and moisture; effective in HeLa cell imaging. | 56 |
| 88–90 | Bacterial imaging | pH-responsive fluorescence; selective bacterial imaging in acidic environments. | 57 |
| 91 | Antibacterial therapy | Multifunctional probe; aggregation-enhanced emission; selectively binds bacterial membranes; dual antibacterial imaging and photodynamic action. | 60 |
| 92 | Antibacterial therapy | Zinc(II)-coordinated probe; non-emissive in water but fluorescent upon aggregation; high singlet oxygen quantum yield (77.6%). | 61 |
| 93–96 | Cancer therapy | BODIPY-based ruthenium(II) and iridium(III) metalla-rectangles; strong DNA and protein interactions; high selectivity and cytotoxicity against cancer cells. | 63 |
| 97 | Photodynamic therapy | Red-emitting iridium(III) trinuclear complex; enhanced singlet oxygen generation; high PDT efficacy in vivo. | 67 |
| 98 | Photodynamic therapy | Near-infrared (NIR) iridium(III) complex; self-assembled into nanoparticles; superior phototoxicity and biocompatibility. | 68 |
| 99 | Photodynamic therapy | Iridium(III)-porphyrin conjugate; long-wavelength absorption (500–700 nm); self-assembled nanoparticles with strong singlet oxygen generation. | 69 |
| 100–101 | Cancer therapy | Trimetallic complexes; aggregation-enhanced emission; effective nucleus accumulation and ROS generation for PDT. | 70 |
| 102 | Security and data encryption | Reversible luminescence; mechanical grinding induces emission color changes; anti-counterfeit applications. | 71 |
| 103–104 | Security and data encryption | PCL behavior; tunable emission via phase changes; potential for re-writable memory devices and secure optical authentication. | 72 |
| 105 | Optoelectronics (NIR OLED, red OLED, white-light OLED) | Strong phosphorescence, aggregation-dependent emission, tunable emission from orange to NIR, high efficiency and stability. | 76 |
| 106 | Optoelectronics (WOLEDs) | AEE behavior, high solid-state luminescence, environmentally friendly zinc(II)-based emitter, efficient charge transport. | 78 |
| 107 | Optoelectronics (WOLEDs) | Aggregation-enhanced emission, optimized blue and white-light emission, high external quantum efficiency. | 79 |
| 108 | NIR optoelectronics | AEE behavior, voltage-independent NIR emission at 710 nm, efficient charge trapping and recombination, high stability. | 80 |
3.1 Applications in chemical sensing, environmental monitoring, and biological imaging
The exploration of aggregation-enhanced emission in various metal complexes has opened up significant possibilities for applications in chemical sensing, environmental monitoring, and biological imaging.40 These innovative materials, often based on luminescent rhenium(I), iridium(III), and other metal complexes, exhibit remarkable changes in their emission characteristics upon aggregation, making them highly sensitive and selective sensors for a wide range of analytes, including hazardous compounds, volatile organic compounds (VOCs), and anions. Below, we discuss the key advances and examples in these fields, highlighting the potential impact on environmental safety, chemical detection, and biomedical applications.Two alkoxy-bridged binuclear rhenium(I) complexes 67, 68 (Fig. 5a) have demonstrated a significant increase in luminescence upon aggregation, with a nearly 500-fold enhancement in emission intensity when exposed to acetonitrile, forming nanoaggregates. This behavior makes these complexes excellent candidates for sensing nitroaromatic compounds like picric acid (PA), which is commonly found in explosives (Fig. 5a). Complex 68, in particular, exhibited selective detection of PA through fluorescence quenching, making it a strong candidate for explosive detection and highlighting its potential in environmental sensing applications such as chemical warfare agent detection.42 In a similar vein, the development of aggregation-enhanced emission has been explored for the selective detection of anions like ClO4−. A dinuclear ionic iridium(III) complex (69·2PF6) (Fig. 5b) was studied, revealing that the exchange of the counterion PF6− with ClO4− in aqueous media led to a “turn-on” phosphorescent response. Notably, the complex cis-69·2ClO4− exhibited a 20-fold increase in photoluminescence intensity in high-water-content environments, which could serve as a robust probe for the selective detection of ClO4−. The ability of this iridium(III) complex to undergo selective ion-exchange-induced AEE represents a novel method for sensing anions and offers promising applications in biological imaging, as demonstrated by preliminary tests in HeLa cells (Fig. 5b).43
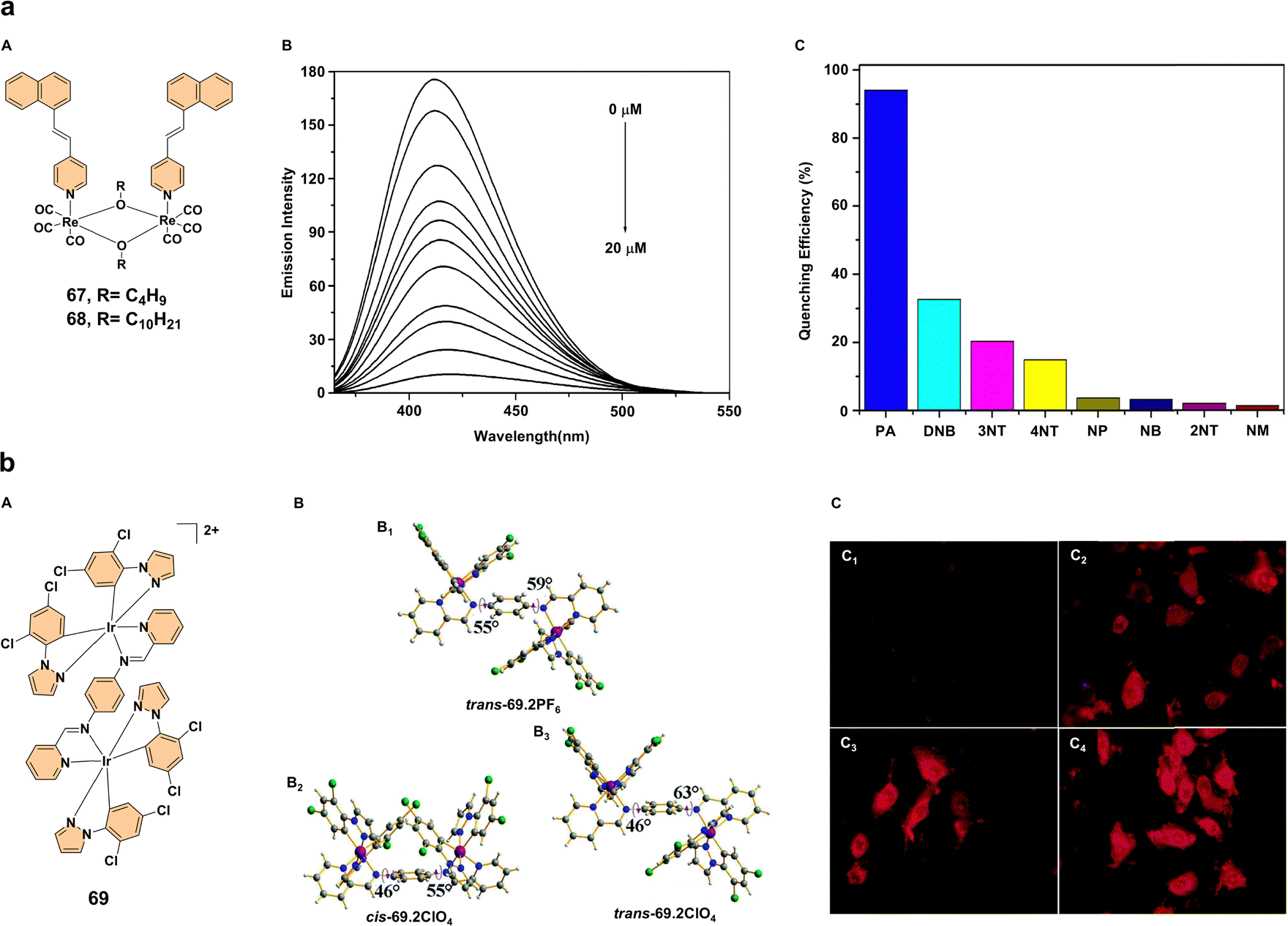 | ||
| Fig. 5 (a) (A) Structures of complexes 67 and 68; (B) emission spectra of nanoaggregates of complex 68 (20 μM) in a dichloromethane/acetonitrile solvent mixture (10:90 v/v) with varying amounts of picric acid (PA). (C) Relative fluorescence quenching of complex 68 upon the addition of 20 equivalents of various nitro compounds: picric acid (PA), 1,4-dinitrobenzene (DNB), 3-nitrotoluene (3NT), 4-nitrotoluene (4NT), nitrophenol (NP), nitrobenzene (NB), 2-nitrotoluene (2NT), and nitromethane (NM). Reproduced with permission.42 Copyright 2013, American Chemical Society. (b) (A) Structure of complex 69; (B) X-ray molecular structures of the dication in (B1) trans-69·2PF6, (B2) cis-69·2ClO4, and (B3) trans-69·2ClO4 crystals, with anions and solvent molecules omitted for clarity. (C) Phosphorescence imaging of HeLa cells incubated with complex 69·2PF6 (10 μM) at 37 °C for 30 minutes (C1) and phosphorescence images of HeLa cells treated with NaClO4·H2O at concentrations of 30, 90, and 200 μM (C2, C3, and C4, respectively) for 15 minutes, followed by the addition of complex 69·2PF6 (10 μM) and incubation at 37 °C for another 30 minutes. Reproduced with permission.43 Copyright 2015, The Royal Society of Chemistry. | ||
These findings highlight the immense potential of polynuclear transition metal complex-based aggregation-enhanced emission systems in advancing chemical sensing technologies, offering highly sensitive and selective detection methods for hazardous substances with applications ranging from environmental monitoring to biological imaging.
