
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrained spiro heterocycles as potential bioisosteres: an update on the synthesis of heteroatom-containing spiro[2.3]hexanes and spiro[3.3]heptanes
Philipp
Natho
 ,
Marco
Colella
and
Renzo
Luisi
*
,
Marco
Colella
and
Renzo
Luisi
*
Department of Pharmacy-Drug Sciences, University of Bari “A. Moro”, Via E. Orabona 4, 70125 Bari, Italy. E-mail: Renzo.Luisi@uniba.it
First published on 15th April 2025
Abstract
The interest of medicinal chemists in strained spiro heterocycles has continuously risen, given their potential as non-classical three-dimensional bioisosteres, as it has been shown that their inherent structural characteristics can impose beneficial physicochemical properties on lead compounds (e.g., metabolic stability, lipophilicity). In particular, strained spiro heterocycles containing at least one four-membered ring are in demand, as the inclusion of a small ring results in a more rigid and denser molecular space, whereas the inclusion of a heteroatom allows for placement of an exit vector orthogonal to the neighbouring carbon-centered exit vectors. The continuous development of new strained spiro heterocycles, their site-specific functionalisation, and their application as bioisostere is thus imperative. This review provides an overview of progress since 2014 with a particular focus on heteroatom-containing spiro[2.3]hexanes and spiro[3.3]heptanes.
 Philipp Natho | Philipp Natho has expertise in synthetic methodology development and natural product synthesis. Philipp received his PhD in 2021 from Imperial College London developing methodologies for the expansion and functionalisation of strained cyclic systems and applying this technology to natural product synthesis under the supervision of Prof. Phil Parsons. He also worked on the synthesis of highly substituted pyridines under the supervision of Prof. Rick Danheiser at the Massachusetts Institute of Technology. After a brief period working as a management consultant, Philipp is now a Marie Skłodowska-Curie Postdoctoral Research Fellow in the Renzo Luisi group at the Università degli Studi di Bari Aldo Moro. |
 Marco Colella | Marco Colella is researcher of Organic Chemistry at the University of Bari (Italy). He received his MSc (summa cum laude) in Chemistry and Pharmaceutical Technology at the University of Bari (Italy) in 2016. In 2020, he obtained the PhD in Chemical and Molecular Sciences under the supervision of Prof. Renzo Luisi. His research activity is focused on the use of flow microreactor technology applied to organometallic chemistry. In 2019, he was a visiting scholar in the group of Prof. Aiichiro Nagaki (Kyoto). In 2023, he was visiting researcher in the group of Prof. Timothy Noël (University of Amsterdam). He is the recipient of the 2020 CINMPIS award for the best PhD thesis. |
 Renzo Luisi | Renzo Luisi is full professor of Organic Chemistry at the University of Bari (Italy). The research activity focuses on the chemistry of hetero-substituted organolithiums, the development of new synthetic methodologies, and the use of flow technology. He obtained the PhD in 2000 under the guidance of Professor Saverio Florio. He has been visiting student at the Roger Adams Lab at Urbana Champaign in the group of Prof. Peter Beak and a visiting professor at the University of Manchester in the group of Jonathan Clayden. He is a RSC fellow and recipient of the 2014 CINMPIS award Innovation in Organic Synthesis, and 2022 award of the Italian Chemical Society for the Development of Synthetic Methodologies. |
1. Introduction
Since the introduction of scaffold hopping,1 escape from flatland,2,3 and conformational restriction concepts,4 synthetic chemists have been on a quest to replace classic aromatic building blocks with novel three-dimensional and sp3-rich alternatives in medicinal drug discovery programs (Fig. 1(A)). To this end, spirocyclic compounds have, over the last two decades, gained particular popularity in medicinal chemistry programs, as their potential for biological activity has been proven by numerous spirocycle-containing natural products so their inclusion into lead compounds is imperative.5,6 Their inherent structural characteristics (high sp3-fraction, three-dimensionality, good balance between rigidity and flexibility) meet the requirements of modern drug design concepts.7 In addition, their well-defined exit vectors allow the precise positioning of substituents in three-dimensional space (cf. two-dimensional space for flat moieties) which often leads to improved physicochemical properties of lead compounds, including reduced lipophilicity, improved metabolic stability, or enhanced target selectivity.7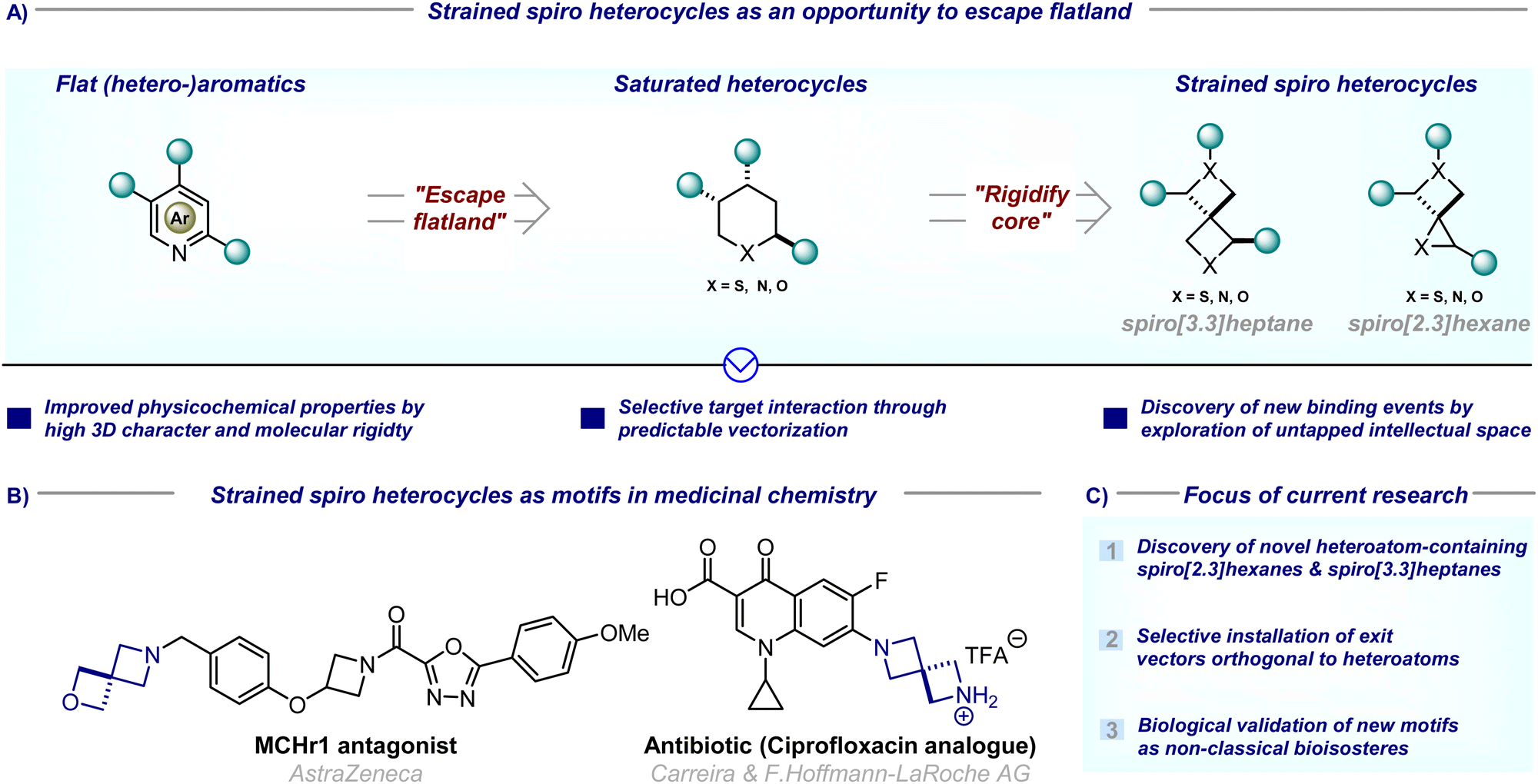 | ||
| Fig. 1 (A) Strained spiro heterocycles as an opportunity to escape flatland; (B) strained spiro heterocycles as motifs in medicinal chemistry; (C) focus of current research. | ||
