
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafast, cytocompatible mineralization of calcium phosphate in the formation of stratified nanoshells of artificial spores†
Duc Tai
Nguyen
a,
Sang Yeong
Han
a,
Hyunwoo
Choi
a,
Nayoung
Kim
a,
Gulaim A.
Seisenbaeva
 b,
Vadim G.
Kessler
b and
Insung S.
Choi
*a
b,
Vadim G.
Kessler
b and
Insung S.
Choi
*a
aDepartment of Chemistry, KAIST, Daejeon 34141, Republic of Korea. E-mail: ischoi@kaist.ac.kr
bDepartment of Molecular Sciences, SLU, Uppsala 75007, Sweden
First published on 3rd April 2025
Abstract
Spatially controlled confinement of catalytic enzymes within nanoshells holds substantial potential for applications in bioreactors, synthetic cells, and artificial spores. The utilization of amorphous calcium phosphate (CaP) precursors enables the extremely rapid (<5 seconds) construction of thick (∼400 nm) CaP nanoshells, stratified with distinct enzymes, on various tannic acid-primed substrates. Saccharomyces cerevisiae cells are nanoencapsulated with enzyme-embedded, multilayered CaP nanoshells in a cytocompatible manner, providing an advanced chemical tool for interfacing living cells with functional entities in a spatially controlled configuration.
Sporulation is an adaptive-survival tactic of many bacteria and fungi characterized by the formation of a robust, protective physicochemical barrier around the cell in response to nutrient deprivation or other adverse environmental conditions.1,2 Inspired by biological sporulation, durable cell-in-nanoshell structures, referred to as artificial spores, have been developed by chemically interfacing individual living cells with external materials in a cytocompatible fashion.3–6 Artificial spores not only provide cytoprotection against harsh environmental stresses but also allow for the chemical manipulation of cellular metabolic activities, empowering the nanoencapsulated cells with exogenous functions.7,8
Artificial spores can be constructed with various cytocompatible materials, encompassing both organic and inorganic species, to ensure cell viability during the process of single-cell nanoencapsulation (SCNE).9–11 Inspired by the silica (SiO2) and calcium carbonate (CaCO3) exoskeletons found in certain unicellular organisms, such as diatoms and coccolithophores, bioinspired mineralization has been employed to fabricate artificial nanoshells composed of SiO2,12–15 CaCO3,16,17 titanium dioxide (TiO2),18,19 or calcium phosphate (CaP).20 The mild reaction conditions for and the robustness of mineral nanoshells make bioinspired mineralization a powerful approach for the protection and functionalization of living cells at the single-cell level.
Although artificial-spore approaches with Ca minerals have been demonstrated, their applications and methodological advancements remain limited, largely due to the complexity, as well as the heterogeneity and low efficiency, of thin-film formation from Ca minerals. In nature, cell-mediated mineralization actively controls the nucleation sites, crystal growth, crystallinity, and morphology of biominerals.21 However, in bioinspired mineralization, it is extremely challenging to replicate the controlled, closed, and isolated microsystems of living cells due to the stochastic nature of nucleation, particularly in the case of polymorphic CaP. Therefore, the open systems for chemical biomineralization have thus far strictly required the optimization of various factors, including solution pH, temperature, ionic strength, influx rate of metal ions, nucleation sites, and crystal growth duration, to achieve successful Ca-mineral SCNE.
