
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A strong H-bond between a cysteine and the catalytic center of a [NiFe]-hydrogenase†
Chara
Karafoulidi-Retsou
 ,
Sagie
Katz
,
Stefan
Frielingsdorf
,
Oliver
Lenz
,
Ingo
Zebger
* and
Giorgio
Caserta
*
,
Sagie
Katz
,
Stefan
Frielingsdorf
,
Oliver
Lenz
,
Ingo
Zebger
* and
Giorgio
Caserta
*
Institut für Chemie, Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: ingo.zebger@tu-berlin.de; giorgio.caserta@tu-berlin.de
First published on 14th March 2025
Abstract
Infrared spectroscopy at cryogenic temperatures was used to monitor protonation changes on an H+-accepting, nickel-coordinating active site cysteine of the H2/H+-cycling membrane-bound [NiFe]-hydrogenase from Cupriavidus necator. Surprisingly, we identified another cysteine in the outer coordination sphere forming a strong H-bond with a cysteine thiolate coordinating both nickel and iron of the catalytic center.
[NiFe]-hydrogenases catalyze the reversible cleavage of dihydrogen into protons and electrons at a bioinorganic heterobimetallic [NiFe] center. This active site is anchored to the large catalytic subunit of the hydrogenase heterodimeric functional unit, known as the hydrogenase module, by four cysteine residues coordinating the nickel ion. Two of these cysteines act as bridging ligands between the Ni and Fe ions. The Fe is further ligated by two cyanides (CN−) and one carbonyl (CO) ligand (Fig. 1a).1,2 During H2 activation, protons are transferred to nearby H+-accepting residues, while electrons are shuttled to redox partners via an array of Fe–S clusters. The catalytic cycle involves several intermediates (Fig. 1b), whose detailed structures have not yet been fully elucidated, despite ongoing efforts of various research groups.3–5 Vibrational spectroscopic techniques, in particular infrared (IR) spectroscopy, are valuable tools in hydrogenase research.6–8 The power of IR spectroscopy arises from the presence of the CO and CN− ligands (Fig. 1a),4,9 which absorb in a frequency regime unaffected by protein or solvent bands, and thus, represent valuable marker bands that react sensitively to redox, acid–base and structural changes taking place at or near the [NiFe] site.3 Recently, the applicability of IR spectroscopy on hydrogenases has been further expanded, enabling the identification of individual amino acids as both coordinating and localized in the vicinity of the active site (e.g., within H-bonding distance).10–12 In 2019, the Hirota group was the first to use IR difference spectroscopy at cryogenic temperatures to detect the S–H stretching vibration of the so-called Nia-L intermediate (Fig. 1b) in the [NiFe]-hydrogenase from Desulfovibrio vulgaris Miyazaki F (DvMF).11 Such absorptions are difficult to discern due to their inherently low molar absorption coefficients. The IR data for DvMF hydrogenase were interpreted as transient protonation of a Ni-coordinating cysteine residue that acts as a H+ acceptor in the photolysis of the bridging hydride of the Nia-C intermediate (Fig. 1b).13 Recently, we have further extended the mechanistic knowledge of the Nia-C → Nia-L transformation.14 For the O2-tolerant regulatory hydrogenase from Cupriavidus necator (CnRH), we observed that this transformation initially forms the metastable Nia-L1 species, which then rapidly converts into the well-characterized Nia-L2 species. Importantly, in both Nia-L intermediates, a Ni-bound cysteine thiolate is protonated and the Nia-C → Nia-L1 → Nia-L2 conversions were accompanied by subtle structural rearrangements in the 2nd coordination sphere of the active site. Although a protonated cysteine is now generally accepted as an integral part of the Nia-L intermediate, the corresponding low-temperature IR absorptions have only been detected for CnRH and DvMF hydrogenase, while the IR spectroscopic