
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic reductive incorporation of carbon dioxide into double bonds†
Mark J.
Deeprose
 * and
Thorsten
Bach
*
* and
Thorsten
Bach
*
Technische Universität München, School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Lichtenbergstraße 4, 85747 Garching, Germany. E-mail: thorsten.bach@ch.tum.de
First published on 3rd April 2025
Abstract
Carbon dioxide (CO2) is a challenging molecule to incorporate into olefins due to its inherent inert properties. Silanes have now been shown to rapidly react selectively with CO2, catalysed by the presence of a caesium base, to form formate salts which can undergo a hydrogen atom transfer (HAT) to give the desired CO2˙− adducts in the presence of activated olefins.
In the past century, and particularly in the past decade, research into the photofixation of carbon dioxide (CO2) and its conversion to a useful chemical feedstock has rapidly expanded due to novel developments in visible-light photocatalysis.1 The need for transforming an inert molecule into a high value chemical C1 source is well-studied and has a high impact especially in modern times where the movement away from fossilised solar energy is in desperate need.2 One example of this is the carboxylation of readily available olefins through a CO2˙−. This can be achieved in one of two ways; by single electron reduction (SER) of CO2 gas, or by hydrogen atom transfer (HAT) of formate salts (Scheme 1).
 | ||
| Scheme 1 (a) Single electron reduction of CO2 and subsequent reaction with activated and non-activated olefins (b) Hydrogen atom transfer of formate salts to generate CO2˙−. | ||
The drawback of incorporating CO2 by SER is the high reduction potential required to generate CO2˙− (Scheme 1a). This involves a photocatalyst that has a strongly negative redox potential, and a substrate with a reduction potential that is lower than −2.2 V vs. SCE.3 For this reason, substrates that are involved in these reactions are often non-activated to avoid the competing reduction reaction,4 or in the case of activated olefins, a radical precursor/acceptor is often utilised to initiate the reaction.5 Additionally, formation of oxalate from an excess of CO2˙− in solution adds to the inefficiency of the reaction conditions. An alternative approach to the formation of CO2˙− can be envisioned to be possible by HAT of formate salts. This process has been utilised successfully by the Wickens group for CO2 integration into both activated and non-activated olefins (Scheme 1b).6 Furthermore, formate salts have been successfully utilised in difunctionalization of olefins, through two consecutive SER processes, and the Maiti group has recently reported formate being generated in situ from CO2 and a silane.7,8
Silanes are highly selective reducing agents in organic synthesis and have been previously illustrated to successfully fixate CO2 and convert it to a range of different products (Scheme 2).9 However, in a lot of these examples high heat or a catalyst is required to enable the reaction. Our work aims to combine the highly selective silane reactivity towards CO2, with the facile HAT of the generated formate, to selectively produce CO2˙− in the presence of reactive olefins. To begin our studies, we focused on detecting the formation of silyl formate in the absence of heat or catalyst. Following a previous study towards fixation of CO2, we employed phenyl silane in DMSO under an atmosphere of CO2.10 As a modification to the reported procedure, we performed this reaction at room temperature and found that after two hours only 25% of the silylformate had been formed (see the ESI,† for details). As previous work has reported, an increase in reaction rate is observed with the addition of formate or acetate salts, one equivalent caesium acetate was added to the reaction.10 Pleasingly, the addition of the base had a dramatic effect on the reaction rate, giving 160% (per eq silane) of formate being detected by 1H NMR within a few minutes. With this result in hand, focus was turned to the HAT of the generated formate salt and the subsequent incorporation of CO2. As established in previous literature, oxygen (O2) would be vital as a sacrificial oxidant in the HAT process.6 We chose 1,1-diphenylethylene 1a as a suitable substrate due to its effectiveness as a radical acceptor and its resistance to polymerisation. Upon visible-light irradiation in the presence of a thiol, and a combined atmosphere of air/CO2 (1/1), 63% of the desired product 2a was observed (Table 1, entry 1).