Within the broad area of spirocycles, those bearing a heteroatom and at least one four-membered ring are particularly attractive for medicinal discovery campaigns, as the inclusion of a small ring results in a more rigid and denser molecular space, whereas the inclusion of a heteroatom allows for relative facile functionalisation/placement of an exit vector orthogonal to the neighbouring carbon-centered exit vectors.7 In addition, this novel chemical space is relatively underexplored, allowing the development of novel lead compounds without overlapping with existing patents, thus relieving the challenge of ever-narrowing intellectual property space in drug development.
Spiro[3.3]heptanes containing at least one ring heteroatom have received special attention due to their proven track record as non-classical bioisosteres of saturated heteroatom-containing six-membered rings (Fig. 1(B)). For example, piperidine, piperazine, and oxane are the respectively third, fourth, and seventh-most frequently used ring systems in small molecule drugs so suitable bioisosteric replacement is highly sought after.8 Given promising initial results,9–11 significant effort has been devoted to the discovery of new spirocycles, and their application as bioisosteres. In addition, methodologies that allow the installation of exit vectors at positions orthogonal to heteroatoms were particularly sought-after as these are synthetically more challenging compared to the installation of exit vectors off heteroatoms (Fig. 1(C)).
In contrast, heteroatom-containing spiro[2.3]hexanes – the lower homologues incorporating one three-membered ring – have hitherto received comparatively less attention. This is surprising given that they often exhibit similar physicochemical properties, and have the added advantage of populating different, less widely intellectually claimed chemical space.12 Particularly the incorporation of a cyclopropyl ring – the sixth most common ring in small molecule drugs8 – can enhance biological target binding due to the decreased conformational flexibility, and increase three-dimensional vector space.13 In addition, the high ring-strain-induced bond strength renders it stable to oxidative metabolism. The exploration of this chemical space as potential bioisosteres is hence imperative (Fig. 1(C)).
This review thus provides an overview of major advances from the last decade (since 2014) in the synthesis of known heteroatom-containing spiro[2.3]hexanes and spiro[3.3]heptanes, and discusses new spirocycles which have been reported for the first time. Where possible, their use as bioisosteres is highlighted. Given the brevity of this highlight, reports on all-carbon spirocycles are not included, as the use of these motifs as non-classical benzene bioisosteres is discussed in depth elsewhere.14–16
2. Discussion
2.1. Spiro[3.3]heptanes
 | ||
| Scheme 1 (A) Molecular structure of 2,6-diazaspiro[3.3]heptane; (B) application as piperazine bioisostere; (C) synthetic routes towards 2,6-diazaspiro[3.3]heptane. | ||
Although this presents a viable route towards unsubstituted 2,6-diazaspiro[3.3]heptane, the use of Tebbe reagent precludes (asymmetric) functionalisation on the constructed azetidine ring in the same transformation. However, the asymmetric synthesis of substituted 2,6-diazaspiro[3.3]heptanes is desirable for introducing an exit vector orthogonal to the heteroatom substituent. With this objective in mind, Reddy and co-workers reported an asymmetric synthesis of 1-substituted 2,6-diazaspiro[3.3]heptanes in a three-step one-pot approach starting from methyl 1-Boc-azetidine-3-carboxylate 5 (Scheme 1(C)-ii).19 Enolate formation by LiHMDS and addition to (hetero-)aromatic and aliphatic Davis–Ellman imines 6 afforded sulfinamide 7. A subsequent reduction/protection/intramolecular substitution sequence afforded the desired spirocycles in up to 85% yield and generally good diastereoselectivity (>9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr), even on a multigram scale.
:1 dr), even on a multigram scale.
The configurational arrangement of the nitrogen atoms in 2,6-diazospiro[3.3]heptane allows an interesting spirocyclic bis-β-lactam to be conceived. Indeed, Dar’in and co-workers recently reported an attractive cascade to this new sub-class of 2,6-diazospiro[3.3]heptane (Scheme 1(C)-iii).20 Starting from 3-diazotetramic acids 8, a microwave-assisted Wolff rearrangement produces the ring-contracted ketene intermediate 9, which undergoes a Staudinger-type [2+2]-cycloaddition with imines to the desired spirocycle. Notably, the cycloaddition step was found to be highly diastereoselective in most cases (>95:5 dr) for the cis-products, although the presence of an electron-withdrawing substituent in the imine structure can lead to a decrease in the diastereoselectivity. A series of potentially medicinally useful and synthetically interesting strained and highly substituted bis-spirocyclic motifs are thus accessible in up to 95% yield.