analysis of the O2-tolerant [NiFe]-hydrogenase 1 from Escherichia coli (EcHyd1) did not reveal any change in the protonation state of the corresponding thiolate side chain.15 Here, we intended to verify the protonation of the corresponding Ni-bound cysteine using the membrane-bound [NiFe]-hydrogenase from C. necator (CnMBH), which is structurally and functionally closely related to EcHyd1. As-isolated oxidized CnMBH resides predominantly in the Nir-B resting state with a hydroxy ligand in the bridging position of the active site.16 In the corresponding IR spectrum, this intermediate is characterized by a stretching vibration of the CO ligand (νCO) at 1948 cm−1 and stretching vibrations of the CN− ligands (νCN) at 2081 and 2098 cm−1 (Fig. S1, ESI†). After 30-min reduction with H2 at pH 5.5 and 283 K, the corresponding IR spectrum shows predominantly signals of the Nia-C intermediate (Fig. S2, ESI†). In contrast, IR data recorded in a 280–210 K temperature window (Fig. 2a) show the presence of a temperature-dependent equilibrium between the Nia-C (νCO at 1957 cm−1 and νCN at 2075 and 2097 cm−1) and the Nia-SR′ (νCO at 1925 cm−1 and νCN at 2049 and 2071 cm−1) state, the latter carrying one H+ and one e− more (Fig. 1b). The effect of temperature on the enrichment of certain hydrogenase intermediates has been observed previously,17,18 and recently, temperature-dependent changes in the Nia-C ⇄ Nia-SR equilibrium have been reported for the NAD+-reducing [NiFe]-hydrogenase from Hydrogenophilus thermoluteolus TH-1.19 Our IR data of CnMBH also included small amounts of Nia-L species even without light exposure (Fig. 2a and Fig. S2, ESI†), which is in agreement with the current view that Nia-L is a genuine intermediate of the catalytic cycle.20–22 Upon illumination of the sample at 95 K (460 nm LED), Nia-C quantitatively converts to Nia-L species, characterized by a νCO at 1911 cm−1 (Fig. 2b, traces 1 and 2). The photoconversion is more clearly visible in the corresponding “light-minus-dark” difference spectrum (Fig. 2b, trace diff (1–2)). A closer inspection of the IR spectrum in the region of the νCN absorptions (2120–2020 cm−1) reveals that two distinct Nia-L species are generated upon irradiation, which show almost overlapping CO absorptions at 1911 cm−1, but distinct CN− bands at 2040, 2045, and 2077 cm−1 (the latter partially overlapping with a νCN of Nia-C at 2073 cm−1). The observed Nia-L species are comparable to the recently resolved Nia-L1 and Nia-L2 intermediates of CnRH, where the two νCO bands are also close (ΔνCO ≈ 3 cm−1), while the corresponding (symmetric and antisymmetric) νCN showed larger differences (Fig. S3, ESI†).14 Interestingly, for CnMBH, two distinct bands at 2516 (negative, Nia-C) and 2523 cm−1 (positive, Nia-L) were observed in the S–H (νSH) region of the spectrum (Fig. 3a), indicating one or more protonated cysteines.11,14,23 Both νSH absorptions appear in a spectral region of strongly H-bonded S–H side groups,23 and exhibit significantly higher intensities than those observed for the Nia-L1/L2 intermediates of CnRH (Fig. S3, ESI†) and DvMF.11,14 Based on all spectroscopic, computational and structural data for [NiFe]-hydrogenases reported so far,4,5 a protonated Ni-binding cysteine (Cys597 for CnMBH) was not expected for the Nia-C state. However, CnMBH contains an additional cysteine residue (Cys81) close to the active site, which is not present in other extensively characterized [NiFe]-hydrogenases such as EcHyd1, CnRH, DvMF hydrogenase and the soluble hydrogenase-1 from Pyrococcus furiosus (PfSH1) (Fig. 3b, c and Fig. S4, ESI†). From the web database HydDB for hydrogenase classification,24 we retrieved more than 200 sequences of [NiFe]-hydrogenase large subunits of the subgroup 1d (O2-tolerant H2-uptake [NiFe]-hydrogenases, ESI†) to which CnMBH and EcHyd1 belong.