Typical Ca-mineral processes proceed through the mixing of a Ca2+ solution with a HPO42− or PO43− solution at controlled pH and reaction times, which can extend to tens of minutes or hours for the formation and growth of amorphous Ca minerals on the cell surface, always carrying a high risk of unwanted nucleation in solution. For localized heterogeneous nucleation, the substrates have been pretreated with Ca2+-adsorbing polymers, such as poly(acrylic acid)22 and polyvinylpyrrolidone,17 to induce surface seeding. In addition, capping agents (e.g., triethylamine23 and polyphenols24) or zwitterion polymers25 are often required to prevent the overgrowth of nanoparticles and their transformation to hydroxyapatite. Recent advances include the use of zeolitic imidazolate framework-8, which promotes exclusive surface growth of CaP through ion exchange between the surface Zn2+ and Ca2+ in solution.24 However, the stochasticity, randomness, and inflexibility of current biomineralization protocols still limit their applications in cell-surface engineering, particularly when the incorporation of drugs, genetic materials, and enzymes is beneficial in advanced cell therapies. Hence, it is of the utmost importance to develop a rapid, highly efficient, reproducible, and versatile methodology for bioinspired CaP or other mineralization. In this work, we remarkably shorten the CaP mineralization process by simply interfacing an amorphous CaP precursor (a-preCaP) with tannic acid (TA)-primed surfaces (Fig. 1a). The formation of CaP-mineral shells around individual living cells is completed within 5 s through rapid heterogeneous nucleation of a-preCaP. In addition, the large adsorption surface area of the a-preCaP clusters enables the hierarchical incorporation of external functional biomolecules, such as enzymes, into the multilayered CaP shells, providing the catalytic empowerment of living cells in SCNE.8,26
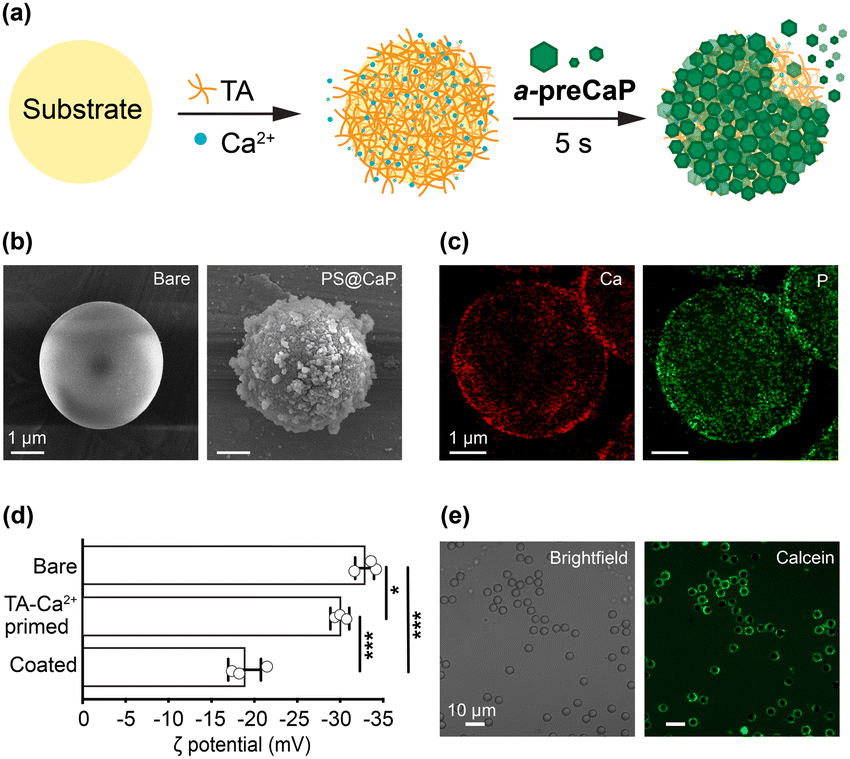 | ||
| Fig. 1 (a) Schematic illustration of the a-preCaP protocol. (b) FE-SEM images of bare PS and PS@CaP. (c) TEM-EDX Ca and P mapping of PS@CaP. (d) Zeta (ζ) potentials of bare PS, TA-Ca2+-primed PS, and PS@CaP. Data are represented as mean ± SD (unpaired, two-sided Student's t-test; n = 3; ***p < 0.001, *p < 0.05). (e) CLSM images of PS@CaP stained with calcein. | ||
The mechanisms on CaP formation in organisms remain uncertain. Biological CaP is often amorphous or low crystalline, and its deposition is suggested to proceed through an amorphous precursor phase, consisting of CaP clusters, rather than the ion association pathway in classical nucleation theory.27,28 CaP minerals have been shown to evolve from pre-nucleation clusters of multiple Ca2+ and PO43− ions into larger post-nucleation clusters and loosely packed polymeric assemblies before crystallization.29,30 These nanometric structures are metastable and are often regarded as the building blocks of CaP precipitation. We utilized the concept of the amorphous precursor phase to initiate and accelerate the localized