A prime example of AEE-active materials in environmental sensing is the neutral dinuclear iridium(III) complex 70 (Fig. 6a), which exhibits not only AEE but also piezochromic luminescence (PCL) and vapochromism. This complex undergoes a reversible color change in its phosphorescence when exposed to high-polarity VOCs, such as chloroalkanes, tetrahydrofuran, and ethyl acetate (Fig. 6a). This behavior makes it an excellent candidate for real-time VOC detection. The high sensitivity to aggregation state changes, combined with exceptional stability in the air, allows this iridium(III) complex to function effectively as a monitoring device for environmental safety, particularly in detecting VOCs.45 Another example comes from a rhenium(I)-based complex 71 (Fig. 6b), which demonstrates AEE enhancement. This complex selectively detects nitroaromatic explosives, such as picric acid (PA) and 2,4,6-trinitrotoluene (TNT), through fluorescence quenching via a photo-induced electron transfer mechanism (Fig. 6b). The complex has been successfully used for on-site detection, such as on paper strips and in soil, demonstrating its practical application in environmental sensing. This system highlights the importance of AEE-active metal complexes in detecting hazardous materials like explosives in real-world settings.46 The self-assembly of complexes 72–74 (Fig. 6c) also exemplifies the potential of metal-based complexes in environmental sensing. These complexes exhibit aggregation-enhanced emission in tetrahydrofuran solutions when water is gradually added. The aggregates formed during this process show significant emission enhancement, and their morphology, influenced by the water content, affects the emission properties. Transmission electron microscopy studies reveal that at lower water content, smaller, rectangular aggregates form, while larger, amorphous aggregates appear at higher water content. These metallacycles have been effectively used as selective luminescent sensors for detecting nitroaromatic explosives like picric acid and nitro-containing antibiotics. The ability of these rhenium(I)-based metallacycles to undergo morphology-driven emission enhancement and selectively detect hazardous pollutants highlights their potential for environmental monitoring, offering a promising approach to public safety.47
 | ||
| Fig. 6 (a) (A) Structure of complex 70; (B) photographic images illustrating the detection of volatile organic compounds (VOCs) using complex 70 under UV light after exposure to various VOCs and mechanical grinding. Reproduced with permission.45 Copyright 2017, The Royal Society of Chemistry. (b) (A) Structure of complex 71; (B) bar graph showing fluorescence intensity quenching of complex 71 after the addition of picric acid (PA) and trinitrotoluene (TNT), with an error margin of ±3%. Excitation wavelength: 405 nm. Categories: 1. Aggregate, 2. TNT, 3. PA, 4. 2-Nitrophenol (2-NP), 5. 3-Nitroacetophenone (3-NAcP), 6. 3-Nitrobenzaldehyde (3-NBAc), 7. 4-Nitrobenzaldehyde (4-NBA), 8. bromonitrobenzene (Br-NB), 9. 4-Nitrophenol (4-NP), 10. nitrobenzene (NB), 11. 2-Nitroaniline (2-NAn), and 12. dinitrophenol (DNP); (C) emission spectra of aggregated complex 71 (10−5 M) upon gradual addition of different volumes of solutions containing (C1) PA and (C2) TNT (10−4 M), with an excitation wavelength of 405 nm. Reproduced with permission.46 Copyright 2021, The Royal Society of Chemistry. (c) Structures of complexes 72–81. | ||
These innovations demonstrate the critical role of AEE-active polynuclear transition metal-based complexes in advancing real-time, on-site environmental monitoring, with the potential to address emerging environmental and safety challenges through further refinement and integration of these technologies.
A common theme across various studies on platinum(II) complexes is the use of aggregation-enhanced emission to amplify luminescent properties upon binding to biological targets. This phenomenon serves as the foundation for designing advanced biosensors tailored to specific applications. Our group reported two isomeric trimetallic complexes 75 and 76 (Fig. 6c), that demonstrated distinct aggregation behaviors and guanine-binding capacities due to their structural differences. Complex 75, with Pt-di-2-picolylamine units arranged in different planes, exhibited pronounced aggregation and a unique 1:2 Pt-5′-GMP binding mode, enabling interactions with two guanine residues per platinum(II) center. This aggregation was further linked to Pt⋯Pt stacking interactions, as revealed by low-temperature emission studies. Moreover, complex 75 exhibited enhanced hydrolytic stability (t1/2 = 55.5 minutes) compared to complex 76 (t1/2 = 16.9 minutes), highlighting its robustness and potential for biomolecular sensing applications. In contrast, the planar geometry of complex 76 resulted in limited aggregation and a simpler 1:1 Pt-5′-GMP binding mode, demonstrating how structural differences can influence functional properties. These findings emphasize the importance of aggregation in determining both the sensitivity and robustness of platinum(II) complexes as biosensors.50 Building on this work, our group explored the utility of square planar platinum(II) complexes (77–80) (Fig. 6c) as luminescent biosensors for DNA detection. These complexes exhibited aggregation-enhanced emission, with exceptionally bright red emission upon binding to DNA. For instance, complex 77 showed strong aggregation behavior in the presence of DNA, resulting in visible photoluminescence under 360 nm UV light. It also demonstrated exceptional hydrolytic stability (t1/2 = 67.62 minutes) and intercalative binding to DNA, as confirmed by UV-visible, circular dichroism (CD) spectroscopy, and emission titration. Notably, complex 79, modified with an electron-donating –NMe2 group, exhibited a significant fluorescence enhancement (∼40-fold), highlighting the impact of chemical modifications on photophysical properties. These results highlight the role of aggregation in amplifying the sensing capabilities of platinum(II) complexes for DNA detection.51 Additionally, a switchable luminescent bioprobe based on AEE was developed by F. Christopher Pigge and collaborators using a neutral dinuclear platinum(II) complex 81 (Fig. 6c). This complex exhibited near-infrared aggregation-enhanced emission at 785 nm in aqueous solutions. Its luminescence was quenched upon binding to G-quadruplex DNA and restored upon DNA degradation by DNAse I, enabling a label-free, sensitive, and rapid enzymatic assay. This assay demonstrated a remarkable detection limit of 0.002 U mL−1 and suitability for high-throughput screening, establishing it as an effective tool for real-time enzymatic activity monitoring and the identification of DNAse I inhibitors. These findings highlight the versatility of AEE-based platinum(II) complexes in chemical biology and clinical applications.52
Taken together, these studies illustrate the transformative potential of polynuclear platinum(II) complexes in biomolecular sensing. Their structural adaptability, aggregation-driven emission enhancements, and diverse functional applications position them as powerful tools for advancing diagnostics and therapeutic development.
One of the promising approaches in biological imaging involves the use of fluorescent complexes 82 and 83 (Fig. 7a). These compounds, which feature a zinc(II)-dipicolylamine core, exhibit strong fluorescence upon binding to bilayer membranes that are enriched with anionic phospholipids. This unique property makes these compounds effective stains for imaging bacterial membranes, particularly in Gram-negative and Gram-positive bacteria like Escherichia coli and Staphylococcus aureus (Fig. 7a). The fluorescence of these compounds is specifically localized to the bacterial membrane, allowing clear distinction from intracellular DNA, a feature that enhances their specificity in imaging. Moreover, these complexes exhibit high selectivity for bacterial cells over mammalian cells, even in complex biological environments such as human saliva. This selectivity arises from differences in membrane surface charge rather than lipid mixing, making these compounds valuable tools for bacterial imaging in diverse settings. These findings suggest that zinc(II)-dipicolylamine complexes hold promise as diagnostic tools for bacterial infections and could be pivotal in advancing imaging techniques for clinical applications.55 Another exciting development in biological imaging is the use of aggregation-enhanced emission exhibited by a series of binuclear copper(I) complexes 84–87 (Fig. 7b). These complexes exhibit excellent stability in air and moisture due to the protective coordination environment provided by soft P-donors and aromatic N atoms. Structurally, complex 87 uniquely adopts both eclipsed and staggered conformations in a 1:1 ratio in the crystal, and the stability order of the complexes in solution follows 85 > 86 > 87 > 84, with 85 being the most stable due to its small P–Cu–P angle, which reduces steric repulsions. UV-visible spectra reveal ligand-centered π–π* and metal-to-ligand charge transfer (MLCT) transitions, while dissociation studies indicate that complex 84 and complex 87 dissociate more readily in dilute solutions, whereas complex 85 remains stable. The complexes exhibit solid-state phosphorescence with emission peaks ranging from 543 to 566 nm (green to yellow luminescence), lifetimes between 2.51–9.26 μs, and quantum yields up to 0.174 (highest for complex 86). The observed AEE effect in dichloromethane/hexane mixtures suggests that luminescence increases upon aggregation, likely due to restricted intramolecular motion. When tested in living HeLa cells, complexes 84–87 successfully labeled the cells with green luminescence under 405 nm excitation, highlighting their potential for bioimaging applications. Among these complexes, complex 86 exhibited the brightest emission, making it particularly suitable for cellular imaging due to its enhanced photoluminescent properties (Fig. 7b). This study demonstrate the first use of aggregation-enhanced emission copper(I) complexes in biological imaging, opening the door for further research into their application in live cell imaging, monitoring cellular activities, and potentially serving as diagnostic tools in medical research.56 Our research group reported that dinuclear iridium(III)–platinum(II) complexes 88–90 (Fig. 7c) exhibit aggregation-enhanced emission and have emerged as promising tools for bacterial imaging. These complexes, specifically designed with an iridium(III) center coordinated to a platinum(II) terpyridine unit, exhibit unique photophysical properties that enable selective staining of bacterial cells. The AEE of these complexes is significantly influenced by pH and ionic strength, with notable emission enhancement observed in acidic environments, such as those mimicking the human gut. These complexes demonstrated excellent bacterial staining in E. coli at low pH (1.2), while showing minimal staining at neutral and alkaline pH levels (Fig. 7c). The complexes' ability to aggregate in response to increased ionic strength or decreased pH makes them ideal candidates for bioimaging in physiological conditions, providing non-invasive, rapid, and reliable methods for studying bacterial morphology and interactions in natural environments like the human gut.57
 | ||
| Fig. 7 (a) (A) Structures of complexes 82 and 83; (B) imaging of E. coli cells co-stained with complexes 82 or 83 and 7AAD. Frames B1 and B3 show the blue and green fluorescence emission of complexes 82 and 83, respectively, while frames B2 and B4 display overlays of 7AAD co-staining, revealing the relative positions of the cell membrane and DNA (magnification 1500×); (C) imaging of S. aureus cells stained with complex 82 (C1) and complex 83 (C2). Reproduced with permission.55 Copyright 2006, The Royal Society of Chemistry. (b) (A) Structures of complexes 84–87; (B) confocal luminescence imaging of HeLa cells incubated with 10 μM solutions of complexes 84–87 in a dimethyl sulfoxide/phosphate-buffered saline mixture (1:99, v/v) for 15 minutes. Panels B1–B4 correspond to complexes 84, 85, 86, and 87, respectively. Reproduced with permission.56 Copyright 2014, American Chemical Society. (c) (A) Structures of complexes 88–90; (B) pH-dependent staining of E. coli (DH5α) cells using complexes 88, 89, and 90, with fluorescence observed at a wavelength of 630 nm (scale bar: ∼5 μm). Reproduced with permission.57 Copyright 2023, The Royal Society of Chemistry. | ||
These findings highlight the immense potential of aggregation-enhanced emission-active polynuclear transition metal complexes in biological imaging, offering innovative tools for bacterial detection and cellular visualization, with promising implications for medical diagnostics, microbiological research, and real-time imaging in complex biological environments.