 | ||
| Scheme 2 (A) Molecular structure of 2-oxa-6-azaspiro[3.3]heptane; (B) application as morpholine bioisostere; (C) scalable route towards 2-oxa-6-azaspiro[3.3]heptane. | ||
 | ||
| Scheme 3 (A) Molecular structure of 2-azaspiro[3.3]heptane; (B) application as piperidine bioisostere; (C) synthetic routes towards 2-azaspiro[3.3]heptane; (D) synthetic application of 2-azaspiro[3.3]heptane. | ||
Building on this work, an asymmetric variant was reported by Reddy and co-workers (Scheme 3(C)-ii).25 To this end, ethyl cyclobutanecarboxylate 20 was deprotonated with LiHMDS, and the produced anion was quenched with a Davis–Ellman's imine 6. Subsequent ester reduction, N-tosyl protection, and intramolecular substitution afford the spirocyclic azetidine with high diastereoselectivity (>98:2 dr) and yield, independent of the electronic nature of the aromatic substituent. Further analogues can be prepared from known 2-azaspiro[3.3]heptane-1-carboxylic acid,26 by a Minisci-type photoredox-mediated α-heteroarylation of N-Boc protected 2-azaspiro[3.3]heptane,27 or a nickel-catalyzed regio- and enantioselective syn-hydrometalative 4-exo-trig cyclization of alkynones.28
Elaboration on the 2-azaspiro[3.3]heptane scaffold in the last decade focused on the modular diversification of the 5- and 6-position (Scheme 3(C)-iii).29,30 To this end, three conceptually related approaches were reported by Brown31 and Tortosa,32,33 leveraging the functionalisation of the alkene moiety in a cyclobutene precursor 21. This allowed for the stereoselective diboration, Cu-/Pd-co-catalysed arylboration, or Cu-catalysed regioselective monoborylation. Notably, the newly installed BPin-motifs served as valuable synthetic handles for further structural diversification, including Suzuki–Miyaura cross-coupling, oxidation, amination, or allyl-group installation. For example, Matteson homologation of the Bpin-group, followed by double oxidation afforded the formylated 2-azaspiro[3.3]heptane derivative (Scheme 3(D)). A further five-step sequence consisting of Aldol reaction, dehydration, hydrogenation, and N-protecting group interconversion afforded the homospiro-derivative of FDA-approved Alzheimer drug Donepezil; showcasing the synthetic utility of this motif for bioisostere incorporation.
:1 dr). Particularly the borylated derivatives provided a versatile platform for further synthetic derivatisation (Scheme 4(D)). For example, oxidation of the Bpin-substitutent with sodium perborate affords the cyclobutanol motif, present in TDO2 inhibitor shown in Scheme 4(B) in 81% yield. Alternatively, the Bpin-analogue can be converted into the corresponding BF3K-salt by treatment with potassium hydrogen difluoride, which can be cross-coupled under nickel-/photoredox catalysis conditions to a pydridyl bromide to afford a drug-resembling derivative. Notably, the 2-oxaspiro[3.3]heptane is tolerant to these conditions.
 | ||
| Scheme 4 (A) Molecular structure of 2-oxaspiro[3.3]heptane; (B) incorporation in pharmaceutical candidates; (C) synthetic routes towards 2-oxaspiro[3.3]heptane; (D) synthetic application of 2-oxaspiro[3.3]heptanes. | ||
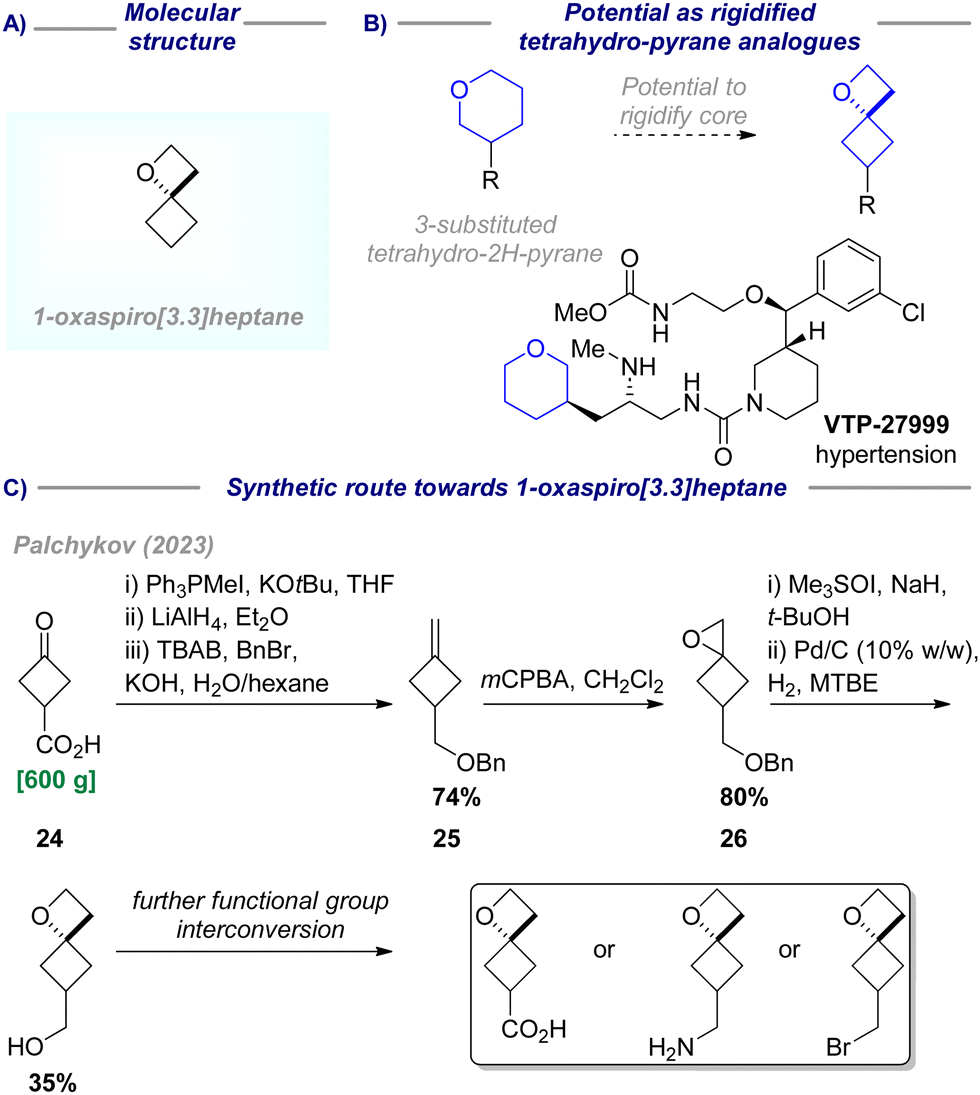 | ||
| Scheme 5 (A) Molecular structure of 1-oxaspiro[3.3]heptane; (B) potential as tetrahydro-2H-pyrane analogue; (C) synthetic route towards 1-oxaspiro[3.3]heptane. | ||
 | ||
| Scheme 6 (A) Molecular structure of 1-azaspiro[3.3]heptane; (B) potential as piperidine analogue; (C) synthetic routes towards 1-azaspiro[3.3]heptane. | ||
Also, the latter concept commencing from the intact azetidine ring to construct the cyclobutyl moiety has been reported (Scheme 6(C)-ii).40 In this case, thermal [2+2]-cycloaddition between nitroalkenes 31 and olefin 30, prepared by a straight-forward three-step aldol condensation protocol from azetidinone, provided the desired spirocycles promoted by Schreiner's thiourea catalyst. Good diastereoselectivity is commonly observed for these highly substituted spiro-compounds, and nitro-alkenes substituted with aliphatic, as well as (hetero-)aromatic substituents are suitable reaction partners. Lower yields tend to be observed when the exocyclic alkene is deactivated by substitution with electron-deficient aromatic rings.