 | ||
| Scheme 2 (a) Reaction of hydrosilanes with CO2 and the subsequent products that can be obtained. (b) Conceptual approach of the present study. | ||
|
|
|||
|---|---|---|---|
| Entry | Change to conditions | NMR yield (%) | Remaining 1a (%) |
| Reactions were performed on a 0.1 mmol scale. Trimethoxybenzene used as an internal standard. Yield of by-product 3a in parentheses. | |||
| 1 | — | 63 | 0 |
| 2 | KOAc | 41 | 0 |
| 3 | NaOAc | 3 | 66 |
| 4 | CsOAc/PhSiH3 (3 eq.) | 58 (25) | 0 |
| 5 | Methyl 4-mercaptobenzoate | 79 | 0 |
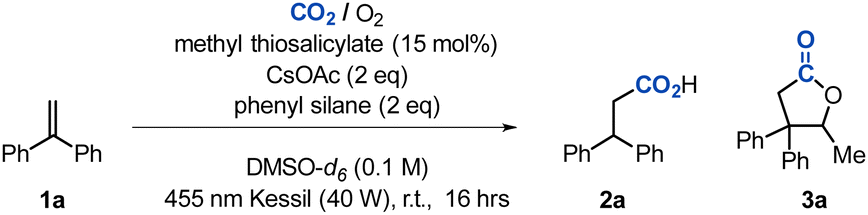
Further optimisation showed that caesium acetate had the broadest reactivity across multiple substrates, and that two equivalents of the silane and base lead to the best yield with DMSO being the most suitable solvent. Upon screening multiple silanes, it was found that all silanes were reactive towards CO2, including polymethylhydrosiloxane (PMHS), with phenyl silane leading to the best product yield. Our optimisation study found, however, that an excess of phenyl silane facilitated the reduction of the caesium acetate base to form ethanal which led to the undesired formation of lactone by-product 3a (Table 1, entry 4). In subsequent experiments, a range of thiol derivatives were tested under the reaction conditions, and found that most thiophenol derivatives were well tolerated, with the exception of sterically hindered or acidic compounds. Methyl 4-mercaptobenzoate achieved the highest yield of 79% for the desired reaction at 15 mol% loading (Table 1, entry 5).
With these results in hand, a series of control reactions were performed. Unexpectedly, it was found that the addition of a photoredox catalyst (4CzIPN) had a detrimental effect on the product yield (Table 2, entry 2). Each individual component of the reaction was found to be essential to the production of 2a (entries 3–8). The reaction conditions were found to be intolerant to the addition of water (entries 10 and 11). Additional experiments confirmed that dry ice could be used as the CO2 source, although a significant amount of lactone 3a was observed (entry 12). The reaction was scaled up to 1 mmol and gave a 70% yield of isolated product (entry 13).
| Entry | Change to conditions | NMR yield (%) | Remaining 1a (%) |
|---|---|---|---|
| Reactions were performed on a 0.1 mmol scale using DMSO-d6 as the solvent (cf.Table 1, entry 5). Yield of by-product 3a in parentheses.a Anhydrous DMSO employed.b 0.2 mmol scale.c Dimethyl 4,4′-disulfanediyldibenzoate as catalyst in the absence of oxygen (GP 2, see ESI).d Yield of isolated product.e Dry ice (approx. 100 mg, 2.2 mmol) added to reaction solution and allowed to warm to r.t. before sealing, no gas purging of reaction headspace. | |||
| 1 | — | 79 | 0 |
| 2 | Presence of 4CzIPN (1 mol%) | 73 | 0 |
| 3 | Absence of CO2 | 0 | 0 |
| 4 | Absence of O2 | 37 | 0 |
| 5 | Absence of thiol | 0 | 30 |
| 6 | Absence of silane | < 1 | 18 |
| 7 | Absence of base | 0 | 63 |
| 8 | Absence of light | 0 | 0 |
| 9 | Non-deuterated DMSO | 84 | 0 |
| 10a | Addition of water (1 eq.) | 65 | 0 |
| 11a | Addition of water (1 M) | 26 | 12 |
| 12a | Dry ice as CO2 sourcece | 50 (22) | 0 |
| 13a | Scale up to 1 mmolc | 70d | 0 |
Oxygen was established to be essential for achieving a high reaction yield, as thiyl radical formation proceeds through an oxidative pathway (Table 2, entry 4). However, upon applying these conditions to a broader substrate scope, it was found that non-substituted styrenes did not show good yields (<30%). This may be due to the generation of singlet oxygen and subsequent oxidation of the activated olefins. To circumvent this problem, oxygen was not introduced into the reaction. Because methyl 4-mercaptobenzoate cannot be oxidized without oxygen, dimethyl 4,4′-disulfanediyldibenzoate was synthesized separately and subsequently introduced into the reaction conditions. This resulted in the highest yield for the optimised substrate (84% isolated, entry 9) and was therefore employed as the optimised conditions for the substrate scope.