3.2. Therapeutic applications
Therapeutic applications of advanced materials in medicine have seen significant progress, particularly in the areas of antibacterial treatment, cancer therapy, and photodynamic therapy (PDT).58 Recent studies focus on multifunctional probes and photosensitizers that offer promising strategies for real-time imaging, bacterial eradication, and effective cancer treatment.One notable development in antibacterial therapy is the creation of a multifunctional probe 91 (Fig. 8a) designed for selective targeting, fluorescence imaging, and photodynamic bacterial killing. This probe demonstrated enhanced fluorescence upon binding to bacterial membranes. It was highly effective against both Gram-positive Bacillus subtilis and Gram-negative Escherichia coli while sparing mammalian cells such as human T-lymphocyte Jurkat and K562 myelogenous leukemia cells (Fig. 8a). The probe utilizes aggregation-enhanced emission and excited-state intramolecular proton transfer (ESIPT) mechanisms, allowing it to target bacterial membranes through its positive charge. Upon light exposure, probe 91 exerts a dual-mode antibacterial action by depolarizing bacterial membranes and generating reactive oxygen species, leading to bacterial death (Fig. 8a). This study highlights the potential of probe 91 as a theranostic tool, enabling both real-time bacterial imaging and photodynamic bacterial killing in vivo, providing an advanced approach to controlling bacterial infections.60 Similarly, zinc(II)-tetradentate-coordinated red-emissive probe 92 (Fig. 8b) was designed to target bacteria selectively while enabling fluorescence imaging and PDT. In contrast to conventional hydrophobic photosensitizers, probe 92 remains non-emissive in aqueous environments but exhibits strong fluorescence when aggregated. It binds to bacterial surfaces, selectively staining Gram-positive Bacillus subtilis and Gram-negative Escherichia coli, making it ideal for real-time imaging without additional washing steps (Fig. 8b). The probe exhibits a high singlet oxygen quantum yield of 77.6%, showing its excellent phototoxicity against bacteria under white light irradiation. In dark conditions, probe 92 demonstrates significant toxicity against Bacillus subtilis, and under light exposure, it efficiently photoinactivates both bacterial strains (Fig. 8b). These findings highlight the potential of probe 92 as a theranostic tool for bacterial imaging and PDT, providing an alternative approach to combating bacterial infections.61
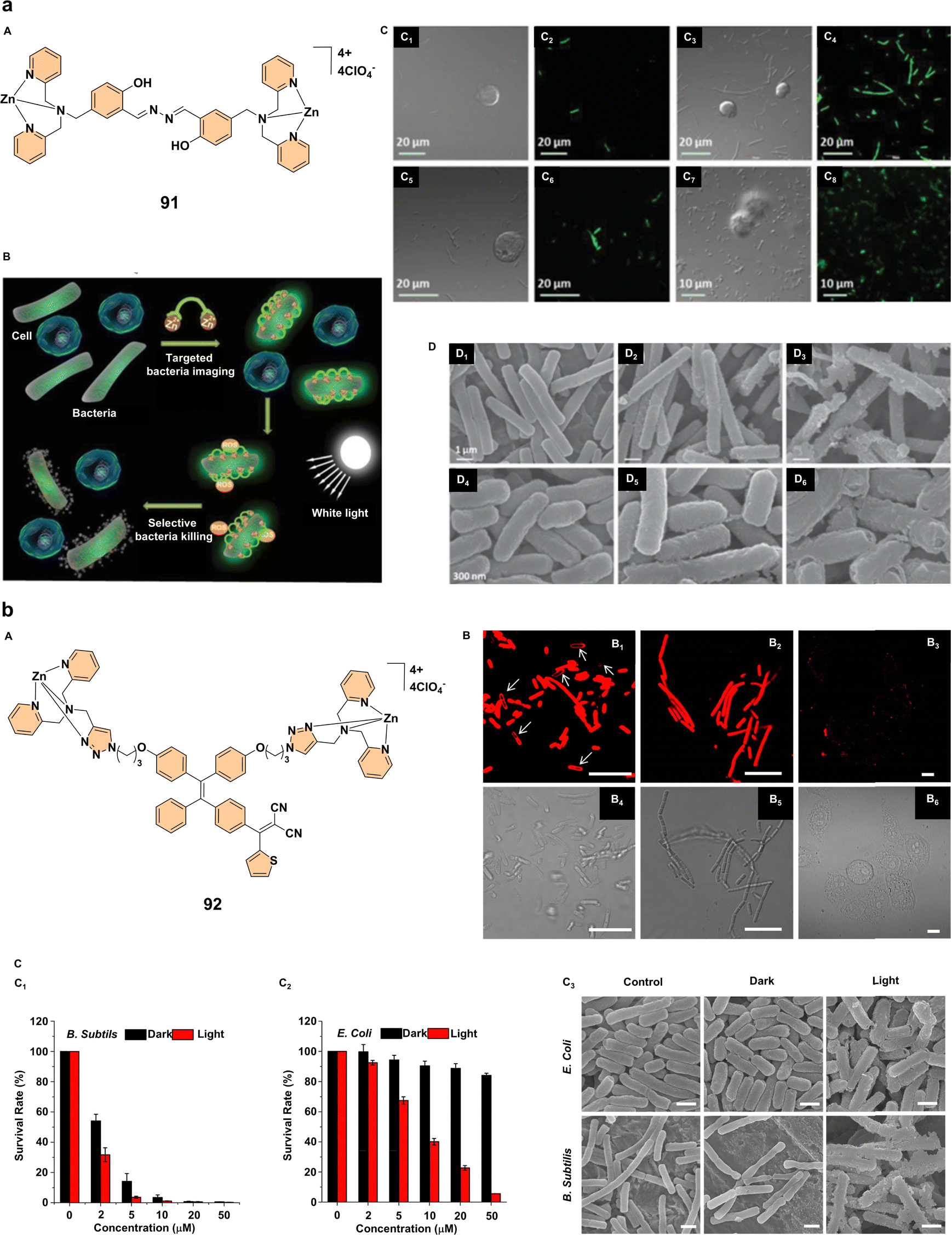 | ||
| Fig. 8 (a) (A) Structure of complex 91; (B) schematic representation of complex 91, illustrating its selective targeting, imaging, and bactericidal activity against bacteria while sparing mammalian cells; (C) confocal laser scanning microscopy (CLSM) images of cells and bacteria incubated with complex 91 (20 × 10−6 M): (C1, C2) B. subtilis and Jurkat T cells; (C3, C4) B. subtilis and K562 cells; (C5, C6) E. coli and Jurkat T cells; (C7, C8) E. coli and K562 cells. The fluorescence of complex 91 is shown in green; (D) morphological analysis of bacteria incubated with complex 91: (D1, D4) B. subtilis and E. coli, respectively, without complex 91 under dark conditions; (D2, D5) with complex 91 (20 × 10−6 M) under dark conditions; (D3, D6) with complex 91 (20 × 10−6 M) and white light irradiation (100 mW cm−2, 6 min). Reproduced with permission.60 Copyright 2015, Wiley-VCH. (b) (A) Structure of complex 92; (B) confocal (B1–B3) and bright-field (B4–B6) images of E. coli (B1, B4), B. subtilis (B2, B5), and HeLa cells (B3, B6) after 30 minutes of incubation with probe 92 (20 μM) without washing. The red fluorescence signal was collected above 590 nm with excitation at 458 nm. All images have a 10 μm scale bar. (C) Survival rates of (C1) B. subtilis and (C2) E. coli treated with complex 92 at varying concentrations, with and without light irradiation. (C3) Scanning electron microscopy images of E. coli and B. subtilis treated with complex 92, with or without light irradiation. All SEM images share a scale bar of 1 μm. Reproduced with permission.61 Copyright 2017, American Chemical Society. | ||
These AEE-active multifunctional probes not only offer promising alternatives to traditional antibiotics but also facilitate more effective and selective antibacterial treatments, with the added benefit of real-time imaging for precise therapeutic intervention.