 | ||
| Fig. 1 Membrane-bound [NiFe]-hydrogenase from Cupriavidus necator (CnMBH) and the consensus catalytic cycle of [NiFe]-hydrogenases. (a) Cartoon representation of CnMBH (PDB: 3RGW (https://www.rcsb.org/structure/3RGW)),25 consisting of the catalytic large subunit (blue) and the electron-transferring small subunit (green), harboring three Fe–S clusters. The NiFe cofactor and the coordinating cysteines are shown in ball and stick representation. Color code: Ni, green; Fe, brown; CO oxygen, red; CN− nitrogen, blue; corresponding carbon atoms, grey; bridging (Sb) and terminal (St) cysteine thiolates, yellow; the vacant bridging position is indicated by an X (magenta) and can be occupied either by hydroxy or hydride ligands. (b) Schematic representation of the generally accepted catalytic cycle of [NiFe]-hydrogenases. Nickel is the redox-active metal and can adopt the oxidation states1+/2+/3+, while the iron maintains an Fe2+ low-spin configuration throughout the catalytic cycle. The proposed catalytic cycle involves the intermediates Nia-S, Nia-SR, Nia-C and Nia-L. Among them, the Nia-SR and Nia-L each comprise three sub-forms,4,13,14 whose structural differences are not fully understood. | ||
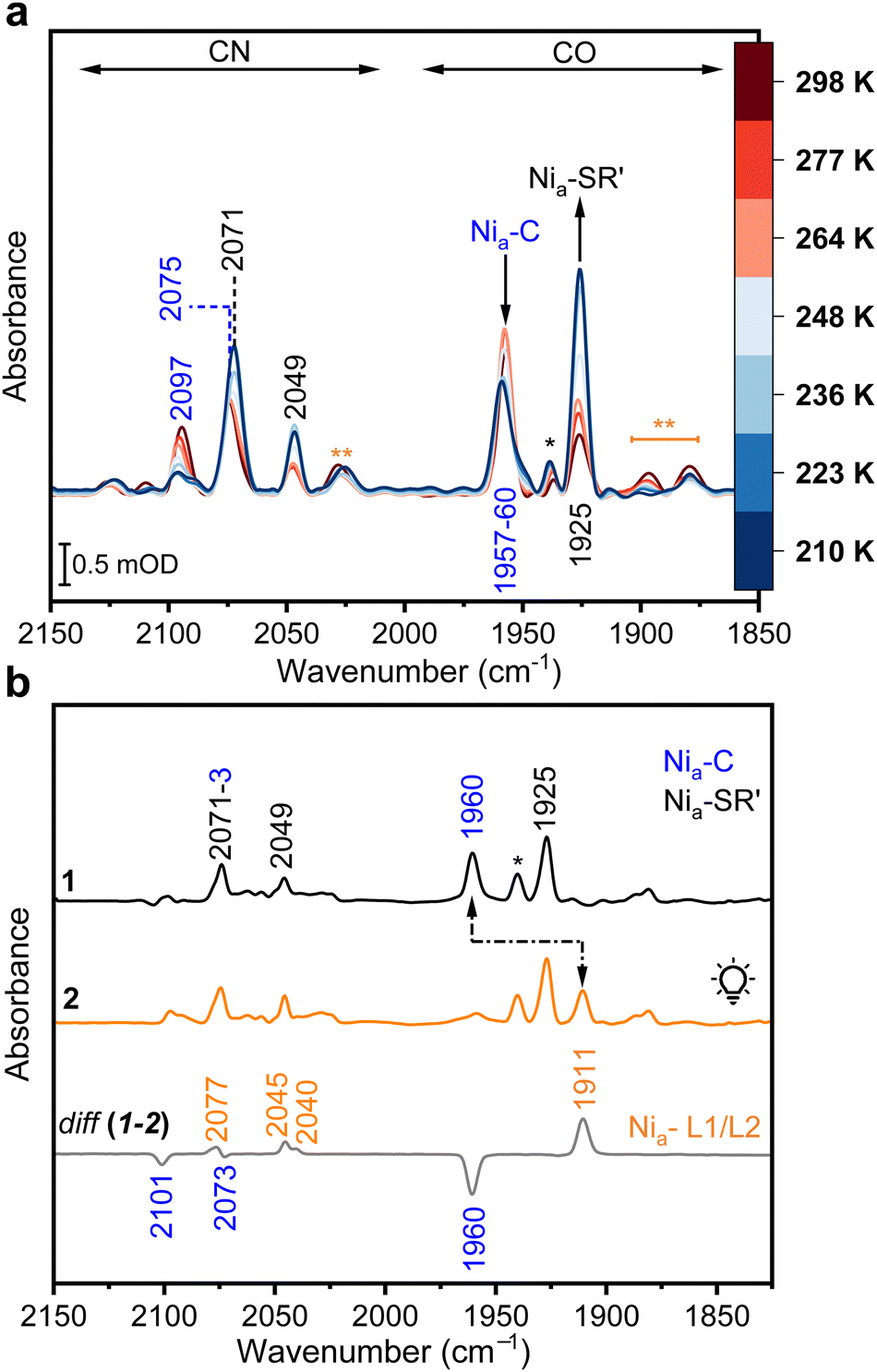 | ||
| Fig. 2 IR spectroscopic characterization of CnMBH. (a) IR absorbance spectra of H2-reduced CnMBH recorded between 210 and 298 K, indicating a temperature-dependent redox equilibrium between the Nia-C and the Nia-SR′ states. Characteristic CO/CN absorptions are labeled with the corresponding wavenumbers. (b) IR spectra of H2-reduced CnMBH at 95 K before (trace 1) and after light exposure (trace 2). Spectral changes are illustrated by black arrows and visualized in more detail by the related difference spectrum (trace diff (1–2)). The absorptions marked with a black asterisk are attributed to remnants of Nia/r-S species (1936 cm−1). νCO signals marked with two orange asterisks at 1895 and 1880 cm−1 are attributed to sub-stoichiometric amounts of Nia-L species enriched under dark conditions. | ||
 | ||
| Fig. 3 An additional cysteine in CnMBH is located near the NiFe site. (a) IR difference spectrum (light-minus-dark) of CnMBH recorded at 95 K. Characteristic absorptions of CO, CN− and SH are labeled with the corresponding wavenumbers. The inset in the upper left shows an enlargement of the SH bands. The minor absorption marked with an asterisk may result from the protonation of the H+-accepting Ni-bound Cys597. (b) Excerpt of a sequence alignment of the large subunits from CnMBH, EcHyd1, CnRH, PfSH1 and DvMF hydrogenase. Two of the four cysteines coordinating the [NiFe] site are highlighted in yellow (ruler positions 82/85), conserved residues are highlighted in red, residues with similar features are framed in blue boxes, secondary structural elements (derived from PDB:3RGW (https://www.rcsb.org/structure/3RGW))25 are shown on top of the alignment. CnMBH carries an additional cysteine (Cys81, ruler position 88), which is also highlighted in yellow. The complete alignment is shown in Fig. S4 (ESI†). (c) [NiFe] active site structure of H2-reduced CnMBH characterized by a bimodal conformation of Cys81.25 The thiolate group in conformation A (Cys81A, 80% occupation) is located at a H-bond distance (dashed cyan lines) from both Cys78 and the CO ligand. | ||
At the position corresponding to Cys81 of CnMBH, polar amino acid residues are found in about 70% of the sequences and cysteine is the second most frequent amino acid (Fig. S5, ESI†). The crystal structure of CnMBH shows that Cys81 is located in a water-filled pocket near the NiFe site and may be part of a proton transfer pathway.16 Previous electrochemical studies have shown that replacing Cys81 in CnMBH results in higher Michaelis constants (KM) for H2,26 suggesting that this residue modulates the substrate affinity in CnMBH. Cys81 adopts a predominant (ca. 80%, Cys81A) conformation in the structure of H2-reduced CnMBH,25 in which its thiolate group is in H-bonding distance to both the bridging Cys78 sulfur atom (3.4 Å) and the CO ligand (3.3 Å) (Fig. 3c). The active site geometry indicates that the sulfur atom of Cys78 is the main acceptor, since the proposed H-bond might adopt the preferred angle of almost 180°.27 Importantly, this H-bond may cause a more polarized S–H bond of Cys81, resulting in higher absorption coefficients for the observed νSH bands. To test this hypothesis, we analysed an MBH variant in which Cys81 had been exchanged for the isosteric serine residue.26 This should lead to an exchange of the thiolate for a hydroxy side group, and should facilitate the spectroscopic assignment without optimally altering the catalytic activity. In fact, the purified CnMBHCys81Ser variant retained H2 oxidation activity with a specific activity of 129.2 ± 9.6 U mg−1, which is ca. 92% of the activity of native CnMBH,28 and is consistent with previous studies.26,29 The IR spectroscopic data of the MBHCys81Ser variant in the as-isolated, H2-reduced and re-oxidized forms, measured at 283 K, are shown in Fig. S6a–c (ESI†). After partial reduction of MBHCys81Ser with H2 (trace b in Fig. S6a, ESI†), the sample was cooled to 95 K and then illuminated with LEDs at 460 nm (Fig. S6b, ESI†). The corresponding IR difference spectra (light-minus-dark, Nia-L-minus-Nia-C) of the MBHCys81Ser variant and that of native CnMBH are depicted in Fig. S7 (ESI†), and a detailed analysis of the active site CO/CN− absorptions is provided as part of the ESI.