heterogeneous deposition of CaP minerals in SCNE. Specifically, we produced a-preCaP by mixing 4.8 mM of CaCl2 and 3.2 mM of K3PO4 in water. Transmission electron microscopy (TEM) images showed an interconnected network of CaP assemblies, each approximately 10–20 nm in size (Fig. S1a, ESI†). Dynamic light scattering (DLS) analysis further indicated the existence of small CaP clusters (average diameter: 4.4 nm) and a large network of CaP assemblies (average diameter: 1000 nm) (Fig. S1b, ESI†). a-preCaP appeared amorphous or ill-defined, as evidenced in the selected area electron diffraction (SAED) pattern (Fig. S1c, ESI†). We observed that CaP minerals were produced in the solution immediately after the addition of a small amount of TA to the a-preCaP solution (Fig. S2a, ESI†). The a-preCaP solution (2 mL) became turbid upon the addition of 100 μL of TA (6 mM). We thought that TA effectively reduced the nucleation energy barrier, thereby facilitating CaP nucleation.31
Following the initial assessment, we investigated the CaP shell formation with abiotic polystyrene (PS) microparticles (sulfonated, diameter: 3.97 μm) as a model. In brief, the PS particles were primed with a mixture of TA (4 mM) and Ca2+ (8 mM) and then subjected to gentle shaking with the a-preCaP solution for 5 s, resulting in the formation of PS@CaP. Field-emission scanning electron microscopy (FE-SEM) images showed a dramatic change in surface morphology, from smooth to particulate (Fig. 1b). Elemental mapping with energy-dispersive X-ray (EDX) analysis of PS@CaP revealed the Ca and P peaks, confirming the formation of the CaP shells (Fig. 1c). In addition, zeta (ζ) potential changes from −32.8 to −30.7 mV after TA-Ca2+ priming, and to −18.9 mV, after CaP deposition, further supported the shell formation (Fig. 1d). The X-ray diffraction (XRD) analysis indicated that the CaP shells were amorphous (Fig. S2b, ESI†). The CaP shells were visualized in the confocal laser-scanning microscope (CLSM) images, after staining the shells with calcein, a green-fluorescent Ca2+ indicator (Fig. 1e). The analyses underscored the rapid and intimate interactions between a-preCaP in solution and TA on the surface, leading to the ultrafast formation of CaP shells.
a-preCaP was also found to strongly interact with proteins. For example, preincubation of the a-preCaP solution with bovine serum albumin-Alexa Fluor 647 (BSA-Alexa 647, final concentration: 10 μg mL−1) inhibited the shell formation on the TA-Ca2+-primed PS surface, with predominant homogeneous nucleation (Fig. S3a, ESI†). However, lowering the BSA-Alexa 647 concentration to 5 μg mL−1 led to heterogeneous nucleation within a short time, suggesting the potential for incorporating proteins into the CaP shell. UV-vis analysis indicated that about 60% of BSA-Alexa 647 was incorporated into the CaP shell within 5 s (Fig. S3b, ESI†). The immobilization efficiency nearly doubled compared with the post-adsorption of BSA-Alexa 647 onto PS@CaP (efficiency: 30%), presumably due to the high surface area of a-preCaP.