 | ||
| Fig. 9 (a) Structures of complex 93–96. (b) (A) Structure of compound 97; (B) synthesis process of 97-NPs; (C) schematic diagram illustrating the use of 97-NPs as photosensitizers for photodynamic therapy; (D) representative images of mice after treatment. The hair on the thigh was shaved just before irradiation. Images were captured on day 14 post-irradiation, showing varying hair growth rates in different mice; (E) tumors harvested from various treatment groups: (E1) treated with saline, (E2) treated with saline and light, (E3) treated with 97-NPs, (E4) treated with 97-NPs and light (100 mg mL−1, 100 μL), with light irradiation at 450 nm, 200 mW cm−2 for 20 minutes. Reproduced with permission.67 Copyright 2019, Wiley-VCH. (c) (A) Structure of compound 98; (B) structure of 98′ and schematic representation of 98-NPs as a photosensitizer for photodynamic therapy. Reproduced with permission.68 Copyright 2020, The Royal Society of Chemistry. | ||
Simultaneously, Photodynamic therapy, a non-invasive treatment that relies on photosensitizers activated by light to generate reactive oxygen species and selectively destroy cancer cells, is gaining attraction.64 Advances in PDT have focused on improving photosensitizers, particularly those based on iridium(III), to enhance their photophysical properties and ROS generation.65 A key advancement in PDT has been the development of red-emitting aggregation-enhanced emission iridium(III) complexes, which offer enhanced properties for PDT.66 One such example is the red-emitting iridium(III) trinuclear complex 97 (Fig. 9b). This complex was synthesized to improve singlet oxygen generation, an essential factor for effective PDT. By increasing the number of iridium(III) centers, complex 97 exhibited enhanced singlet oxygen generation compared to its mononuclear counterparts. When formulated into nanoparticles (97-NPs), these showed brighter emission, higher phosphorescence quantum yields, longer excited-state lifetimes, and better biocompatibility (Fig. 9b). The 97-NPs demonstrated a high molar absorption coefficient, which contributed to potent PDT effects both in vitro and in vivo, effectively inhibiting tumor growth in mouse models with negligible dark toxicity (Fig. 9b). This highlights the promise of multinuclear AEE iridium(III) complexes as potent photosensitizers for cancer treatment, offering an innovative approach for clinical applications.67 Another significant development in PDT is the use of near-infrared aggregation-enhanced emission photosensitizers. A tri-nuclear cationic iridium(III) complex 98 was self-assembled into nanoparticles (98-NPs) to enhance PDT performance (Fig. 9c). The introduction of a rigid 1,3,5-triphenyl benzene bridging ligand extended the π-conjugation, improving the photophysical properties and increasing singlet oxygen generation. The resulting 98-NPs exhibited bright NIR emission, higher 1O2 production capacity, and better water solubility. Compared to a previous photosensitizer (98′), these 98-NPs showed superior phototoxicity (IC50 = 1.4 × 10−6 M). The self-assembled iridium(III) complex 98-NPs demonstrated excellent biocompatibility, efficient cellular uptake, and enhanced PDT efficacy, making them as promising candidates for future clinical applications (Fig. 9c). This approach further emphasizes the potential of iridium(III)-based photosensitizers in PDT, particularly those designed for NIR light activation, which offers deeper tissue penetration.68 The development of cyclometalated iridium(III) complex-porphyrin conjugates is another promising direction in PDT. A tetra-nuclear iridium(III) complex 99 containing a porphyrin unit (Fig. 10a) exhibits long-wavelength absorption (500–700 nm) and near-infrared emission (635–750 nm), overcoming the limitations of traditional iridium(III) complexes, which typically absorb at shorter wavelengths. Additionally, this iridium(III) complex-porphyrin conjugate successfully self-assembles into nanoparticles (99-NPs). The 99-NPs demonstrated excellent biocompatibility, high-efficiency singlet oxygen generation, and low half-maximal inhibitory concentrations (IC50). This makes them strong candidates for PDT under white light irradiation, offering a more versatile and practical treatment approach (Fig. 10a). The enhanced photophysical properties and intracellular 1O2 production suggest that these iridium(III)–porphyrin conjugates could play a significant role in future clinical PDT applications.69 Our group explored the photophysical properties and applications of trimetallic complexes 100 and 101 (Fig. 10b), focusing on their aggregation behavior. Complex 100 exhibits minimal emission in solution at room temperature but shows strong aggregation-enhanced emission at low temperatures and in the solid state, highlighting the role of aggregation in modulating its optical properties. In contrast, complex 101 demonstrates a different aggregation-dependent emission behavior. Additionally, the hydrolyzed tricationic forms of these complexes show enhanced nuclear accumulation, making them promising candidates for photodynamic therapy due to their ability to generate reactive oxygen species upon photoexcitation (Fig. 10b). This study highlights the impact of aggregation on photophysical properties and the potential therapeutic applications of these complexes in cancer treatment.70
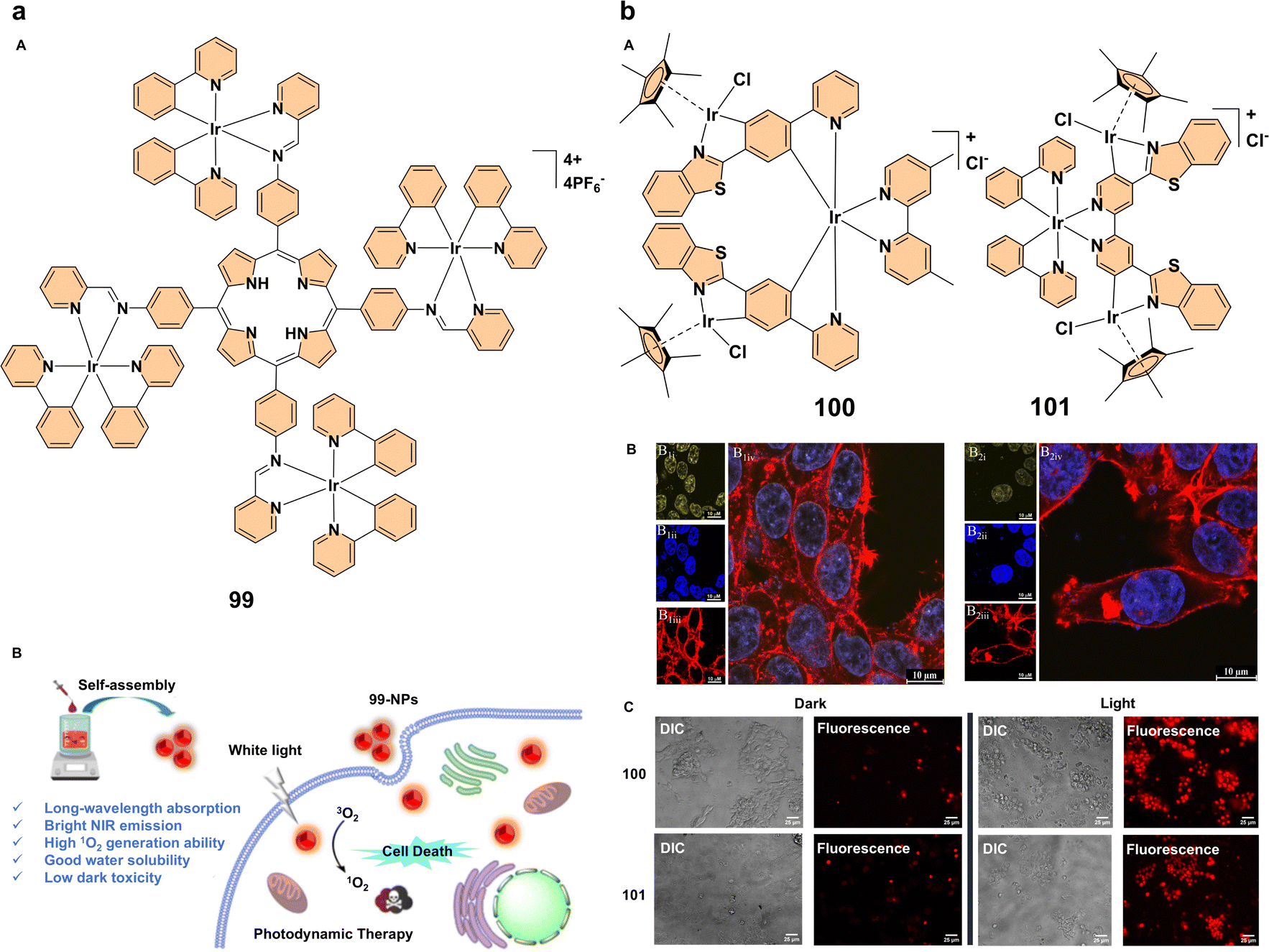 | ||
| Fig. 10 (a) (A) Structure of complex 99; (B) design principle of complex 99 as a photosensitizer for photodynamic therapy. Reproduced with permission.69 Copyright 2023, The Royal Society of Chemistry. (b) (A) structures of complexes 100 and 101; (B) confocal microscopy images of MCF7 cells treated with iridium(III) complexes 100 and 101 after 18 h of incubation (fixed cells, scale bar: 50 μm). Panels B1–B2 show images captured at λex = 405 nm and λem = 550–570 nm (B1i, B2i), DAPI-stained nuclei at λex = 400 nm and λem = 460 nm (B1ii, B2ii), and actin filaments labeled with phalloidin at λex = 647 nm and λem = 660 nm (B1iii, B2iii). Merged images of the different channels for fixed cells are presented in (B1iv, B2iv); (C) differential interference contrast (DIC) and IX81 fluorescence images of viable MCF7 cells after a 24 h treatment with complexes 100 and 101, followed by photoirradiation with 390 nm LED light for 1 h. The left panel shows pre-photoirradiation cells, while the right panel shows post-photoirradiation cells. All images were taken at 60× magnification, and the scale bar is 25 μm. Reproduced with permission.70 Copyright 2023, American Chemical Society. | ||
Together, these innovations in chemotherapeutic agents and photodynamic therapy highlight the potential of ruthenium(II) and iridium(III)-based complexes to revolutionize cancer treatment, offering more selective, effective, and safer alternatives to conventional therapies.
3.3. Security, data encryption, and optoelectronic applications
Aggregation-enhanced emission-active polynuclear transition metal complexes have become pivotal in the development of next-generation photonic devices, particularly organic light-emitting diodes and other light-emitting technologies. Their strong, tunable emission properties, especially in the solid state, enable the design of efficient, durable, and highly functional devices. The following examples highlight the wide-ranging applications of these complexes, demonstrating their interrelationships and more significant potential in security, data encryption, and the development of optoelectronic devices. | ||
| Fig. 11 (a) (A) Structure of complex 102; (B) photographic images showing (B1) the first-level anti-counterfeit trademark, (B2) the second-level anti-counterfeit trademark, and (B3) information encryption and decryption devices. The yellow letters ‘AIPE’ can be seen within the bottom ‘NENU’. Reproduced with permission.71 Copyright 2017, The Royal Society of Chemistry. (b) (A) Structures of complexes 103 and 104; (B) proposed mechanism for the photochemical luminescence of complexes 103 and 104; (C) information encryption and decryption devices using complex 103 (C1) and a reversible data recording device using complex 104 (C2). Reproduced with permission.72 Copyright 2020, Elsevier. | ||
So, these examples highlight the transformative potential of AEE-active complexes in advancing security and data encryption technologies, setting the stage for innovative solutions such as tamper-proof devices, optical authentication systems, and re-writable memory, all driven by their unique photophysical properties.