 | ||
| Scheme 7 (A) Molecular structure; (B) synthetic route. | ||
 | ||
| Scheme 8 (A) Molecular structure of 1,2-diazaspiro[3.3]heptane; (B) potential application as rigidified hexahydropyridazine analogue; (C) synthetic route towards 1,2-diazaspiro[3.3]heptane. | ||
 | ||
| Scheme 9 (A) Molecular structure of 1-oxa-2,6-diazaspiro[3.3]heptane; (B) potential application as rigidified piperazine analogue; (C) synthetic route towards 1-oxa-2,6-diazaspiro[3.3]heptane; (D) synthetic application of 1-oxa-2,6-diazaspiro[3.3]heptane. | ||
2.2. Spiro[2.3]hexanes
 | ||
| Scheme 10 (A) Molecular structure of 1-oxaspiro[2.3]hexane; (B) synthetic application of 1-oxaspiro[2.3]hexane; (C) synthetic route towards 1-oxaspiro[2.3]hexane; (D) synthetic application of 1-oxaspiro[2.3]hexane. | ||
With regards to enantioselective synthesis, Secci and Luisi reported a sequential nucleophilic addition/Payne rearrangement between cyclobutanones 40 and chiral oxiranes 41, leveraging the predictable face-stereoselectivity upon the addition of sterically demanding nucleophiles to C3-substituted cyclobutanones (Scheme 10(C)-i).47 Readily available chiral oxiranes 41 were selectively α-lithiated at the more stable benzylic position using sec-BuLi and TMEDA at −98 °C, and subsequently added to C3-substituted cyclobutanones 40 to afford enantioenriched cyclobutanol intermediates 42 (er > 98:2) in good to excellent yields (72–94%). A subsequent base-initiated stereospecific Payne rearrangement afforded the desired 1-oxaspiro[2.3]hexanes in up to 98% yield without depreciation of the enantiomeric ratio. A chiral phosphoric-acid catalysed enantioselective rearrangement on alkenylcyclobutanols involving the opening of a bromonium ion was reported by Merino.48
The oxirane motif can also be obtained by strain-release of the “spring-loaded” bridge-head bond on bicyclo[1.1.0]butanes (Scheme 10(C)-ii).49 The required carbinol 45 can be obtained by lithiation of bicyclo[1.1.0]butyl sulfoxide 43 with tert-BuLi at −95 °C, followed by addition to ketones or aldehydes 44. In the same pot, a palladium-catalysed cross-coupling with aryl triflates activates the bridge-head bond for strain-release driven opening by the carbinol 45, concomitantly forming the oxirane, as well as the new carbon–carbon bond in a syn-periplanar arrangement. A wide range of aliphatic and aromatic ketones and aldehydes, as well as aryl-triflates can undergo the desired reaction two-step approach. Although the yield is dependent on the steric and electronic properties of the carbonyl and aryl triflate starting materials (31–87%), excellent diastereoselectivity (typically >98:2 dr) is observed in all cases.
The inherent ring strain of 1-oxaspiro[2.3]hexanes can be exploited for further synthetic derivatisation (Scheme 10(D)). Luisi and co-workers showed that a Lewis acid-mediated ring expansion of their 1-oxaspiro[2.3]hexanes readily affords cyclopentanones in excellent yield and good diastereomeric ratio. The thus obtained substitution pattern resembles closely a known histone deacetylase inhibitor. In contrast, Aggarwal and co-workers exploited the ring-strain of the tri-substituted epoxide towards ring-opening functionalisation (e.g., pyrazole) to afford cyclobutanols with a substitution pattern that would be challenging to obtain from the classic approach of nucleophilic addition to cyclobutanones.
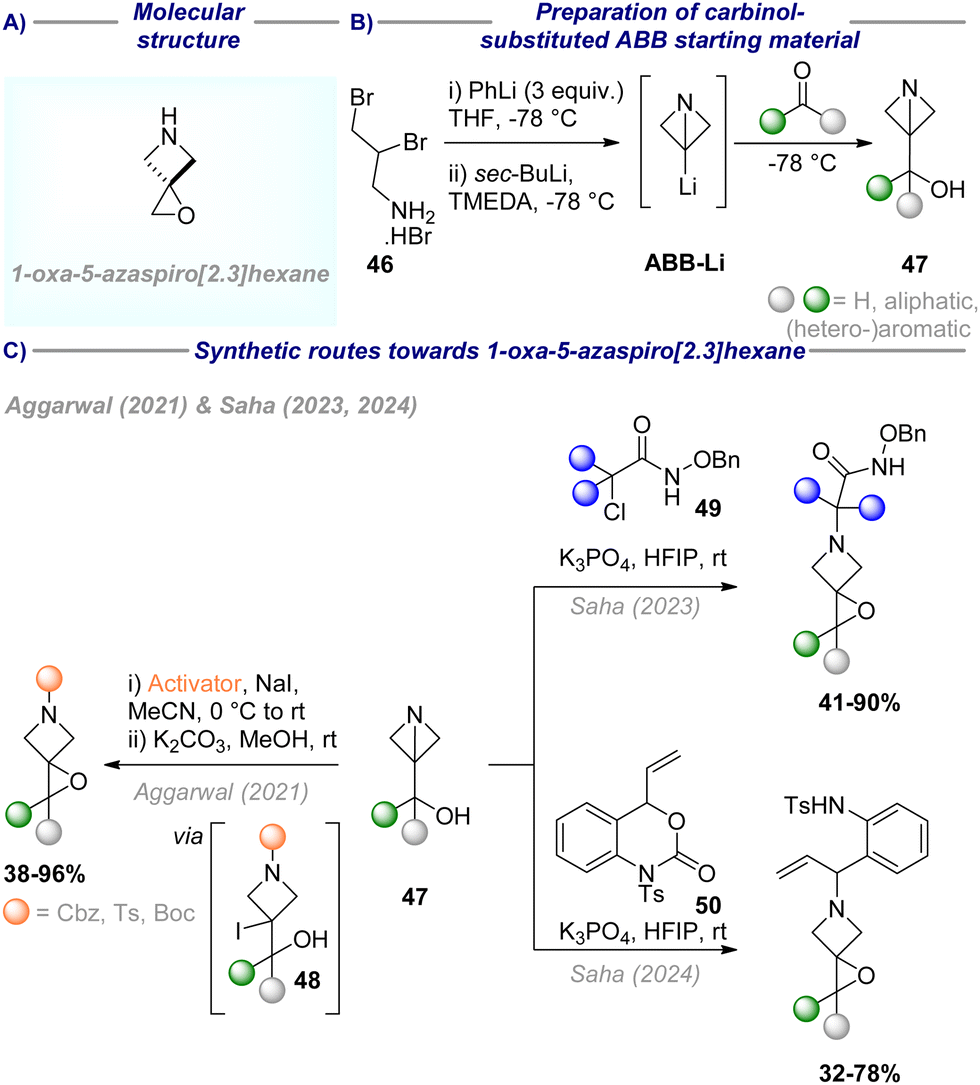 | ||