Next, a range of styrenes were tested under the reaction conditions (Scheme 3a). It was found that most styrenes exhibited reactivity under the optimised reaction conditions (products 2b–2h), but overall gave lower yields than 1,1-diphenylethylene. To identify if a more stable radical intermediate would increase the reaction yield, an alkyl group was added in the α-position but did not improve the product yield. To determine if heterocycles were tolerant to the reaction conditions, 2-vinyl pyridine was employed as a substrate and achieved incorporation of CO2 with a 37% internal yield.
 | ||
| Scheme 3 Reactions were performed on a 0.2 mmol scale. (a) Styrene substrate scope. (b) Acrylate derivative scope. (c) Strecker-type reactions. Due to difficulties with isolation, yields shown in parentheses were obtained by 1H NMR using trimethoxybenzene as an internal standard. | ||
To broaden the substrate scope beyond aryl olefins, a range of acrylate derivatives were tested. The acrylate derivatives presented a higher tolerance in the reaction conditions, giving yields of up to 89%. Generally, substituted acrylates resulted in high yields, possibly due to their resistance to polymerisation (2i–2l, 2n). The reactivity of the CO2˙− in sterically hindered environments was demonstrated as it was even able to form quaternary carbon–carbon bonds (2o). Amide and nitrile functional groups were also shown to be tolerated in these reaction conditions (2m, 2p). Good diastereoselectivity was observed with substrates containing trisubstituted olefins (2k, 2l).
A common method towards the synthesis of amino acids is the Strecker synthesis. This requires the use of highly toxic cyanide salts to form simple amino acids from imines. Incorporation of CO2via a radical anion into imines would broaden the substrate scope and avoid the need for cyanide salts. Previous work in this area has utilised diarylamine compounds to form a carbanion intermediate.11,12 To ascertain if CO2 could be incorporated through our method, imines were synthesised and subjected to the reaction conditions (Scheme 3c, products 4a–4d). Pleasingly, all imines were converted to the desired amino acid in good to quantitative yield. To the best of our knowledge, this is the first instance of amino acids being synthesised from imines via direct addition of CO2˙−. Electron rich olefins did not indicate reactivity towards CO2˙−, and non-activated olefins provided no conversion or low yields (see ESI,† for details). Furthermore, employing bicyclobutanes led to their degradation under the reaction conditions, and reactive halides (allyl bromide) underwent hydrodehalogenation in the presence of phenyl silane.
Our proposed mechanism for the fixation of CO2 and subsequent HAT and radical addition is outlined in Scheme 4a. Initially, phenyl silane coordinates to caesium acetate, activating the silicon–hydrogen bond. This activated intermediate is able to selectively reduce CO2 and produce a silylformate which is able to go through a transmetallation with caesium acetate, forming silylacetate and caesium formate. The silylformate was found to not participate in HAT (Table 2, entry 7). Disulphide 5 is homolytically cleaved from direct irradiation (see ESI,† for UV/Vis spectra) to produce two thiyl radicals which in turn abstract a hydrogen atom from the generated caesium formate 6 to form the desired CO2˙−7. Radical anion 7 reacts with the substrate olefin, leading to the radical adduct carboxylate 8. The generated radical 8 can be subjected to a reduction from another molecule of 7, and the corresponding anion 10 can be protonated. Alternatively, the generated radical 8 can undergo HAT with HSAr6 to produce product 9 and to regenerate a thiyl radical.
 | ||
| Scheme 4 (a) Proposed reaction mechanism. (b) Deuterium labelling studies. | ||
Several conditions were employed to elucidate this proposed mechanism. Firstly, evidence for the addition of CO2˙− was demonstrated by a radical clock test from a cyclopropyl substituted substrate (see the ESI,† for details). The desired product was detected in a 53% yield, suggesting that a radical addition takes place. Secondly, deuterium studies were undertaken to determine the fate of the generated radical 8 (Scheme 4b). When using phenyl silane-d3, a low yield and a low deuterium incorporation was observed. Upon addition of D2O into the reaction conditions, a favoured route was identified, whereby the generated radical 8 undergoes reduction to the anion, and is subsequently protonated.13 These two experiments demonstrate that both processes happen in the reaction mechanism. Furthermore, the removal of radical inhibitors from commercial supplies of olefins was necessary to obtain good reaction yields, further demonstrating the role of radicals in the reaction conditions. A reaction plot of the oxygenated reaction (using methyl 4-mercaptobenzoate) indicated an initial delay in the reaction rate in the first two hours. This can be explained by the need for the thiol catalyst to be oxidised into the disulphide, before homolytic cleavage can take place.6b Disulphide 5 could be synthesised in near quantitative yield by irradiating methyl 4-mercaptobenzoate under an aerobic atmosphere for 3 hours.