Among the key players in OLED technology are platinum complexes, which have gathered attention for their exceptional photophysical properties.75 The dinuclear platinum(III) complex 105 (Fig. 12a) stands out for its exceptional OLED performance, attributed to its strong phosphorescence and aggregation-dependent emission characteristics, which are directly influenced by the interaction between the platinum(III) center and fluorescent chelators. The oxadiazole-thiol ligands, with their donor–acceptor (D–A) character, not only stabilize the d7–d7 platinum(III) electronic configuration but also modulate the charge transfer dynamics via a ligand–metal–metal charge transfer (LMMCT) transition. This results in highly tunable phosphorescence, shifting from orange (618 nm in solution) to NIR (749 nm in the crystalline phase), with a noticeable blue shift upon mechanical grinding, confirming that intermolecular interactions are strongly influenced by the ligand environment. To evaluate its optoelectronic potential, complex 105 was incorporated into three different OLED architectures: device I (NIR OLED): ITO/PEDOT:PSS (40 nm)/PVK (30 nm)/105 (55 nm)/TPBI (60 nm)/CsF (0.8 nm)/Al (120 nm) → 716 nm NIR emission, EQEmax = 5.1%, strong stability with 3.8% EQE at 100 mA cm−2 (Fig. 12a). Device II (red OLED): ITO/PEDOT:PSS (40 nm)/mCP:105 (90:10, 50 nm)/DPEPO (10 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm) → 614 nm emission, EQEmax = 8.7%. Device III (white-light OLED, hybrid): ITO/PEDOT:PSS (40 nm)/CzAcSF:105 (90:10, 25 nm)/DPEPO (10 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm) → dual emission (470 nm, 612 nm), EQEmax = 11.6%. These results confirm that platinum(III) complexes are highly effective phosphorescent emitters, enabling high-performance solution-processed NIR, red, and white OLEDs with remarkable efficiency. The combination of D-A chelators with a dinuclear platinum(III) center ensures efficient charge transport, tunable emission, and superior stability, making them promising candidates for next-generation optoelectronic applications.76
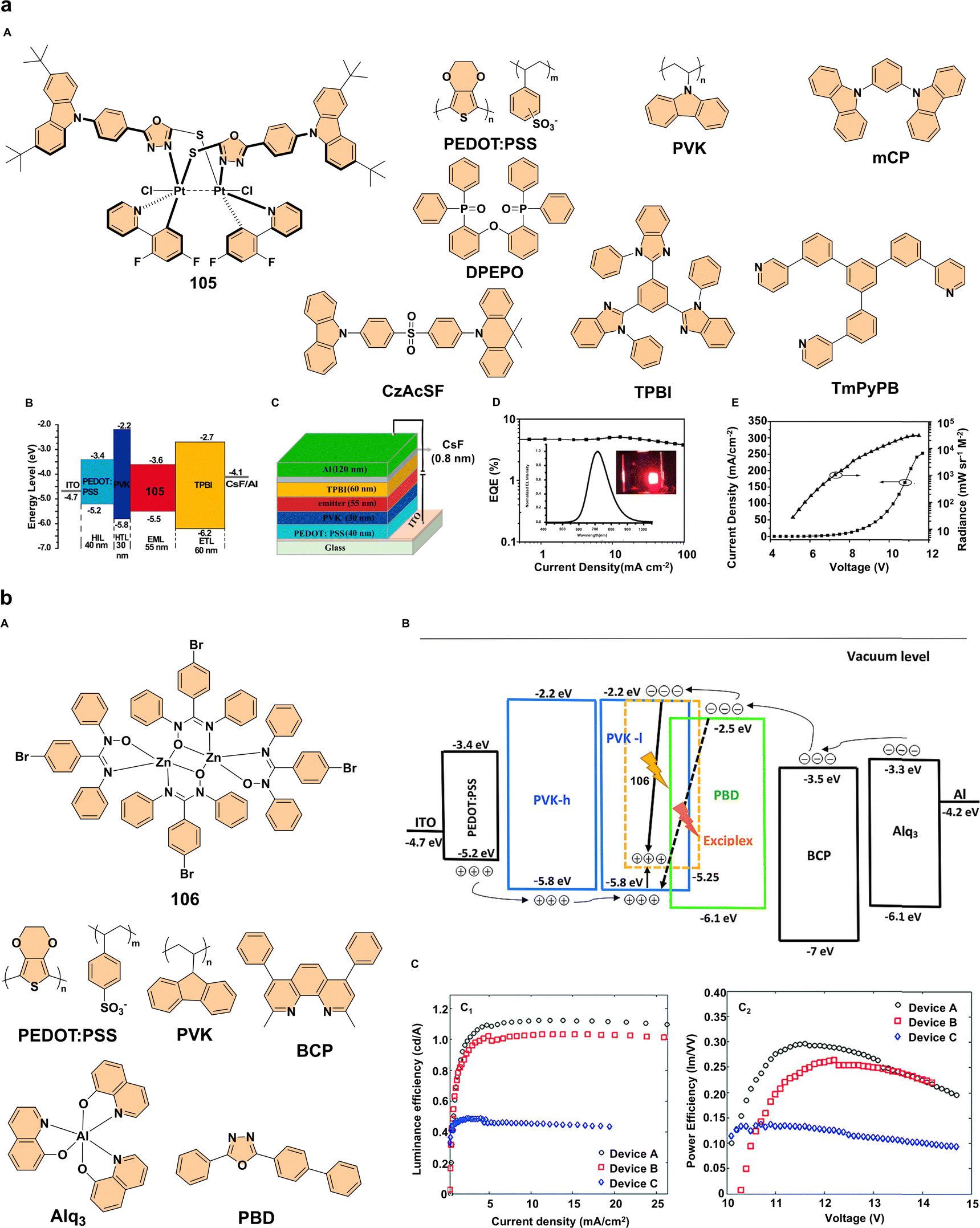 | ||
| Fig. 12 (a) (A) Structure of complex 105 and structures of the materials utilized in OLED fabrication; (B) energy-level diagrams for device I; (C) configuration of device I; (D) electroluminescent characteristics of device I, with insets showing the electroluminescence spectra of device I and a photograph of the device in operation; (E) current density–voltage–radiance (J–V–R) curve of device I, illustrating the structures of the hole injection layer (HIL), hole transport layer (HTL), and electron transport layer (ETL). Reproduced with permission.76 Copyright 2020, American Chemical Society. (b) (A) Structure of complex 106 and structures of the materials utilized in WOLED fabrication; (B) energy level alignment of the components used in the device structure, illustrating the electron and hole injection pathways from the corresponding charge transport materials into the emissive layer. The formation of excitons on complex 106 molecules is shown, along with the creation of exciplexes at the interface between the LUMO of PBD and the HOMO of PVK-l, as indicated by the dashed arrow; (C) C1: luminance efficiency as a function of current density (LE–J) and C2: power efficiency as a function of voltage (PE–V) for devices A, B, and C. Reproduced with permission.78 Copyright 2018, Wiley-VCH. | ||
In addition to traditional OLEDs, white organic light-emitting diodes (WOLEDs) are a crucial area of development for energy-efficient lighting technologies.77 WOLEDs combine multiple emission sources to achieve a full spectrum of light, making them ideal for general lighting applications. A typical WOLED incorporating the dinuclear zinc(II) complex 106 (Fig. 12b) follows a multilayered device architecture designed for efficient charge transport and high luminance. The device structure consists of ITO/PEDOT:PSS (30 nm)/PVK-h (15 nm)/PVK-l:PBD:complex 106 (3, 6, and 12 wt%) (30 nm)/BCP (10 nm)/Alq3 (40 nm)/Al (150 nm)/Ag (50 nm). This architecture was designed to balance charge recombination and enhance device performance. To explore solution-processed WOLEDs based on complex 106, three devices with different doping concentrations (3, 6, and 12 wt%) were fabricated, referred to as devices A, B, and C, respectively. Device A achieved the highest luminance efficiency (LE) of 1.12 cd A−1 at 9.7 mA cm−2, followed by device B (1.03 cd A−1 at 10 mA cm−2) and device C (0.49 cd A−1 at 2.4 mA cm−2). The corresponding maximum power efficiencies (PEs) were 0.30, 0.25, and 0.13 lm W−1 for devices A, B, and C, respectively. Although these values are lower than those of phosphorescent and TADF-based WOLEDs, they are comparable to the best-reported thermally-processed fluorescent zinc(II)-based WOLEDs (0.12–2.1 cd A−1 for LE and 0.038–1.17 lm W−1 for PE). As doping concentration increases, turn-on voltages rise, leading to a decrease in efficiency, as seen in devices B and C (Fig. 12b). The incorporation of complex 106 into WOLEDs enhances device efficiency by leveraging AEE behavior, ensuring high solid-state luminescence while minimizing non-radiative decay and self-quenching. This leads to improved quantum efficiency and prolonged stability. With high luminance and power efficiency, complex 106 serves as a viable alternative to phosphorescent emitters, offering white-light emission suitable for lighting applications. Moreover, by avoiding heavy metals like iridium and platinum, zinc(II)-based WOLEDs provide an environmentally sustainable alternative for next-generation lighting technologies.78
Another significant advancement in OLED technology comes from platinum(II) complexes, particularly the dinuclear platinum(II) complex 107 (Fig. 13), which demonstrates remarkable aggregation-enhanced emission properties. The device structure used in these WOLEDs follows the configuration: ITO/PEDOT:PSS (45 nm)/FIrpic:107:Ir-Tz-1:TCTA:26DCzPPy (40 nm)/TmPyPb (55 nm)/LiF (0.7 nm)/Al (100 nm), where PEDOT:PSS serves as the hole injection layer, TmPyPb functions as the electron transport and hole-blocking layer, and the emissive layer comprises a precise mixture of blue, green, and red emitters to ensure balanced white light emission (Fig. 13). Devices W1, W2, and W3 were designed to optimize blue emission and overall electroluminescent performance. Device W1, despite a higher FIrpic doping concentration, exhibits weak blue emission at low voltages due to hole trapping by complex 107 and Ir-Tz-1, improving at higher voltages and enhancing the color rendering index (CRI). Its peak ηext, ηL, and ηP are 13.5%, 37.4 cd A−1, and 28.2 lm W−1, respectively. To improve blue emission and CRI, device W2 was fabricated with reduced complex 107 doping, and device W3 with increased FIrpic doping, both showing enhanced blue components. Device W2 achieved superior efficiency with ηL and ηP of 42.3 cd A−1 and 31.8 lm W−1 (Fig. 13). This superior EL performance highlights the promise of dinuclear platinum(II) complexes for advancing WOLED technology, particularly for high-performance, broad-spectrum emission applications.79
 | ||
| Fig. 13 (a) Structure of complex 107 and structures of the materials used in WOLEDs. (b) (A) Device configuration; (B) energy level diagram; (C) (C1) J–V–L characteristics of devices W1, W2, and W3. EL efficiency versus luminance for (C2) device W1, (C3) device W2, and (C4) device W3. Reproduced with permission.79 Copyright 2021, The Royal Society of Chemistry. | ||