| Scheme 11 (A) Molecular structure of 1-oxa-5-azaspiro[2.3]hexane; (B) preparation of carbinol-substituted ABB starting material; (C) synthetic routes towards 1-oxa-5-azaspiro[2.3]hexane. | ||
Inspired by this reactivity, Saha sought to further extend the diversity-oriented nature of this approach, by introduction of N-functionalisation beyond classical protecting groups (Scheme 11(C)). Thus, building on this approach, they found that the spiroepoxidation/N-functionalisation sequence could be triggered effectively by (aza)oxyallyl cations, generated in situ from α-halohydroxamate 49.51 Notably, and in contrast to Aggarwal's report, this eliminates the requirement for prior activation of the ABB-core by the formation of an iodohydrin. Various sterically challenging (aza)oxyallyl cations successfully triggered the formation of the desired epoxide in 41–90% yield.
In a further extension of this work, the same authors discovered that also aza-ortho-quinone methide, in situ generated from vinyl benzoxazinanone 50 by HFIP and K3PO4, could serve as an efficient N-activator for the desired sequence.52 This enables the introduction of an alkene moiety, suitable for further synthetic derivatisation.
 | ||
| Scheme 12 (A) Molecular structure of 1,5-dioxaspiro[2.3]hexane; (B) synthetic route towards 1,5-dioxaspiro[2.3]hexane; (C) mechanistic proposal; (D) synthetic application of 1,5-dioxaspiro[2.3]hexanes. | ||
:2 dr).
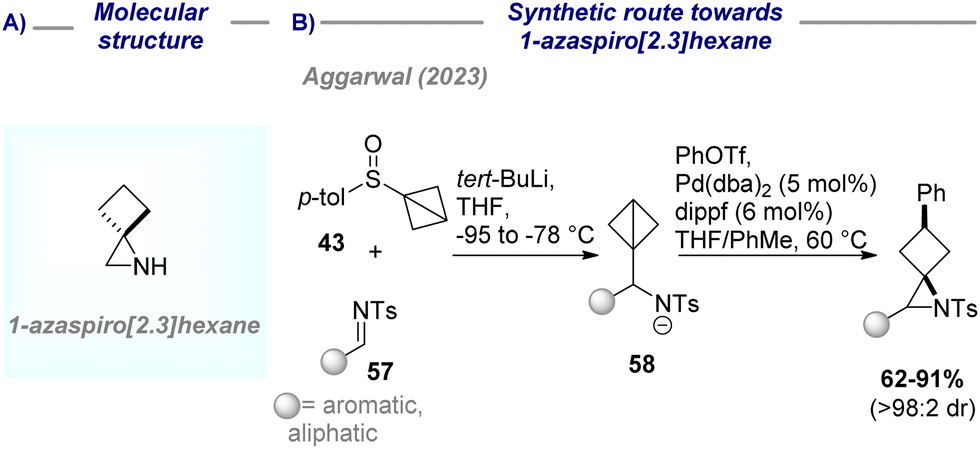 | ||
| Scheme 13 (A) Molecular structure of 1-azaspiro[2.3]hexane; (B) synthetic route towards 1-azaspiro[2.3]hexane. | ||
 | ||
| Scheme 14 (A) Molecular structure of 5-azaspiro[2.3]hexane; (B) potential as bioisostere and medicinal importance; (C) synthetic routes towards 5-azaspiro[2.3]hexanes. | ||
Given the demand for motifs containing exit vectors orthogonal to the heteroatom, the synthesis of conformationally frozen N-Boc protected amino acid 61 was reported by Grygorenko and co-workers, and further derivatised to the monoprotected diamine analogues – promising leads in the design of new saturated ring bioisosteres and peptidomimetics (Scheme 14(C)-ii).57 Beginning from commercially available N-Boc-protected azetidinone 1 the acid-bearing cyclopropyl moiety in spirocycle 61 was introduced by a three-step sequence consisting of Horner–Wadsworth–Emmons olefination (forming ester 60), followed by a low-yielding Corey–Chaikovsky cyclopropanation, and hydrolysis. Albeit low-yielding, the method was found to be scalable, affording the 5-azaspiro[2.3]hexane 61 on a 10 g scale. The amino acid 61 could be converted into the monoprotected diamine 62 by a modified Curtius rearrangement and acid-catalysed Boc-deprotection. In a related work, di Fabio and co-workers reported the synthesis of conformationally frozen analogues of L-glutamic acid.58
Given the importance of the β-lactam scaffold as a privileged structure in drug discovery, also spirocyclic β-lactams have received great interest in the medicinal chemistry community. Thus, also the synthesis of β-lactam-containing 5-azaspiro[2.3]hexanes has received particular attention in the last decade (Scheme 14(C)-iii). To this end, Ma reported that the desired spirocyclic β-lactam could be obtained by carbene addition onto 3-methylene-1,4-diphenylazetidin-2-one 63.59 Specifically, this [2+1]-cycloaddition between azetidine-2-one and unstable 2,2,2-trifluorodiazoethane in DMF catalysed by FeTPPCl (TPP = 5,10,15,20-tetraphenyl-21H,23H-porphine), which promoted the reaction in up to 89% yield and ca. 8:2 dr.