In conclusion, a method towards the facile incorporation of CO2 into activated olefins has been established. The reaction pathway of CO2 silylation and subsequent HAT has been confirmed, and the reaction conditions have been successfully employed across a range of activated olefins and imine double bonds to give the desired products with up to 89% yields of isolated material and quantitative yields by 1H NMR. The reaction was found to not require a photoredox catalyst, which allows for redox active substrates to be included. Similar results can also be achieved using dry ice as the source of CO2 and furthermore the reaction has been revealed to tolerate scale up. Further work is necessary to apply this type of methodology to non-activated olefins, but the initial work demonstrated here suggests that a silane mediated general route towards hydrocarboxylation across double bonds, using CO2 as a C1 source, is possible.
Financial support by the Deutsche Forschungsgemeinschaft (Ba 1372/23) is gratefully acknowledged.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- For a selection of examples on the direct functionalisation of CO2: (a) S. Tazuke and N. Kitmaura, Nature, 1978, 275, 301–302 CrossRef CAS; (b) H. Yuan, Y. Yu, S. Yang, Q. Lei, Z. Yang, B. Lan and Z. Han, Chem. Commun., 2024, 60, 6292–6295 RSC; (c) A. Stoumpidi, A. Trapali, M. Poisson, A. Barrozo, S. Bertaina, M. Orio, G. Charalambidis and A. G. Coutsolelos, ChemCatChem, 2023, 15, e202200856 CrossRef CAS; (d) R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed; (e) C.-H. Lim, S. Ilic, A. Alherz, B. T. Worrell, S. S. Bacon, J. T. Hynes, K. D. Glusac and C. B. Musgrave, J. Am. Chem. Soc., 2019, 141, 272–280 CrossRef CAS PubMed; (f) R. Cauwenbergh, V. Goyal, R. Maiti, K. Natte and S. Das, Chem. Soc. Rev., 2022, 51, 9371–9423 RSC.
- For visible light photochemical valorisation of CO2: (a) S. E. Lee, A. Nasirian, Y. E. Kim, P. T. Fard, Y. Kim, B. Jeong, S.-J. Kim, J.-O. Baeg and J. Kim, J. Am. Chem. Soc., 2020, 142, 19142–19149 CrossRef CAS PubMed; (b) Q. Lei, H. Yuan, J. Du, M. Ming, S. Yang, Y. Chen, J. Lei and Z. Han, Nat. Commun., 2023, 14, 1087 CrossRef CAS PubMed; (c) C. Singh, J. Y. Kim, N.-J. Park, C. U. Kim, R. K. Yadav and J.-O. Baeg, ACS Appl. Mater. Interfaces, 2024, 16, 31085–31097 CrossRef CAS PubMed; (d) J. Lan, X. Lu, B. Ren, F. Duo, X. Niu and J. Si, Org. Biomol. Chem., 2024, 22, 682–693 RSC; (e) H. Rao, C.-H. Lim, J. Bonin, G. M. Miyake and M. Robert, J. Am. Chem. Soc., 2018, 140, 17830–17834 CrossRef CAS PubMed; (f) Y. Qin, R. Cauwenbergh, S. Pradhan, R. Maiti, P. Franck and S. Das, Nat. Commun., 2023, 14, 7604 CrossRef CAS PubMed.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 CrossRef CAS.
- (a) W. Zhang, Z. Chen, Y.-X. Jiang, L.-L. Liao, W. Wang, J.-H. Ye and D.-G. Yu, Nat. Commun., 2023, 14, 3529 CrossRef CAS; (b) L. Song, W. Wang, J.-P. Yue, Y.-X. Jiang, M.-K. Wei, H.-P. Zhang, S.-S. Yan, L.-L. Liao and D.-G. Yu, Nat. Catal., 2022, 5, 832–838 CrossRef CAS; (c) H. Yang, Q. Yang, Y. Yao, P. Gu, J. Sun and S. Sun, Org. Lett., 2024, 26, 6341–6346 CrossRef CAS PubMed.