† Notably, the previously observed νSH bands of native CnMBH are not present in the IR difference spectrum of the MBHCys81Ser variant. Therefore, we conclude that the observed S–H absorptions of native CnMBH (Fig. 3a) originate from the protonated side chain of Cys81. In addition, the observed frequency shift for the νSH bands (2516 → 2523 cm−1) during the Nia-C → Nia-L transformation suggests that the strength of the S–H bond of Cys81 is directly influenced by electronic/structural changes at the [NiFe] site. We propose that photolysis of the bridging hydride induces local changes in the H-bond network near the [NiFe] site, which causes the observed shift of the νSH for Cys81. The initial aim of this study was to analyse the protonation state of Cys597 in CnMBH, and we expect Cys597 to be protonated in the case of the Nia-L1/2 intermediates. In fact, the spectral signals of the Nia-L species of CnMBH are quite similar to those of CnRH, in which the analogous Ni-bound cysteine resides in a protonated state (Fig. S3, ESI†).14 In the case of native CnMBH, the expected S–H absorption of Cys597 might be covered by the intense absorptions of Cys81, leaving only a remnant visible as an absorption at 2530 cm−1 (asterisk in Fig. 3a). Additionally, the simultaneous presence of two Nia-L sub-forms in CnMBH implies a heterogeneous population of Cys597-S–H conformers, which reduces the probability of observing the corresponding S–H spectral features. The rather low enrichment of the Nia-C intermediate, especially in the MBHCys81Ser variant (Fig. S6a, ESI†), might be another reason for the absence of clear signals from Cys597 protonation. Thus, future research will focus on the enrichment of Nia-C and Nia-L1/L2 species in both CnMBH and variants devoid of Cys81 to specifically probe the spectral contribution of residues surrounding the [NiFe] site and the protonation state of Cys597. Our findings emphasize the necessity of a careful interpretation of the absorptions in the spectral region in which S–H signals occur. While the protonation of a Ni-bound cysteine (Cys597 in CnMBH) seemed obvious given its role as a H+ acceptor during catalysis,14,30,31 the presented IR data shifted the focus to the more distant Cys81. Since several O2-tolerant [NiFe]-hydrogenases of subgroup 1d contain a cysteine at this position (Fig. S5, ESI†), our data will support their biophysical investigation and the corresponding interpretation of the IR data. Furthermore, our observations are of general importance in (bio)catalysis as they might offer new insights for the investigation of biological and (semi-)synthetic systems (e.g., nitrogenases,32 Ni-containing rubredoxins,33 de novo-designed hydrogenases34) which operate via transiently protonated thiolate functionalities.