Although synthetically viable, the requirement of a relatively high catalyst loading (10 mol%), and a four-fold excess of unstable 2,2,2-trifluorodiazoethane posing a safety risk, mandate the necessity for more scalable, safer and environmentally friendlier procedures for the synthesis of β-lactam-containing 5-azaspiro[2.3]hexanes. Addressing these concerns, Xiao reported a metal-free synthesis of the desired β-lactam-containing spirocycles from the same azetidine-2-one starting material 63 and ketoesters catalysed by tris(dimethylamino)phosphine at room temperature (Scheme 14(C)-iii).60 These mild conditions enabled the synthesis of various ester-substituted cyclopropyl-containing spirocycles in 76–86% yield and some preference for the trans-diastereomer. Distinct from a classic carbene addition, the mechanism involves the initial formation of the Kukhtin–Ramirez intermediate, formed by oxophilic addition of tris(dimethylamino)phosphine to the ketoesters. This enolate undergoes Michael addition to the α-methylene-β-lactam, followed by 3-exo-tet ring-closure to afford preferably the trans-spirocyclopropyl β-lactam product to minimize steric hindrance.
Bearing in mind the importance of chirality in drug design, an enantioselective variant for the synthesis of β-lactam-containing 5-azaspiro[2.3]hexanes was recently reported by Zhang (Scheme 14-iv).61 In contrast to previously discussed methods, the key step did not involve the (enantioselective) construction of the cyclopropyl ring, but instead a desymmetrization of highly strained spirocyclopropenes 64. These can be prepared by the addition of dibromocarbene to an α-methylene-β-lactam, followed by an elimination/debromination sequence.62 The enantioselective hydroboration was then achieved by treatment with a copper catalyst (10 mol%), and (R)-DTBM-Segphos, a bulky organophosphorus ligand, affording the β-lactam-containing 5-azaspiro[2.3]hexanes in moderate to good yields (50–78%) and high diastereoselectivity (>20:1 dr) and good enantioselectivity (>90:10 er). Importantly, installation of the BPin-handle allowed for further enantiospecific transformation suitable for late-stage structural diversification, accessing for example the corresponding hydroxy-, allyl-, or phenyl group.
 | ||
| Scheme 15 (A) Molecular structure of 4-azaspiro[2.3]hexane; (B) potential application as rigidified piperidine analogue; (C) synthetic routes towards 4-azaspiro[2.3]hexane. | ||
Building on this work, Grygorenko and co-workers showed that from the related N-Boc protected enamine 67, difluorinated and ester-substituted cyclopropanes are accessible on a decagram scale by treatment with Ruppert–Prakash reagent (TMSCF3)/Petasis reagent or ethyl diazoacetate/Cu(acac)2, respectively (Scheme 15(C)-ii).63 Notably, the ethyl ester moiety serves as a handle for the installation of other functional groups, e.g., a free amine group.
 | ||
| Scheme 16 (A) Molecular structure of 4,5-diazaspiro[2.3]hexane; (B) synthetic route towards 4,5-diazaspiro[2.3]hexane; (C) synthetic application of 4,5-diazaspiro[2.3]hexanes. | ||
2.3. Further expected developments and future outlook
Spiro-heterocycles as three-dimensional scaffolds have shown promising results in scaffold-hopping and replacement of flat or non-strained cycles in drug design, due to their well-defined exit vectors, strain-induced reduced entropy and tuneable geometric properties. A number of recently FDA-approved drugs already incorporate well-studied (larger) strained spiro-heterocycles (e.g., revumenib approved by FDA in 2024 as menin-KMT2A PPI inhibitor). Given the promising initial results of recently introduced spiro[2.3]hexane and spiro[3.3]heptane analogues discussed in this highlight, and their prevalence in the patent literature, we expect further developments in this area to transfer these preliminary results into clinical trials and approved drugs.To maximise the utility of heteroatom-containing spiro[2.3]hexanes and spiro[3.3]heptanes in drug discovery, we foresee that several developments are necessary in this field. First, to navigate the crowded intellectual property space, the development of novel spiro[2.3]hexane and spiro[3.3]heptane motifs is imperative. We expect that a particular focus will be placed on motifs incorporating more than two heteroatoms as these are currently relatively underexplored, and provide an opportunity to modulate the geometric constraints, modify exit vector placement, and functionalise with further groups (e.g., on nitrogen). Furthermore, we anticipate that spiro[2.3]hexane and spiro[3.3]heptane analogues with a further reduction in the degree of freedom (e.g., incorporation of unsaturation) will be investigated as these not only further reduce entropy but provide a platform for functionalisation that are precluded for fully saturated analogues. In addition, the development of further enantioselective protocols will be beneficial for drug discovery programs.
From a practical perspective, we anticipate further progress in improving scalability and convergence of protocols to sustain the large quantities required for pre-clinical and clinical trials. We expect that the use of enabling technologies (e.g., flow technology) will support the requirement for more scalable protocols. This will go hand-in-hand with increased commercialisation of such scaffolds for late-stage functionalisation which should catalyse their exploitation and incorporation in compound libraries and drug development campaigns.
Last, the current lack of confidence in which strained spiro heterocycle is suitable for the replacement of classic heterocycles in typical MedChem libraries could hinder its utilization in drug discovery programs. Whereas some bioisosteric replacement is well-established (e.g., oxetane as a bioisostere for dimethyl- or carbonyl-groups), the knowledge base of physico-chemical properties of known and novel spiro[2.3]hexanes and spiro[3.3]heptanes needs to be expanded. This would provide a high-confidence guide, which classic two-dimensional or non-strained three-dimensional heterocycle they could replace in initial lead compounds and accelerate their inclusion in building compound libraries around lead compounds. We expect that in silico-driven clustering approaches could proof useful in accelerating these investigations to compare the molecular properties (e.g., physico-chemical properties, drug-likeness, shape-related indexes etc.) of novel motifs with those of classic heterocycles in typical MedChem libraries.
3. Conclusions
In the last decade, significant progress has been made to tackle the challenges laid out. Several new spiro[2.3]hexane (e.g., 4,5-diazaspiro[2.3]hexane) and spiro[3.3]heptane (e.g., 1-oxa-2,6-diazaspiro[3.3]heptane) motifs were synthesised for the first time. In addition, their applicability as non-classical bioisosteres for saturated heterocycles was investigated, with several cores showing promising initial results for further utilisation (e.g., 4-azaspiro[2.3]hexane as a piperidine bioisostere). Last, significant effort has been devoted to enabling the introduction of orthogonal exit vectors with respect to the heteroatom at various carbon centres along the spiro[2.3]hexane or spiro[3.3]heptane core, which should facilitate selective target interaction for medicinal application by targeted substituent positioning in three-dimensional space. We have also laid out our expected developments for the near future to aid the utility of these scaffolds in drug discovery.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Università degli Studi di Bari Aldo Moro. P. N. acknowledges funding from the European Commission's Horizon Europe research and innovation programme through a Marie Skłodowska-Curie Postdoctoral Fellowship “ExpandFlow” (grant agreement no. 101106497). M. C. and R. L. acknowledge financial support from the European Commission–Horizon Europe Framework, project SusPharma (grant agreement No. 101057430).Notes and references
- Y. Hu, D. Stumpfe and J. Bajorath, J. Med. Chem., 2017, 60, 1238–1246 CrossRef CAS PubMed.