Our ultrafast protocol for CaP shell formation was successfully applied to living Saccharomyces cerevisiae, creating the SCNEd cell, S.cerevisiae@CaP. The excellent cytocompatibility of our protocol was confirmed by the fluorescein diacetate (FDA) assay, which showed no noticeable decrease in viability after SCNE (viability, relative to naïve cells: 97.5%) (Fig. 2a). The CaP shell formation was supported by the changes from −24.6 mV in ζ potential to −20.1 mV after TA-Ca2+ priming and to −7.4 mV after CaP shell formation (Fig. S4, ESI†), as well as by the CLSM analysis, alongside FE-SEM and TEM analyses. TEM images showed the complete CaP shells, about 400 nm thick, after just 5 s of gentle shaking, highlighting the ultrafast CaP-shell formation through the a-preCaP approach (Fig. 2b). As a comparison, we also formed S.cerevisiae@CaP by the conventional method,20 where Ca2+-TA-primed S. cerevisiae was incubated in 800 μL of a Ca2+ solution (6 mM), and 200 μL of a PO43− solution (16 mM) was added dropwise to the cell suspension at 20 μL min−1 (final concentrations: 4.8 mM of CaCl2 and 3.2 mM of K3PO4), resulting in much thinner (∼100 nm), uneven, and incomplete shells (Fig. S5, ESI†). Furthermore, we observed no noticeable heterogeneity in shell formation, whether between batches or within a batch (Fig. S6, ESI†). Our ultrafast, cytocompatible approach can be simply expanded to the SCNE of other cell types, such as probiotic Lactobacillus acidophilus (Fig. S7, ESI†). The thick and durable CaP mineral shells noticeably protected the SCNEd cells from attacks by lytic enzymes (Fig. 2c). The cell density (OD600) of SCNEd S. cerevisiae or L. acidophilus was monitored alongside their naïve counterparts in solutions containing lyticase or lysozyme, respectively. SCNEd cells maintained high cell densities (S. cerevisiae: 97.5%, L. acidophilus: 82%), while only 30% of naïve cells remained after 3 h of reaction, indicating the enhanced resistance of SCNEd cells conferred by the thick CaP shells.
 | ||
| Fig. 2 (a) Live/dead assay: CLSM image and viability of S. cerevisiae before and after SCNE. FDA (green): live and PI (red): dead. Data are represented as mean ± SD (unpaired, two-sided Student's t-test; n = 3; n.s.: not significant). (b) (top) FE-SEM and (bottom) TEM images of naïve and SCNEd S.cerevisiae@CaP. (c) Time-dependent OD600 changes of naïve (black) and SCNEd cells (red) under lytic enzyme attack. (left) S. cerevisiae and (right) L. acidophilus. | ||
As a proof-of-concept for hierarchical protein embedment in CaP shells, two different BSA proteins, BSA-Alexa 488 (green) and BSA-Alexa 647 (red), were separately compartmentalized within the double-layered CaP shells on S. cerevisiae. The shell-forming process was repeated with BSA-adsorbed a-preCaP, producing S.cerevisiae@CaP[layer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1]/[layer2]. CLSM images showed distinct green and red layers surrounding S. cerevisiae in S.cerevisiae@CaP[BSA-Alexa 488]/[BSA-Alexa647] and S.cerevisiae@CaP[BSA-Alexa647]/[BSA-Alexa488] (Fig. 3a). Z-stack CLSM images of S.cerevisiae@CaP[BSA-Alexa488]/[BSA-Alexa647] captured the spatial arrangement of red and green layers, confirming the orderly compartmentalization of BSA-Alexa 488 and BSA-Alexa 647 within the double-layered CaP shells (Fig. 3b). Line profiles of the shells revealed a 200–300 nm separation between the green and red fluorescence peaks, further validating the compartmentalization of the proteins. UV-vis analysis determined incorporation efficiencies of 91.2% and 81.5% for the first and second layers, respectively. It is to note that the hierarchical compartmentalization of proteins within mineral-based nanoshells remains elusive. In addition to proteins, DNA was immobilized within the CaP shells, as evidenced by the red fluorescence of propidium iodine in CLSM images (Fig. S8, ESI†). Moreover, our protocol enabled the formation of multilayered shells, as demonstrated by the triple-layered CaP shell in S.cerevisiae@CaP[BSAAlexa647]/[BSAAlexa488]/[DNA-Hoechst] (Fig. S9, ESI†). These results underscored the potential of our system for embedding various functional entities within multilayered CaP shells in a hierarchical manner, benefitting the development of cascade reaction systems.8,32–34