The potential of AEE-active complexes extends beyond visible-light applications to specialized fields such as medical imaging and communication technologies, particularly with near-infrared organic light-emitting diodes (NIR-PLEDs). These devices leverage the unique properties of materials that emit in the NIR spectrum, enabling applications in bioimaging and secure communications. The iridium(III) complex 108 (Fig. 14A) exemplifies this shift toward NIR-PLEDs. This complex demonstrates aggregation-enhanced emission behavior, where its emission intensity significantly increases in the solid state or specific solvent mixtures. This property makes complex 108 highly suitable for NIR-PLEDs, expanding the range of aggregation-enhanced luminescence into the infrared region and enhancing the versatility of AEE-active materials for targeted applications. The NIR-PLED device has a structure of ITO/PEDOT:PSS (40 nm)/PVK:OXD7:complex 108 (65:30:5 wt%) (120 nm)/LiF (1 nm)/Al (100 nm), where ITO serves as the anode, PEDOT:PSS as the hole-injecting layer, and the PVK-OXD7 blend with the dinuclear iridium(III) complex 108 acts as the emitting layer (Fig. 14B). LiF and Al function as the electron injection layer and cathode, respectively. The device structure enables efficient electroluminescence, as evidenced by its voltage-independent emission spectrum peaking at 710 nm (Fig. 14C). At 15 V, NIR electroluminescence initiates (Von = 5 W sr−1 cm−2), with irradiance (Le) and current density (J) increasing as voltage rises. At 21 V, the device achieves Lemax = 41.4 W sr−1 cm−2 at Jmax = 3.7 mA cm−2. Notably, the NIR-PLED exhibits Le-regulated performance, with ηEQE peaking at 1.08% at 14.9 W sr−1 cm−2 and showing minimal roll-off (<2%). The performance of the device depends on the interaction between the iridium(III) center and chelating ligands in complex 108. Generally, iridium(III) complexes act as efficient phosphorescent emitters due to strong spin–orbit coupling, enabling triplet-state emission. The chelating ligands in complex 108 stabilize the iridium(III) center and tune emission properties while enhancing charge transport. AEE improves luminescence efficiency by restricting intramolecular motion, reducing triplet–triplet annihilation, and boosting stability. Additionally, Förster energy transfer between the PVK-OXD7 host and complex 108 ensures efficient charge trapping and recombination, optimizing electroluminescence for advanced NIR-PLEDs. These findings highlight the potential of AEE-active dinuclear iridium(III) complexes in advancing NIR-PLED technology, offering a promising pathway for the development of high-efficiency, stable, and solution-processable electroluminescent devices for next-generation applications.80
 | ||
| Fig. 14 (A) Structure of complex 108 and structures of the materials used in OLED fabrication; (B) device architecture and energy level diagram; (C) C1: normalized electroluminescent spectra. C2: current density (J, mA cm−2) versus applied bias voltage (V) and irradiance (Le, W sr−1 m−2) versus applied bias voltage (V). C3: ηEQE as a function of Le for the NIR-PLED utilizing the dinuclear complex 108. Reproduced with permission.80 Copyright 2020, Elsevier. | ||
Therefore, the advancements in AEE-active complexes, including zinc(II), iridium(III), platinum(II) and platinum(III)-based systems, demonstrate their immense potential in revolutionizing optoelectronic applications by enabling highly efficient, stable, and solution-processable OLEDs and WOLEDs. These innovations open the way for next-generation lighting, display, and communication technologies, offering enhanced performance, energy efficiency, and sustainability.
4. Future directions and challenges
The field of aggregation-enhanced emission in polynuclear transition metal complexes presents exciting opportunities for advancing material science, but several challenges must be addressed to unlock their full potential. While these complexes offer unique photophysical properties, further research is necessary to enhance their stability, efficiency, and functional versatility across various applications.One of the major challenges is improving the thermal and photostability of PTMCs, which often degrade under prolonged exposure to high temperatures, harsh environmental conditions, or sustained illumination. This limitation restricts their viability in long-term applications, OLEDs, PDT, and security-based luminescent materials. Future research should focus on designing robust ligand architectures and supramolecular frameworks that provide steric protection, enhance electronic stability, and minimize decomposition pathways. The development of self-healing luminescent materials or stimuli-responsive PTMCs capable of adaptive reorganization under environmental stress could also enhance their longevity and practical utility.
Another critical area is the optimization of luminescence efficiency and energy transfer mechanisms. Despite their promise, AEE-active PTMCs often suffer from non-radiative decay pathways in the solid state, which limit their quantum yields. Strategies such as fine-tuning coordination environments, incorporating heterometallic centers, and developing novel bridging ligands could significantly improve luminescence performance.
Scalability and cost-effective synthesis remain pressing concerns. The reliance on expensive noble metals, such as iridium, platinum, and gold, poses economic and environmental challenges. Research into earth-abundant metal alternatives (e.g., iron, copper, and zinc complexes) could reduce costs while maintaining desirable optical properties. Moreover, advancements in green chemistry approaches, including solvent-free synthesis, mechanochemical reactions, and bio-derived ligands, could enhance sustainability while reducing toxic byproducts.
Interdisciplinary approaches will play a crucial role in expanding the functional versatility of AEE-active PTMCs. Future research could explore their integration into hybrid organic–inorganic systems, metal–organic frameworks (MOFs), nanostructures, and bio-compatible scaffolds to create multifunctional materials with enhanced adaptability. For instance, embedding PTMCs into flexible substrates or stimuli-responsive films could facilitate wearable optoelectronic devices, smart luminescent coatings, and real-time biosensors.
Emerging applications in advanced sensing, bioimaging, and quantum communication further highlight the need for fundamental studies on excited-state dynamics, emission tuning, and environmental responsiveness. Expanding the scope of PTMCs in near-infrared bioimaging, theranostics, and deep-tissue phototherapy could revolutionize biomedical applications. Additionally, their potential in secure communication, encryption-based optical storage, and multi-level anti-counterfeiting technologies remains an underexplored yet promising avenue.
By addressing these challenges and harnessing emerging technologies, AEE-active PTMCs have the potential to drive innovations in sustainable energy, precision medicine, quantum electronics, and next-generation photonic devices, ultimately transforming the landscape of functional luminescent materials.
5. Conclusion
Aggregation-enhanced emission in polynuclear transition metal complexes presents a new method to address the limitations of traditional luminescent materials, especially the issue of aggregation-caused quenching. By leveraging mechanisms such as restriction of inter- and intra-molecular motions, metal–metal interactions, and π–π stacking, these complexes exhibit enhanced luminescence in aggregated states, enabling their application across diverse fields. From chemical sensing and bioimaging to energy-efficient lighting and security technologies, AEE-active PTMCs offer unparalleled versatility and functionality. However, challenges such as stability, efficiency, and sustainable synthesis must be addressed to fully realize their potential. Through advanced ligand engineering, computational design, and innovative hybrid material integration, these hurdles can be overcome. The insights gained from studying AEE mechanisms and their applications not only broaden the scope of material science but also establish new directions for groundbreaking technologies. With continued research and development, AEE-active PTMCs are well-positioned to contribute significantly to energy-efficient technologies, advanced therapeutics, and a more sustainable future.Data availability
This review does not include primary research findings, software, or code, and no new data were generated or analyzed.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. D. expresses his heartfelt appreciation to IISER Kolkata for providing the necessary research infrastructure.Notes and references
- (a) L. C.-C. Lee and K. K.-W. Lo, Chem. Rev., 2024, 124, 8825–9014 CrossRef CAS; (b) L. Ravotto and P. Ceroni, Coord. Chem. Rev., 2017, 346, 62–76 CrossRef CAS; (c) J. N. Demas and B. A. DeGraff, Coord. Chem. Rev., 2001, 211, 317–351 CrossRef CAS; (d) X. Li, Y. Xie and Z. Li, Chem. – Asian J., 2021, 16, 2817–2829 CrossRef CAS PubMed; (e) S. Li, L. Zhou and H. Zhang, Light: Sci. Appl., 2022, 11, 177 CrossRef CAS PubMed; (f) X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Mater. Chem. C, 2013, 1, 3376–3390 RSC; (g) Y. Hasegawa, T. Nakagawa and T. Kawai, Coord. Chem. Rev., 2010, 254, 2643–2651 CrossRef CAS.
- (a) B. Ruan, X. Kang, B. Guo, D.-D. Deng, J.-J. Tian, K. He, X.-Y. Wang, S. Pu and Z. Chen, J. Mol. Struct., 2024, 1309, 138171 CrossRef CAS; (b) Y. Zhang, Y. Huang, R. Miao and H. Chen, Small Struct, 2023, 4, 2300157 CrossRef CAS.
- (a) Z. He, C. Ke and B. Z. Tang, ACS Omega, 2018, 3, 3267–3277 CrossRef CAS PubMed; (b) M. H. Chua, B. Y. K. Hui, K. L. O. Chin, Q. Zhu, X. Liu and J. Xu, Mater. Chem. Front., 2023, 7, 5561–5660 RSC.