- F. Lovering, MedChemComm, 2013, 4, 515 RSC.
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed.
- P. de Sena, M. Pinheiro, D. A. Rodrigues, R. do Couto Maia, S. Thota and C. A. M. Fraga, Curr. Top. Med. Chem., 2019, 19, 1712–1733 CrossRef PubMed.
- E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Molecules, 2019, 24, 4165 CrossRef CAS PubMed.
- Y. Zheng, C. M. Tice and S. B. Singh, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed.
- E. M. Carreira and T. C. Fessard, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed.
- J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- M. Golden, D. Legg, D. Milne, A. Bharadwaj M., K. Deepthi, M. Gopal, N. Dokka, S. Nambiar, P. Ramachandra, U. Santhosh, P. Sharma, R. Sridharan, M. Sulur, M. Linderberg, A. Nilsson, R. Sohlberg, J. Kremers, S. Oliver and D. Patra, Org. Process Res. Dev., 2016, 20, 675–682 CrossRef CAS.
- K. Ploj, L. Benthem, D. Kakol-Palm, P. Gennemark, L. Andersson, M. Bjursell, J. Börjesson, L. Kärrberg, M. Månsson, M. Antonsson, A. Johansson, S. Iverson, B. Carlsson, A. Turnbull and D. Lindén, Br. J. Pharmacol., 2016, 173, 2739–2751 CrossRef CAS PubMed.
- J. A. Burkhard, B. Wagner, H. Fischer, F. Schuler, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 3524–3527 CrossRef CAS PubMed.
- A. Malashchuk, A. V. Chernykh, M. Y. Perebyinis, I. V. Komarov and O. O. Grygorenko, Eur. J. Org. Chem., 2021, 6570–6579 CrossRef CAS.
- T. T. Talele, J. Med. Chem., 2016, 59, 8712–8756 CrossRef CAS PubMed.
- P. K. Mykhailiuk, Org. Biomol. Chem., 2019, 17, 2839–2849 RSC.
- J. Tsien, C. Hu, R. R. Merchant and T. Qin, Nat. Rev. Chem., 2024, 8, 605–627 CrossRef CAS PubMed.
- K. Prysiazhniuk, O. P. Datsenko, O. Polishchuk, S. Shulha, O. Shablykin, Y. Nikandrova, K. Horbatok, I. Bodenchuk, P. Borysko, D. Shepilov, I. Pishel, V. Kubyshkin and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2024, 63, e202316557 CrossRef CAS PubMed.
- S. W. Reilly, L. N. Puentes, K. Wilson, C.-J. Hsieh, C.-C. Weng, M. Makvandi and R. H. Mach, J. Med. Chem., 2018, 61, 5367–5379 CrossRef CAS PubMed.
- T. D. Weinhold, J. A. Law and J. H. Frederich, Adv. Synth. Catal., 2024, 366, 2214–2219 CrossRef CAS.
- L. R. Reddy, Y. Waman, K. Nayak, K. Baharooni and S. Kotturi, Org. Lett., 2019, 21, 3481–3484 CrossRef CAS PubMed.
- V. Krivovicheva, I. Lyutin, G. Kantin and D. Dar’in, J. Org. Chem., 2024, 89, 3585–3589 CrossRef CAS PubMed.
- G. Wuitschik, M. Rogers-Evans, A. Buckl, M. Bernasconi, M. Märki, T. Godel, H. Fischer, B. Wagner, I. Parrilla, F. Schuler, J. Schneider, A. Alker, W. B. Schweizer, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4512–4515 CrossRef CAS PubMed.
- R. van der Haas, J. Dekker, J. Hassfeld, A. Hager, P. Fey, P. Rubenbauer and E. Damen, Synthesis, 2017, 2394–2401 CAS.
- J. A. Burkhard, C. Guérot, H. Knust, M. Rogers-Evans and E. M. Carreira, Org. Lett., 2010, 12, 1944–1947 CrossRef CAS PubMed.
- A. A. Kirichok, I. Shton, M. Kliachyna, I. Pishel and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2017, 56, 8865–8869 CrossRef CAS PubMed.
- L. R. Reddy, Y. Waman, P. Kallure, K. S. Nalivela, Z. Begum, T. Divya and S. Kotturi, Chem. Commun., 2019, 55, 5068–5070 RSC.
- A. Kirichok and T. Yegorova, Fr.-Ukr. J. Chem., 2023, 11, 31–38 CrossRef CAS.
- C. Bosset, H. Beucher, G. Bretel, E. Pasquier, L. Queguiner, C. Henry, A. Vos, J. P. Edwards, L. Meerpoel and D. Berthelot, Org. Lett., 2018, 20, 6003–6006 CrossRef CAS PubMed.
- X. Li, J. Deng, J. Long, Y. Fu, Y. Zheng and W. Liu, Angew. Chem., Int. Ed., 2025, 64, e202415164 CrossRef CAS PubMed.
- A. Chernykh, A. Tkachenko, I. Feskov, C. Daniliuc, N. Tolmachova, D. Volochnyuk and D. Radchenko, Synlett, 2016, 1824–1827 CAS.
- O. O. Grygorenko and K. P. Melnykov, Chem. Heterocycl. Compd., 2019, 55, 692–694 CrossRef CAS.
- A. K. Simlandy, M.-Y. Lyu and M. K. Brown, ACS Catal., 2021, 11, 12815–12820 CrossRef CAS PubMed.
- L. Nóvoa, L. Trulli, A. Parra and M. Tortosa, Angew. Chem., Int. Ed., 2021, 60, 11763–11768 CrossRef PubMed.
- L. Nóvoa, L. Trulli, I. Fernández, A. Parra and M. Tortosa, Org. Lett., 2021, 23, 7434–7438 CrossRef PubMed.
- X. Lin, P. Yuen, R. Mendonca, B. Parr, R. Pastor, Z. Pei, L. Gazzard, F. Jaipuri, S. Kumar and X. Li, et al., Preparation of Imidazoisoindole Compounds as TDO2 Inhibitors, US2019016726, 2019 Search PubMed.