1]/[layer2]. CLSM images showed distinct green and red layers surrounding S. cerevisiae in S.cerevisiae@CaP[BSA-Alexa 488]/[BSA-Alexa647] and S.cerevisiae@CaP[BSA-Alexa647]/[BSA-Alexa488] (Fig. 3a). Z-stack CLSM images of S.cerevisiae@CaP[BSA-Alexa488]/[BSA-Alexa647] captured the spatial arrangement of red and green layers, confirming the orderly compartmentalization of BSA-Alexa 488 and BSA-Alexa 647 within the double-layered CaP shells (Fig. 3b). Line profiles of the shells revealed a 200–300 nm separation between the green and red fluorescence peaks, further validating the compartmentalization of the proteins. UV-vis analysis determined incorporation efficiencies of 91.2% and 81.5% for the first and second layers, respectively. It is to note that the hierarchical compartmentalization of proteins within mineral-based nanoshells remains elusive. In addition to proteins, DNA was immobilized within the CaP shells, as evidenced by the red fluorescence of propidium iodine in CLSM images (Fig. S8, ESI†). Moreover, our protocol enabled the formation of multilayered shells, as demonstrated by the triple-layered CaP shell in S.cerevisiae@CaP[BSAAlexa647]/[BSAAlexa488]/[DNA-Hoechst] (Fig. S9, ESI†). These results underscored the potential of our system for embedding various functional entities within multilayered CaP shells in a hierarchical manner, benefitting the development of cascade reaction systems.8,32–34
 | ||
| Fig. 3 (a) CLSM images and line profiles of (left) S.cerevisiae@CaP[BSA-Alexa488]/[BSA-Alexa647] and (right) S.cerevisiae@CaP[BSA-Alexa647]/[BSA-Alexa488]. (b) Reconstructed 3D z-stacked CLSM images of S.cerevisiae@CaP[BSA-Alexa488]/[BSA-Alexa647]. Green: BSA-Alexa 488; red: BSA-Alexa 647. (c) (left) Schematic for cascade reactions of GOx and HRP. (right) Graph of reaction rates for different combination and order of GOx and HRP in S.cerevisiae@CaP shells. Data are represented as mean ± SD (unpaired, two-sided Student's t-test; n = 3; **p < 0.01, *p < 0.05). | ||
Two enzymes—glucose oxidase (GOx) and horseradish peroxidase (HRP)—were compartmentalized within double-layered CaP shells on S. cerevisiae cells, with the order and combination of enzymes varied in each layer. In this system, GOx catalyzes the glucose oxidation, generating hydrogen peroxide (H2O2) as a byproduct. HRP subsequently utilizes H2O2 to oxidize 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) as a probe into ABTS˙+, producing a characteristic dark green radical (λmax = 414 nm). As a negative control, naïve cells showed no significant change in ABTS˙+ absorbance over 4 h, confirming the lack of enzymatic activity (Fig. S10a, ESI†). In contrast, SCNEd cells functionalized with GOx and HRP demonstrated robust cascade reactions, evidenced by a marked increase in ABTS˙+ absorbance (Fig. S10b, ESI†). Notably, S.cerevisiae@CaP[HRP]/[GOx] achieved a 3.53-fold higher reaction rate compared with S.cerevisiae@CaP[GOx]/[HRP], highlighting the critical role of spatial enzyme arrangement within CaP multilayers for efficient substrate channeling (Fig. 3c).8,26 Furthermore, S.cerevisiae@CaP[HRP]/[GOx] showed a 1.52-fold rate increase compared with S.cerevisiae@CaP[GOx_HRP]/[GOx_HRP], where both enzymes were co-mixed in each layer. These results showed the potential of hierarchical enzyme compartmentalization within CaP shells to enhance the catalytic efficiency of multicompartmental systems by regulating the directional trafficking of substances and substrates. We also stored naïve and S.cerevisiae@CaP[GOx_HRP] cells at 4 °C for 7 days without any added nutrients, showing no noticeable decrease in cell viability compared with naïve cells, although the reaction rates decreased linearly over time, presumably due to leaching (Fig. S11, ESI†).