- (a) D. Sengottuvelu, V. Kachwal, P. Raichure, T. Raghav and I. R. Laskar, ACS Appl. Mater. Interfaces, 2020, 12, 31875–31886 CrossRef CAS; (b) Y. Wang, H. Liu, Z. Chen and S. Pu, Spectrochim. Acta, Part A, 2021, 245, 118928 CrossRef CAS PubMed; (c) X. Yan, P. Zhu, Z. Zhou, H. Yang, H. Lan and S. Xiao, RSC Adv., 2019, 9, 35872–35877 RSC; (d) A. Mondal, E. Ahmmed, S. Chakraborty, A. Sarkar, S. Lohar and P. Chattopadhyay, ChemistrySelect, 2020, 5, 147–155 CrossRef CAS.
- (a) B. Das and P. Gupta, Coord. Chem. Rev., 2024, 504, 215656 CrossRef CAS; (b) Z. Huang and J. J. Wilson, Eur. J. Inorg. Chem., 2021, 1312–1324 CrossRef CAS; (c) R. Ziessel, M. Hissler, A. El-ghayoury and A. Harriman, Coord. Chem. Rev., 1998, 178–180, 1251–1298 CrossRef CAS; (d) J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472–5473 RSC; (e) R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS; (f) B. Das and P. Gupta, Chem. – Asian J., 2021, 16, 2495–2503 CrossRef CAS PubMed.
- M. Goel and M. Jayakannan, J. Phys. Chem. B, 2010, 114, 12508–12519 CrossRef CAS PubMed.
- (a) T. Wu, J. Huang and Y. Yan, Chem. – Asian J., 2019, 14, 730–750 CrossRef CAS PubMed; (b) M. Dharmarwardana, B. M. Otten, M. M. Ghimire, B. S. Arimilli, C. M. Williams, S. Boateng, Z. Lu, G. T. McCandlessa, J. J. Gassensmith and M. A. Omary, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2106572118 CrossRef CAS PubMed; (c) Q. Li and Z. Li, Adv. Sci., 2017, 4, 1600484 CrossRef PubMed.
- M. A. Omary, R. M. Kassab, M. R. Haneline, O. Elbjeirami and F. P. Gabbaï, Inorg. Chem., 2003, 42, 2176–2178 CrossRef CAS PubMed.
- C. Cunha, A. Pinto, A. Galvão, L. Rodríguez and J. S. S. de Melo, Inorg. Chem., 2022, 61, 6964–6976 CrossRef CAS PubMed.
- S.-Y. Yang, Y. Chen, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2024, 53, 5366–5393 RSC.
- (a) H.-S. Lo, S.-K. Yip, N. Zhu and V. W.-W. Yam, Dalton Trans., 2007, 4386–4389 RSC; (b) X. Zheng, M. H.-Y. Chan, A. K.-W. Chan, S. Cao, M. Ng, F. K. Sheong, C. Li, E. C. Goonetilleke, W. W. Y. Lam, T.-C. Lau, X. Huang and V. W.-W. Yam, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2116543119 CrossRef CAS PubMed.
- V. W.-W. Yam, K. H.-Y. Chan, K. M.-C. Wong and B. W.-K. Chu, Angew. Chem., Int. Ed., 2006, 45, 6169–6173 CrossRef CAS PubMed.
- M.-X. Zhu, W. Lu, N. Zhu and C.-M. Che, Chem. – Eur. J., 2008, 14, 9736–9746 CrossRef CAS PubMed.
- Y. Hu, K. H.-Y. Chan, C. Y.-S. Chung and V. W.-W. Yam, Dalton Trans., 2011, 40, 12228–12234 RSC.
- S. Y.-L. Leung and V. W.-W. Yam, Chem. Sci., 2013, 4, 4228–4234 RSC.
- H.-L. Au-Yeung, S. Y.-L. Leung and V. W.-W. Yam, Chem. Commun., 2018, 54, 4128–4131 RSC.
- J. Chen, L. Ao, C. Wei, C. Wang and F. Wang, Chem. Commun., 2019, 55, 229–232 RSC.
- J. Song, H. Xiao, B. Zhang, L. Qu, X. Zhou, P. Hu, Z.-X. Xu and H. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202302011 CrossRef CAS PubMed.
- (a) Z. Lei, J.-Y. Zhang, Z.-J. Guana and Q.-M. Wang, Chem. Commun., 2017, 53, 10902–10905 RSC; (b) T. P. Seifert, V. R. Naina, T. J. Feuerstein, N. D. Knöfela and P. W. Roesky, Nanoscale, 2020, 12, 20065–20088 RSC.
- W.-X. Ni, M. Li, J. Zheng, S.-Z. Zhan, Y.-M. Qiu, S. W. Ng and D. Li, Angew. Chem., Int. Ed., 2013, 52, 13472–13476 CrossRef CAS PubMed.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179–183 CrossRef CAS PubMed.
- K. Fujisawa, S. Yamada, Y. Yanagi, Y. Yoshioka, A. Kiyohara and O. Tsutsumi, Sci. Rep., 2015, 5, 7934 CrossRef CAS PubMed.
- Z. Chen, Z. Li, L. Yang, J. Liang, J. Yin, G.-A. Yu and S. H. Liu, Dyes Pigm., 2015, 121, 170–177 CrossRef CAS.
- F. C.-M. Leung and V. W.-W. Yam, Eur. J. Inorg. Chem., 2017, 5271–5278 CrossRef CAS.
- Z. Chen, G. Liu, S. Pu and S. H. Liu, Dyes Pigm., 2018, 152, 54–59 CrossRef CAS.
- Z. Chen, D. Wu, X. Han, J. Liang, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2014, 50, 11033–11035 RSC.
- (a) Z. Zhao, B. He and B. Z. Tang, Chem. Sci., 2015, 6, 5347–5365 RSC; (b) L.-L. Yang, H. Wang, J. Zhang, B. Wu, Q. Li, J.-Y. Chen, A.-L. Tang, J. W. Y. Lam, Z. Zhao, S. Yang and B. Z. Tang, Nat. Commun., 2024, 15, 999 CrossRef CAS PubMed; (c) M. Dommett, M. Rivera and R. Crespo-Otero, J. Phys. Chem. Lett., 2017, 8, 6148–6153 CrossRef CAS PubMed.
- C. H. Shin, J. O. Huh, S. J. Baek, S. K. Kim, M. H. Lee and Y. Do, Eur. J. Inorg. Chem., 2010, 3642–3651 CrossRef CAS.
- G. Li, X. Ren, G. Shan, W. Che, D. Zhu, L. Yan, Z. Su and M. R. Bryce, Chem. Commun., 2015, 51, 13036–13039 RSC.
- C. Li, B. Zhang, Y. Dong, Y. Li, P. Wang, Y. Yu, L. Cheng and L. Cao, Dalton Trans., 2020, 49, 8051–8055 RSC.
- J. Liang, Z. Chen, L. Xu, J. Wang, J. Yin, G.-A. Yu, Z.-N. Chen and S. H. Liu, J. Mater. Chem. C, 2014, 2, 2243–2250 RSC.
- Z. Chen, J. Zhang, M. Song, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2015, 51, 326–329 RSC.
- C. Xu, H. Shen, T.-M. Liu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, iScience, 2023, 26, 106568 CrossRef CAS PubMed.
- G. Li, Y. Wu, G. Shan, W. Che, D. Zhu, B. Song, L. Yan, Z. Sua and M. R. Bryce, Chem. Commun., 2014, 50, 6977–6980 RSC.
- D. G. Congrave, A. S. Batsanov and M. R. Bryce, Dalton Trans., 2018, 47, 16524–16533 RSC.
- J. Sturala, M. K. Etherington, A. N. Bismillah, H. F. Higginbotham, W. Trewby, J. A. Aguilar, E. H. C. Bromley, A.-J. Avestro, A. P. Monkman and P. R. McGonigal, J. Am. Chem. Soc., 2017, 139, 17882–17889 CrossRef CAS PubMed.
- B. Manimaran, P. Thanasekaran, T. Rajendran, R.-J. Lin, I.-J. Chang, G.-H. Lee, S.-M. Peng, S. Rajagopal and K.-L. Lu, Inorg. Chem., 2002, 41, 5323–5325 CrossRef CAS PubMed.
- A. Mukhopadhyay, S. Mahata and N. Goswami, J. Phys. Chem. Lett., 2024, 15, 8510–8519 CrossRef CAS PubMed.
- E. Q. Procopio, M. Mauro, M. Panigati, D. Donghi, P. Mercandelli, A. Sironi, G. D’Alfonso and L. D. Cola, J. Am. Chem. Soc., 2010, 132, 14397–14399 Search PubMed.
- P. Alam, C. Climent, P. Alemany and I. R. Laskar, J. Photochem. Photobiol., C, 2019, 41, 100317 CrossRef CAS.
- (a) A. A. Bhosle, M. Banerjee and A. Chatterjee, Sens. Diagn., 2024, 3, 745–782 RSC; (b) L. Li, X. Lyu, S. Liang and Z. Liu, Dyes Pigm., 2023, 220, 111651 CrossRef CAS.
- V. Sathish, A. Ramdass, Z.-Z. Lu, M. Velayudham, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Phys. Chem. B, 2013, 117, 14358–14366 CrossRef CAS PubMed.
- G. Li, W. Guan, S. Du, D. Zhu, G. Shan, X. Zhu, L. Yan, Z. Su, M. R. Bryce and A. P. Monkman, Chem. Commun., 2015, 51, 16924–16927 Search PubMed.
- (a) V. Sathish, A. Ramdass, P. Thanasekaran, K.-L. Lu and S. Rajagopal, J. Photochem. Photobiol., C, 2015, 23, 25–44 CrossRef CAS; (b) A. Fermi, P. Ceroni and I. R. Laskar, Dalton Trans., 2023, 52, 10637–10638 Search PubMed.
- Y. Jiang, G. Li, D. Zhu, Z. Su and M. R. Bryce, J. Mater. Chem. C, 2017, 5, 12189–12193 Search PubMed.
- A. Mukherjee, S. Bhattacharya and M. Chakravarty, Dalton Trans., 2021, 50, 9144–9157 RSC.
- M. Nurnabi, S. Gurusamy, J.-Y. Wu, C.-C. Lee, M. Sathiyendiran, S.-M. Huang, C.-H. Chang, I. Chao, G.-H. Lee, S.-M. Peng, V. Sathish, P. Thanasekaran and K.-L. Lu, Dalton Trans., 2023, 52, 1939–1949 RSC.