- F. Wu, Preparation of C-Glycoside Derivatives as Sodium Glucose Linked Co-Transporter (SGLT) Inhibitors for Treatment of Diabetes, US2014128331, 2014 Search PubMed.
- P. R. D. Murray, W. M. M. Bussink, G. H. M. Davies, F. W. van der Mei, A. H. Antropow, J. T. Edwards, L. A. D’Agostino, J. M. Ellis, L. G. Hamann, F. Romanov-Michailidis and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 4055–4063 CrossRef CAS PubMed.
- L. Jia, R. D. Simpson, J. Yuan, Z. Xu, W. Zhao, S. Cacatian, C. M. Tice, J. Guo, A. Ishchenko, S. B. Singh, Z. Wu, B. M. McKeever, Y. Bukhtiyarov, J. A. Johnson, C. P. Doe, R. K. Harrison, G. M. McGeehan, L. W. Dillard, J. J. Baldwin and D. A. Claremon, ACS Med. Chem. Lett., 2011, 2, 747–751 CrossRef CAS PubMed.
- Y. K. Kozyriev and V. A. Palchykov, Tetrahedron Lett., 2023, 122, 154515 CrossRef CAS.
- A. A. Kirichok, H. Tkachuk, Y. Kozyriev, O. Shablykin, O. Datsenko, D. Granat, T. Yegorova, Y. P. Bas, V. Semirenko, I. Pishel, V. Kubyshkin, D. Lesyk, O. Klymenko-Ulianov and P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2023, 62, e202311583 CrossRef CAS PubMed.
- Z. Dobi, T. Holczbauer and T. Soós, Eur. J. Org. Chem., 2017, 1391–1395 CrossRef CAS.
- M. L. G. Borst, C. M. J. Ouairy, S. C. Fokkema, A. Cecchi, J. M. C. A. Kerckhoffs, V. L. de Boer, P. J. van den Boogaard, R. F. Bus, R. Ebens, R. van der Hulst, J. Knol, R. Libbers, Z. M. Lion, B. W. Settels, E. de Wever, K. A. Attia, P.-J. Sinnema, J. M. de Gooijer, K. Harkema, M. Hazewinkel, S. Snijder and K. Pouwer, ACS Comb. Sci., 2018, 20, 335–343 CrossRef CAS PubMed.
- A. K. Pancholi, G. P. Iacobini, G. J. Clarkson, D. W. Porter and M. Shipman, J. Org. Chem., 2018, 83, 491–498 CrossRef CAS PubMed.
- E. Graziano, P. Natho, M. Andresini, F. Mastrolorito, I. Mahdi, E. Mesto, M. Colella, L. Degennaro, O. Nicolotti and R. Luisi, Adv. Synth. Catal., 2024, 366, 3894–3902 CAS.
- P. Musci, M. Colella, M. Andresini, A. Aramini, L. Degennaro and R. Luisi, Chem. Commun., 2022, 58, 6356–6359 RSC.
- P. Natho and R. Luisi, Tetrahedron Green Chem., 2023, 2, 100015 CrossRef.
- M. Spennacchio, P. Natho, M. Andresini and M. Colella, J. Flow Chem., 2024, 14, 43–83 CrossRef CAS.
- A. Cocco, M. G. Rubanu, M. L. Sechi, A. Frongia, P. Mastrorilli, L. Degennaro, M. Colella, R. Luisi and F. Secci, Org. Biomol. Chem., 2021, 19, 1945–1949 RSC.
- E. Capel, M. Rodríguez-Rodríguez, U. Uria, M. Pedron, T. Tejero, J. L. Vicario and P. Merino, J. Org. Chem., 2022, 87, 693–707 CrossRef CAS PubMed.
- B. Wölfl, N. Winter, J. Li, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2023, 62, e202217064 CrossRef PubMed.
- C. H. U. Gregson, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2021, 60, 7360–7365 CrossRef CAS PubMed.
- V. Jaiswal, S. Mondal, B. Singh, V. P. Singh and J. Saha, Angew. Chem., Int. Ed., 2023, 62, e202304471 CrossRef CAS PubMed.
- B. Singh, P. Sasmal, A. Taites, S. Hazra and J. Saha, Org. Lett., 2024, 26, 9558–9563 CrossRef CAS PubMed.
- P. Natho, M. Colella, M. Andresini, L. Degennaro and R. Luisi, Org. Lett., 2024, 26, 3032–3036 CrossRef CAS PubMed.
- P. Natho, M. Colella, A. Vicenti, G. Romanazzi, F. Ullah, N. S. Sheikh, A. J. P. White, F. Pasca and R. Luisi, Angew. Chem., Int. Ed., 2025, e202424346 CAS.
- B. M. DeMuynck, L. Zhang, E. K. Ralph and D. A. Nagib, Chem, 2024, 10, 1015–1027 CAS.
- S. Laval, W. Dayoub, L. Pehlivan, E. Métay, A. Favre-Reguillon, D. Delbrayelle, G. Mignani and M. Lemaire, Tetrahedron, 2014, 70, 975–983 CrossRef CAS.
- A. Malashchuk, A. V. Chernykh, M. Y. Perebyinis, I. V. Komarov and O. O. Grygorenko, Eur. J. Org. Chem., 2021, 6570–6579 CrossRef CAS.
- B. Bechi, D. Amantini, C. Tintori, M. Botta and R. di Fabio, Beilstein J. Org. Chem., 2014, 10, 1114–1120 CrossRef PubMed.
- S. Li, W.-J. Cao and J.-A. Ma, Synlett, 2016, 673–678 Search PubMed.
- S.-Q. Luo, W. Liu, B.-F. Ruan, S.-L. Fan, H.-X. Zhu, W. Tao and H. Xiao, Org. Biomol. Chem., 2020, 18, 4599–4603 RSC.
- C. Zhou, Y. Liang, Y. Li, M. Huang, Z. Luo, K.-F. Yang, Z. Li, G.-Q. Lai and P. Zhang, Org. Lett., 2024, 26, 1941–1946 CrossRef CAS PubMed.
- P. An, H.-Y. Wu, T. M. Lewandowski and Q. Lin, Chem. Commun., 2018, 54, 14005–14008 RSC.
- S. Galavskyy, A. Chernykh, O. Liashuk, D. Lesyk, S. V. Shishkina, D. Kliukovskyi, D. M. Volochnyuk, S. V. Ryabukhin and O. O. Grygorenko, J. Org. Chem., 2024, 89, 18477–18486 CrossRef CAS PubMed.
- P. Kumar, O. Zainul and S. T. Laughlin, Org. Biomol. Chem., 2018, 16, 652–656 RSC.
| This journal is © The Royal Society of Chemistry 2025 |