In summary, the a-preCaP strategy enables facile and ultrafast CaP-based nanoshell formation on various substrates, generating thick (∼400 nm) CaP nanoshells around tannic acid-primed substrates in under 5 s. Beyond cytocompatibility, a-preCaP could adsorb biomolecules, enabling spatially controlled compartmentalization of target molecules within the CaP nanoshells. We have demonstrated that the combination and arrangement of GOx and HRP within the double-layered shells significantly affect the enzymatic cascade performance, emphasizing the critical role of spatial organization in multi-enzyme systems. The multi-compartment, multifunctional artificial spores hold promise for advanced cell therapies, cell factories, cell microrobots, and targeted delivery systems.
This work was supported by the Basic Science Research Program through the National Research Foundation (NRF) of Korea (RS-2024-00335713), under the framework of the international cooperation program managed by the NRF of Korea (2020K2A9A2A12000250), and the Swedish Foundation for International Cooperation in Research and Higher Education (STINT MG 2019-8464).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. T. McKenney, A. Driks and P. Eichenberger, Nat. Rev. Microbiol., 2013, 11, 33–44 CrossRef CAS PubMed.
- P. Setlow, Microbiol. Spectrum, 2014, 2, 5 CrossRef PubMed.
- S. H. Yang, D. Hong, J. Lee, E. H. Ko and I. S. Choi, Small, 2013, 9, 178–186 CrossRef CAS PubMed.
- D. Hong, M. Park, S. H. Yang, J. Lee, Y.-G. Kim and I. S. Choi, Trends Biotechnol., 2013, 31, 442–447 CrossRef CAS PubMed.
- J. H. Park, S. H. Yang, J. Lee, E. H. Ko, D. Hong and I. S. Choi, Adv. Mater., 2014, 26, 2001–2010 CrossRef CAS PubMed.
- J. H. Park, D. Hong, J. Lee and I. S. Choi, Acc. Chem. Res., 2016, 49, 792–800 CrossRef CAS PubMed.
- J. Oh, N. Kumari, D. Kim, A. Kumar and I. S. Lee, Nat. Commun., 2024, 15, 5773 CrossRef CAS PubMed.
- H. Lee, J. Park, N. Kim, W. Youn, G. Yun, S. Y. Han, D. T. Nguyen and I. S. Choi, Adv. Mater., 2022, 34, 2201247 CrossRef CAS PubMed.
- W. Youn, J. Y. Kim, J. Park, N. Kim, H. Choi, H. Cho and I. S. Choi, Adv. Mater., 2020, 32, 1907001 CrossRef CAS PubMed.
- C. Li, M. Feng, B. Li, X. Feng, Y. Zhang and B. Wang, ACS Nano, 2025, 19, 2890–2899 CrossRef CAS PubMed.
- A. Belluati, D. Happel, M. Erbe, N. Kirchner, A. Szelwicka, A. Bloch, V. Berner, A. Christmann, B. Hertel, R. Pardehkhorram, A. Reyhani, H. Kolmar and N. Bruns, Nanoscale, 2023, 15, 19486–19492 Search PubMed.
- M. M. Maciel, T. R. Correia, V. M. Gaspar, J. M. M. Rodrigues, I. S. Choi and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2009619 Search PubMed.
- J. Lee, J. Choi, J. H. Park, M.-H. Kim, D. Hong, H. Cho, S. H. Yang and I. S. Choi, Angew. Chem., Int. Ed., 2014, 53, 8056–8059 Search PubMed.
- S. H. Yang, E. H. Ko, Y. H. Jung and I. S. Choi, Angew. Chem., Int. Ed., 2011, 50, 6115–6118 Search PubMed.
- S. H. Yang, K.-B. Lee, B. Kong, J.-H. Kim, H.-S. Kim and I. S. Choi, Angew. Chem., Int. Ed., 2009, 48, 9160–9163 Search PubMed.
- R. F. Fakhrullin and R. T. Minullina, Langmuir, 2009, 25, 6617–6621 CrossRef CAS PubMed.