- (a) Y.-N. Niu, X.-H. Jin, L.-L. Liao, H. Huang, B. Yu, Y.-M. Yu and D.-G. Yu, Sci. China: Chem., 2021, 64, 1164–1169 CrossRef CAS; (b) J. Hou, A. Ee, H. Cao, H. Ong, J. Xu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 17220–17224 CrossRef CAS PubMed; (c) Q. Fu, Z.-Y. Bo, J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Nat. Commun., 2019, 10, 3592 Search PubMed; (d) J. Ye, M. Miao, H. Huang, S. Yan, Z. Yin, W. Zhou and D. Yu, Angew. Chem., Int. Ed., 2017, 56, 15416–15420 CrossRef CAS PubMed; (e) V. R. Yatham, Y. Shen and R. Martin, Angew. Chem., Int. Ed., 2017, 56, 10915–10919 CrossRef CAS PubMed.
- (a) S. N. Alektiar and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 13022–13028 CrossRef CAS PubMed; (b) S. N. Alektiar, J. Han, Y. Dang, C. Z. Rubel and Z. K. Wickens, J. Am. Chem. Soc., 2023, 145, 10991–10997 CrossRef CAS PubMed.
- (a) S. Wang, P. Xu and X. Zhu, ChemCatChem, 2023, 15, e202300695 CrossRef CAS; (b) P. Xu, S. Wang, H. Xu, Y.-Q. Liu, R.-B. Li, W.-W. Liu, X.-Y. Wang, M.-L. Zou, Y. Zhou, D. Guo and X. Zhu, ACS Catal., 2023, 13, 2149–2155 CrossRef CAS; (c) T.-T. Zhao, X.-G. Zhang, W.-B. He and P.-F. Xu, Green Chem., 2023, 25, 8539–8543 RSC; (d) P. Xu, W.-W. Liu, T.-Z. Hao, Y.-Q. Liu, H.-X. Jiang, J. Xu, J.-Y. Li, L. Yin, S.-L. Zhu and X. Zhu, J. Org. Chem., 2024, 89, 9750–9754 CrossRef CAS PubMed; (e) F. Zhang, X. Y. Wu, P. P. Gao, H. Zhang, Z. Li, S. Ai and G. Li, Chem. Sci., 2024, 15, 6178–6183 RSC.
- P. Ghosh, S. Maiti, A. Malandain, D. Raja, O. Loreau, B. Maity, T. K. Roy, D. Audisio and D. Maiti, J. Am. Chem. Soc., 2024, 146, 30615–30625 CrossRef CAS PubMed.
- (a) K. Motokura, R. A. Pramudita and Y. Manaka, J. Jpn. Pet. Inst., 2019, 62, 255–263 CrossRef CAS; (b) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS PubMed; (c) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660–10661 CrossRef CAS PubMed; (d) T. T. Metsänen and M. Oestreich, Organometallics, 2015, 34, 543–546 CrossRef; (e) C. Das Neves Gomes, O. Jacquet, C. Villiers, P. Thuéry, M. Ephritikhine and T. Cantat, Angew. Chem., Int. Ed., 2012, 51, 187–190 CrossRef CAS PubMed; (f) R. A. Pramudita and K. Motokura, Green Chem., 2018, 20, 4834–4843 RSC; (g) S. Itagaki, K. Yamaguchi and N. Mizuno, J. Mol. Catal. A: Chem., 2013, 366, 347–352 CrossRef CAS; (h) A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2012, 51, 2981–2984 CrossRef PubMed; (i) M. L. Scheuermann, S. P. Semproni, I. Pappas and P. J. Chirik, Inorg. Chem., 2014, 53, 9463–9465 CrossRef CAS PubMed; (j) L. González-Sebastián, M. Flores-Alamo and J. J. Garcıa, Organometallics, 2013, 32, 7186–7194 CrossRef.
- H. Wu, H. Li, W. Zhao, P. Sudarsanam and S. Yang, Fuel, 2022, 326, 125074 CrossRef CAS.
- T. Murata, M. Hiyoshi, M. Ratanasak, J. Hasegawa and T. Ema, Chem. Commun., 2020, 56, 5783–5786 RSC.
- (a) X. Fan, X. Gong, M. Ma, R. Wang and P. J. Walsh, Nat. Commun., 2018, 9, 4936 CrossRef PubMed; (b) W.-W. Liu, P. Xu, H.-X. Jiang, M.-L. Li, T.-Z. Hao, Y.-Q. Liu, S.-L. Zhu, K.-X. Zhang and X. Zhu, ACS Catal., 2024, 14, 10053–10059 CrossRef CAS.
- C. U. Brzezinski, A. R. LeBlanc, M. G. Clerici and W. M. Wuest, Org. Lett., 2024, 26, 5534–5538 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00604j |
| This journal is © The Royal Society of Chemistry 2025 |