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008-390540038 (“Unifying Systems in Catalysis-UniSysCat”). O. L. and I. Z. are thankful for financial support from EU Horizon 2020/McGEA/Proposal ID 101183014 HORIZON-MSCA-2023-SE-01-01. The authors acknowledge Marius Horch and Yvonne Rippers for helpful discussions.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Caserta, S. Hartmann, C. Van Stappen, C. Karafoulidi-Retsou, C. Lorent, S. Yelin, M. Keck, J. Schoknecht, I. Sergueev, Y. Yoda, P. Hildebrandt, C. Limberg, S. DeBeer, I. Zebger, S. Frielingsdorf and O. Lenz, Nat. Chem. Biol., 2023, 19, 498–506 CrossRef CAS PubMed.
- J. Fritsch, O. Lenz and B. Friedrich, Nat. Rev. Microbiol., 2013, 11, 106–114 CAS.
- S. T. Stripp, B. R. Duffus, V. Fourmond, C. Léger, S. Leimkühler, S. Hirota, Y. Hu, A. Jasniewski, H. Ogata and M. W. Ribbe, Chem. Rev., 2022, 122, 11900–11973 CrossRef CAS.
- P. A. Ash, R. Hidalgo and K. A. Vincent, ACS Catal., 2017, 7, 2471–2485 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- C. M. Silveira, L. Zuccarello, C. Barbosa, G. Caserta, I. Zebger, P. Hildebrandt and S. Todorovic, Molecules, 2021, 26, 4852 CAS.
- G. Caserta, L. Zuccarello, C. Barbosa, C. M. Silveira, E. Moe, S. Katz, P. Hildebrandt, I. Zebger and S. Todorovic, Coord. Chem. Rev., 2022, 452, 214287 CAS.
- B. L. Greene, G. E. Vansuch, B. C. Chica, M. W. W. Adams and R. B. Dyer, Acc. Chem. Res., 2017, 50, 2718–2726 CAS.
- S. T. Stripp, ACS Catal., 2021, 11, 7845–7862 CAS.
- M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, J. Am. Chem. Soc., 2019, 141, 17394–17403 Search PubMed.
- H. Tai, K. Nishikawa, Y. Higuchi, Z. Mao and S. Hirota, Angew. Chem., Int. Ed., 2019, 58, 13285–13290 Search PubMed.
- H. Tai, S. Hirota and S. T. Stripp, Acc. Chem. Res., 2021, 54, 232–241 Search PubMed.
- H. Tai, K. Nishikawa, S. Inoue, Y. Higuchi and S. Hirota, J. Phys. Chem. B, 2015, 119, 13668–13674 CAS.
- A. F. T. Waffo, C. Lorent, S. Katz, J. Schoknecht, O. Lenz, I. Zebger and G. Caserta, J. Am. Chem. Soc., 2023, 145, 13674–13685 Search PubMed.
- P. A. Ash, S. E. T. Kendall-Price, R. M. Evans, S. B. Carr, A. R. Brasnett, S. Morra, J. S. Rowbotham, R. Hidalgo, A. J. Healy, G. Cinque, M. D. Frogley, F. A. Armstrong and K. A. Vincent, Chem. Sci., 2021, 12, 12959–12970 CAS.