- (a) Q. Huang, N. Li, H. Zhang, C. Che, F. Sun, Y. Xiong, T. D. Canady and B. T. Cunningham, Lab Chip, 2020, 20, 2816–2840 RSC; (b) S. K. Sharma, N. Sehgal and A. Kumar, Curr. Appl. Phys., 2003, 3, 307–316 CrossRef.
- S. Adhikari, P. Nath, A. Das, A. Datta, N. Baildya, A. K. Duttaroy and S. Pathak, Biomed. Pharmacother., 2024, 171, 116211 CrossRef CAS PubMed.
- B. Das, S. T. Borah, S. Ganguli and P. Gupta, Chem. – Eur. J., 2020, 26, 14987–14995 CrossRef CAS PubMed.
- B. Das and P. Gupta, Dalton Trans., 2021, 50, 10225–10236 RSC.
- M. T. Gabr and F. C. Pigge, Molecules, 2019, 24, 4390 CrossRef CAS PubMed.
- (a) C. Zhu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Appl. Bio Mater., 2018, 1, 1768–1786 CrossRef CAS PubMed; (b) M. Maruthi and S. K. Kalangi, Prog. Mol. Biol. Transl. Sci., 2021, 184, 61–79 CAS.
- (a) N. A. Yusoh, M. R. Gill and X. Tian, Chem. Soc. Rev., 2025 10.1039/D4CS01193G; (b) B. Das and P. Gupta, Coord. Chem. Rev., 2025, 522, 216209 CrossRef CAS; (c) B. Das and S. Biswas, Dalton Trans., 2025, 54, 1731–1749 RSC.
- W. M. Leevy, J. R. Johnson, C. Lakshmi, J. Morris, M. Marquez and B. D. Smith, Chem. Commun., 2006, 1595–1597 RSC.
- X.-L. Xin, M. Chen, Y.-B. Ai, F.-L. Yang, X.-L. Li and F. Li, Inorg. Chem., 2014, 53, 2922–2931 CrossRef CAS PubMed.
- S. T. Borah, B. Das, P. Biswas, A. I. Mallick and P. Gupta, Dalton Trans., 2023, 52, 2282–2292 Search PubMed.
- (a) S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed; (b) Y. Wu, S. Li, Y. Chen, W. He and Z. Guo, Chem. Sci., 2022, 13, 5085–5106 RSC; (c) Y. Li, B. Liu, H. Shi, Y. Wang, Q. Sun and Q. Zhang, Dalton Trans., 2021, 50, 14498–14512 RSC; (d) B. Das, P. Biswas, A. I. Mallick and P. Gupta, Chem. – Eur. J., 2024, 30, e202400646 CrossRef CAS PubMed; (e) V. Vitali, S. Zineddu and L. Messori, RSC Adv., 2025, 15, 748–753 RSC.
- (a) B. Wu, Kenry and F. Hu, Interdiscip. Med., 2024, 2, e20230038 CrossRef; (b) C. He, P. Feng, M. Hao, Y. Tang, X. Wu, W. Cui, J. Ma and C. Ke, Adv. Funct. Mater., 2024, 34, 2402588 CrossRef CAS.
- M. Gao, Q. Hu, G. Feng, N. Tomczak, R. Liu, B. Xing, B. Z. Tang and B. Liu, Adv. Healthcare Mater., 2015, 4, 659–663 CrossRef CAS PubMed.
- G. Feng, C.-J. Zhang, X. Lu and B. Liu, ACS Omega, 2017, 2, 546–553 CrossRef CAS PubMed.
- (a) B. Liu, H. Zhou, L. Tan, K. T. H. Siu and X.-Y. Guan, Signal Transduction Targeted Ther., 2024, 9, 175 CrossRef PubMed; (b) L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 201 CrossRef PubMed; (c) A. Mondal, B. Das, S. Karmakar, S. Pani, S. Khan, P. Gupta and J. D. Sarma, J. Med. Chem., 2024, 67, 20559–20570 CrossRef CAS PubMed.
- G. Gupta, A. Das, N. B. Ghate, T. Kim, J. Y. Ryu, J. Lee, N. Mandal and C. Y. Lee, Chem. Commun., 2016, 52, 4274–4277 RSC.
- (a) Y. Yang, S. Jiang, S. G. Stanciu, H. Peng, A. Wu and F. Yang, Mater. Horiz., 2024, 11, 5815–5842 RSC; (b) M. Overchuk, R. A. Weersink, B. C. Wilson and G. Zheng, ACS Nano, 2023, 17, 7979–8003 CrossRef CAS PubMed; (c) L. Yu, Z. Liu, W. Xu, K. Jin, J. Liu, X. Zhu, Y. Zhang and Y. Wu, Acta Pharm. Sin. B, 2024, 14, 1111–1131 CrossRef CAS PubMed.
- (a) P. Szymaszek, M. Tyszka-Czochara and J. Ortyl, Eur. J. Med. Chem., 2024, 276, 116648 CrossRef CAS PubMed; (b) H. Huang, S. Banerjee and P. J. Sadler, ChemBioChem, 2018, 19, 1574–1589 CrossRef CAS PubMed.
- (a) L. C.-C. Lee and K. K.-W. Lo, Small Methods, 2024, 8, 2400563 CrossRef CAS PubMed; (b) G. Vigueras, G. Gasser and J. Ruiz, Dalton Trans., 2025, 54, 1320–1328 RSC; (c) B. Das, Small, 2024, 21, 2410338 CrossRef PubMed.
- L. Zhang, Y. Li, W. Che, D. Zhu, G. Li, Z. Xie, N. Song, S. Liu, B. Z. Tang, X. Liu, Z. Su and M. R. Bryce, Adv. Sci., 2019, 6, 1802050 CrossRef PubMed.
- L. Li, L. Zhang, X. Tong, Y. Li, Z. Yang, D. Zhu, Z. Su and Z. Xie, Dalton Trans., 2020, 49, 15332–15338 RSC.
- Z. Wang, L. Li, W. Wang, R. Wang, G. Li, H. Bian, D. Zhu and M. R. Bryce, Dalton Trans., 2023, 52, 1595–1601 RSC.
- B. Das, S. Gupta, A. Mondal, K. J. Kalita, A. I. Mallick and P. Gupta, J. Med. Chem., 2023, 66, 15550–15563 CrossRef CAS PubMed.
- Y. Jiang, G. Li, W. Che, Y. Liu, B. Xu, G. Shan, D. Zhu, Z. Su and M. R. Bryce, Chem. Commun., 2017, 53, 3022–3025 RSC.
- J. Xie, D. Li, Y. Duan, Y. Geng, T. Yang, G. Li, D. Zhu and Z. Su, Dyes Pigm., 2020, 172, 107855 CrossRef CAS.
- (a) O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed; (b) K. Zhou, B. Qi, Z. Liu, X. Wang, Y. Sun and L. Zhang, Adv. Funct. Mater., 2024, 34, 2411671 Search PubMed.
- (a) G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed; (b) S. S. Swayamprabha, D. K. Dubey, Shahnawaz, R. A. K. Yadav, M. R. Nagar, A. Sharma, F.-C. Tung and J.-H. Jou, Adv. Sci., 2021, 8, 2002254 Search PubMed; (c) S.-J. Zou, Y. Shen, F.-M. Xie, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Mater. Chem. Front., 2020, 4, 788–820 RSC.
- (a) Y. H. Jung, G. S. Lee, S. Muruganantham, H. R. Kim, J. H. Oh, J. H. Ham, S. B. Yadav, J. H. Lee, M. Y. Chae, Y.-H. Kim and J. H. Kwon, Nat. Commun., 2024, 15, 2977 CrossRef CAS PubMed; (b) S. A. Katkova, D. O. Kozina, K. S. Kisel, M. A. Sandzhieva, D. A. Tarvanen, S. V. Makarov, V. V. Porsev, S. P. Tunik and M. A. Kinzhalov, Dalton Trans., 2023, 52, 4595–4605 RSC; (c) G. Li, X. Zhao, T. Fleetham, Q. Chen, F. Zhan, J. Zheng, Y.-F. Yang, W. Lou, Y. Yang, K. Fang, Z. Shao, Q. Zhang and Y. She, Chem. Mater., 2020, 32, 537–548 CrossRef CAS.
- X. Wu, D.-G. Chen, D. Liu, S.-H. Liu, S.-W. Shen, C.-I. Wu, G. Xie, J. Zhou, Z.-X. Huang, C.-Y. Huang, S.-J. Su, W. Zhu and P.-T. Chou, J. Am. Chem. Soc., 2020, 142, 7469–7479 CrossRef CAS PubMed.
- (a) J. Ren, D. Zhang, X. Li, Z. Zhang, Y. Wu, J. Su, H. Zeng and B. Xu, ACS Mater. Lett., 2025, 7, 1260–1268 CrossRef CAS; (b) H. Xiang, R. Wang, J. Chen, F. Li and H. Zeng, Light: Sci. Appl., 2021, 10, 206 CrossRef CAS PubMed; (c) Y. Liu, F. Zhu, Y. Wang and D. Yan, Light: Sci. Appl., 2024, 13, 86 CrossRef CAS PubMed; (d) H. Dong, H. Jiang, J. Wang, Y. Guan, J. Hua, X. Gao, B. Bo and J. Wang, Org. Electron., 2018, 62, 524–529 CrossRef CAS.
- M. Cibian, A. Shahalizad, F. Souissi, J. Castro, J. G. Ferreira, D. Chartrand, J.-M. Nunzi and G. S. Hanan, Eur. J. Inorg. Chem., 2018, 4322–4330 CrossRef CAS.
- X. Chen, Y. Sun, X. Zhao, X. Deng, X. Yang, Y. Sun, G. Zhou and Z. Wu, Mater. Chem. Front., 2021, 5, 4160–4173 RSC.
- Y. He, G. Fu, W. Li, B. Wang, T. Miao, M. Tan, W. Feng and X. Lü, J. Lumin., 2020, 218, 116847 CrossRef CAS.
Footnote |
| † This work is dedicated to Prof. Amitava Das on his 65th birthday, in recognition of his exceptional contributions to the field of chemistry and his distinguished career upon his retirement from IISER Kolkata. |
| This journal is © The Royal Society of Chemistry 2025 |