- Z. Geng, X. Wang, F. Wu, Z. Cao and J. Liu, Sci. Adv., 2023, 9, eade0997 Search PubMed.
- E. H. Ko, Y. Yoon, J. H. Park, S. H. Yang, D. Hong, K.-B. Lee, H. K. Shon, T. G. Lee and I. S. Choi, Angew. Chem., Int. Ed., 2013, 52, 12279–12282 CrossRef CAS PubMed.
- W. Youn, E. H. Ko, M.-H. Kim, M. Park, D. Hong, G. A. Seisenbaeva, V. G. Kessler and I. S. Choi, Angew. Chem., Int. Ed., 2017, 56, 10702–10706 Search PubMed.
- B. Wang, P. Liu, W. Jiang, H. Pan, X. Xu and R. Tang, Angew. Chem., Int. Ed., 2008, 120, 3616–3620 Search PubMed.
- X. Yan, Q. Zhang, X. Ma, Y. Zhong, H. Tang and S. Mai, Jpn. Dent. Sci. Rev., 2023, 59, 181–190 CrossRef PubMed.
- H. L. Kim, Y. S. Shin and S. H. Yang, CrystEngComm, 2022, 24, 1344–1351 RSC.
- Z. Liu, C. Shao, B. Jin, Z. Zhang, Y. Zhao, X. Xu and R. Tang, Nature, 2019, 574, 394–398 Search PubMed.
- X. Zhan, Z. Wen, X. Chen, Q. Lei, Y. Chen, L. Zhou, G. Zheng, F. Kong, J. Guo, Y. Duan, Y. Lai, P. Yin, C. J. Brinker, H. Chen and W. Zhu, Cell Rep. Phys. Sci., 2022, 3, 101103 Search PubMed.
- M. Xu, F. Ji, Z. Qin, D. Dong, X. Tian, R. Niu, D. Sun, F. Yao and J. Li, CrystEngComm, 2018, 20, 2374–2383 RSC.
- S. Yang, W. Youn, H. B. Rheem, S. Y. Han, N. Kim, S. Han, P. Schattling, B. Städler and I. S. Choi, Angew. Chem., Int. Ed., 2025, 64, e202415823 Search PubMed.
- D. Gebauer, A. Völkel and H. Cölfen, Science, 2008, 322, 1819–1822 CrossRef CAS PubMed.
- A. Dey, P. H. H. Bomans, F. A. Müller, J. Will, P. M. Frederik, G. de With and N. A. J. M. Sommerdijk, Nat. Mater., 2010, 9, 1010–1014 Search PubMed.
- W. J. E. M. Habraken, J. Tao, L. J. Brylka, H. Friedrich, L. Bertinetti, A. S. Schenk, A. Verch, V. Dmitrovic, P. H. H. Bomans, P. M. Frederik, J. Laven, P. van der Schoot, B. Aichmayer, G. de With, J. J. DeYoreo and N. A. J. M. Sommerdijk, Nat. Commun., 2013, 4, 1507 Search PubMed.
- E. Turhan, I. Goldberga, C. Pötzl, W. Keil, J.-M. Guigner, M. F. T. Haßler, H. Peterlik, T. Azaïs and D. Kurzbach, J. Am. Chem. Soc., 2024, 146, 25614–25624 CrossRef CAS PubMed.
- S.-Y. Jung, H. Hwang, H.-S. Jo, S. Choi, H.-J. Kim, S.-E. Kim and K. Park, Int. J. Mol. Sci., 2021, 22, 4614 Search PubMed.
- S. Y. Han, N. Kim, G. Yun, H. Lee and I. S. Choi, Nat. Commun., 2023, 14, 6828 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, Y. Su, P. Ouyang, J. Ge and Z. Liu, Chem. Commun., 2014, 50, 12465–12468 Search PubMed.
- H. Xu, Z. Liu and H. Liang, Adv. Compos. Mater., 2024, 7, 246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00522a |
| This journal is © The Royal Society of Chemistry 2025 |