- S. Frielingsdorf, J. Fritsch, A. Schmidt, M. Hammer, J. Löwenstein, E. Siebert, V. Pelmenschikov, T. Jaenicke, J. Kalms, Y. Rippers, F. Lendzian, I. Zebger, C. Teutloff, M. Kaupp, R. Bittl, P. Hildebrandt, B. Friedrich, O. Lenz and P. Scheerer, Nat. Chem. Biol., 2014, 10, 378–385 CAS.
- G. Caserta, V. Pelmenschikov, C. Lorent, A. F. Tadjoung Waffo, S. Katz, L. Lauterbach, J. Schoknecht, H. Wang, Y. Yoda, K. Tamasaku, M. Kaupp, P. Hildebrandt, O. Lenz, S. P. Cramer and I. Zebger, Chem. Sci., 2021, 12, 2189–2197 CAS.
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CAS.
- C. Karafoulidi-Retsou, C. Lorent, S. Katz, Y. Rippers, H. Matsuura, Y. Higuchi, I. Zebger and M. Horch, Angew. Chem., Int. Ed., 2024, e202409065 Search PubMed.
- R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed., 2015, 54, 7110–7113 CrossRef CAS PubMed.
- H. Tai, K. Nishikawa, M. Suzuki, Y. Higuchi and S. Hirota, Angew. Chem., Int. Ed., 2014, 53, 13817–13820 Search PubMed.
- B. L. Greene, G. E. Vansuch, C.-H. Wu, M. W. W. Adams and R. B. Dyer, J. Am. Chem. Soc., 2016, 138, 13013–13021 Search PubMed.
- V. A. Lorenz-Fonfria, Chem. Rev., 2020, 120, 3466–3576 Search PubMed.
- D. Søndergaard, C. N. S. Pedersen and C. Greening, Sci. Rep., 2016, 6, 34212 CrossRef PubMed.
- J. Fritsch, P. Scheerer, S. Frielingsdorf, S. Kroschinsky, B. Friedrich, O. Lenz and C. M. T. Spahn, Nature, 2011, 479, 249–252 CrossRef CAS.
- M. Ludwig, J. A. Cracknell, K. A. Vincent, F. A. Armstrong and O. Lenz, J. Biol. Chem., 2009, 284, 465–477 CrossRef CAS PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- J. Fritsch, S. Löscher, O. Sanganas, E. Siebert, I. Zebger, M. Stein, M. Ludwig, A. L. De Lacey, H. Dau, B. Friedrich, O. Lenz and M. Haumann, Biochemistry, 2011, 50, 5858–5869 CrossRef CAS PubMed.
- M. Saggu, M. Ludwig, B. Friedrich, P. Hildebrandt, R. Bittl, F. Lendzian, O. Lenz and I. Zebger, ChemPhysChem, 2010, 11, 1215–1224 CrossRef CAS PubMed.
- R. M. Evans, N. Krahn, J. Weiss, K. A. Vincent, D. Söll and F. A. Armstrong, J. Am. Chem. Soc., 2024, 146, 16971–16976 CrossRef CAS PubMed.
- A. Sirohiwal, A. P. Gamiz-Hernandez and V. R. I. Kaila, J. Am. Chem. Soc., 2024, 146, 18019–18031 CrossRef CAS PubMed.
- K. Sengupta, J. P. Joyce, L. Decamps, L. Kang, R. Bjornsson, O. Rüdiger and S. DeBeer, J. Am. Chem. Soc., 2025, 147, 2099–2114 CrossRef CAS PubMed.
- J. W. Slater, S. C. Marguet, M. E. Gray, H. A. Monaco, M. Sotomayor and H. S. Shafaat, ACS Catal., 2019, 9, 8928–8942 CrossRef CAS.
- S. Malayam Parambath, D. Prakash, W. Swetman, A. Surakanti and S. Chakraborty, Chem. Commun., 2023, 59, 13325–13328 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplementary figures, supplementary discussion, and supplementary references. See DOI: https://doi.org/10.1039/d5cc00646e |
| This journal is © The Royal Society of Chemistry 2025 |