
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the role of superoxide radicals in CdS quantum dot instability†
Miaoli
Gu
a,
Xianglin
Xiang
 a,
Bei
Cheng
a,
Jiaguo
Yu
*b and
Liuyang
Zhang
*b
a,
Bei
Cheng
a,
Jiaguo
Yu
*b and
Liuyang
Zhang
*b
aState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, Wuhan 430070, P. R. China
bLaboratory of Solar Fuel, Faculty of Materials Science and Chemistry, China University of Geosciences, 68 Jincheng Street, Wuhan 430078, P. R. China. E-mail: yujiaguo93@cug.edu.cn; zhangliuyang@cug.edu.cn
First published on 21st March 2025
Abstract
Photocorrosion hinders CdS development, this study shows that after 2 hours of irradiation in oxygen, H2O2 yield of CdS reached 1.4 mmol g−1, while it was nearly undetectable under nitrogen. Capture experiments and electron paramagnetic resonance confirm that O2 acts as an electron acceptor, forming radicals that oxidize and deactivate CdS.
With the rapid development of nanotechnology, quantum dots (QDs), as an important nanomaterial in photocatalytic energy systems, demonstrate significant value in clean energy production, environmental remediation, and sustainable development.1 At the heart of these systems lies the effective capture and conversion of solar energy into chemical or electrical energy, enabling a wide spectrum of applications, including water splitting for hydrogen production, carbon dioxide reduction, and the degradation of organic pollutants.2 Among various semiconductor nanomaterials, sulfide-based photocatalysts, such as CdS, have emerged as promising candidates for solar-driven hydrogen production. The narrow bandgap (∼2.4 eV) of CdS facilitates efficient solar energy absorption, while its low work function enhances the transfer of photogenerated electrons. These properties result in high quantum efficiency and relatively stable photocatalytic performance, making CdS and similar sulfide semiconductors prime candidates for photocatalytic hydrogen evolution.3 However, despite these advantages, achieving the goal of overall water splitting—simultaneous production of both hydrogen and oxygen—remains a significant challenge. One of the key obstacles is the susceptibility of sulfide catalysts to photodegradation, which severely impacts their long-term stability and performance.4
Most literature reports suggest that photodegradation in sulfides is predominantly driven by photogenerated holes (h+), which participate in oxidation reactions with sulfide ions (S2−) under light exposure. In CdS, this interaction leads to the formation of by-products like sulfate (SO42−) or sulfur (S0),5 which disrupt the crystalline integrity of the material. As a result, active sites are gradually lost, leading to a marked decrease in efficiency. Additionally, these oxidation by-products can form a passivating layer on the catalyst surface, impeding charge transfer and accelerating catalyst deactivation. These degradation pathways not only diminish the catalyst's catalytic efficiency but also substantially reduce its operational lifespan, making long-term stability a critical challenge for practical applications.
Despite extensive research on the photocatalytic performance of CdS, a key gap remains in understanding how the corrosion of CdS-based catalysts under varying atmospheric conditions impacts their degradation mechanisms and long-term stability. While most studies focus on performance metrics, few address the impact of corrosion on solution properties and structural changes over time. Additionally, the role of oxygen in exacerbating these effects remains unclear. This underscores the need for further investigation into how CdS corrosion contributes to functional deterioration and material segregation in different environments.
We observed that photodegradation of CdS quantum dots modified with mercaptopropionic acid (CdS-MPA QDs) in oxygen-rich conditions caused a significant change in the solution, transitioning from a transparent state to visible aggregation and phase separation. This segregation, which has not been previously reported, indicates severe structural failure of the CdS material, providing direct evidence that corrosion not only reduces photocatalytic efficiency but also triggers physical segregation, potentially accelerating catalyst deactivation.
To better understand the photodegradation mechanisms underlying the instability of sulfide-based photocatalysts, water-soluble CdS QDs were synthesized as a prototype using solvothermal and ligand exchange methods, followed by a series of comparative experiments.6 A particularly striking observation was that photodegradation of CdS occurred more rapidly in oxygen-rich environments. Under oxygenated conditions, the catalyst displayed pronounced instability, with its activity diminishing over a short period. This observation suggests that oxygen plays a pivotal role in accelerating surface oxidation reactions, thereby enhancing the interaction between photogenerated reactive species and the sulfide material, leading to faster structural deterioration.
Further structural characterization using transmission electron microscopy (TEM) revealed a well-defined nanoparticle morphology for CdS-MPA QDs (Fig. 1a), with a size distribution ranging from 3 to 5 nm. High-resolution TEM imaging (Fig. 1b) confirmed the highly crystalline nature of CdS-MPA QDs, with a lattice spacing of 3.32 Å, corresponding to the (111) facet of CdS-MPA QDs.5d,7
 | ||
| Fig. 1 (a) Low and (b) high magnification TEM images of CdS-MPA QDs, the inset in (b) shows the lattice fringe. | ||
To investigate the role of atmospheric conditions on the stability of CdS-MPA QDs under light irradiation, we conducted a series of experiments using a 5 W LED light source (wavelength range: 400–800 nm) in different atmospheric environments (Fig. S1, ESI†). Under nitrogen conditions, the CdS-MPA QDs catalyst exhibited remarkable stability, with no significant structural or color changes observed after 5 hours of irradiation. The solution remained highly transparent (as shown in Fig. S1a, ESI†), and the absorption spectrum showed negligible alteration (Fig. 2a), indicating that CdS-MPA QDs maintain their stability in the absence of oxygen. Furthermore, no H2O2 was detected (Fig. 2b).
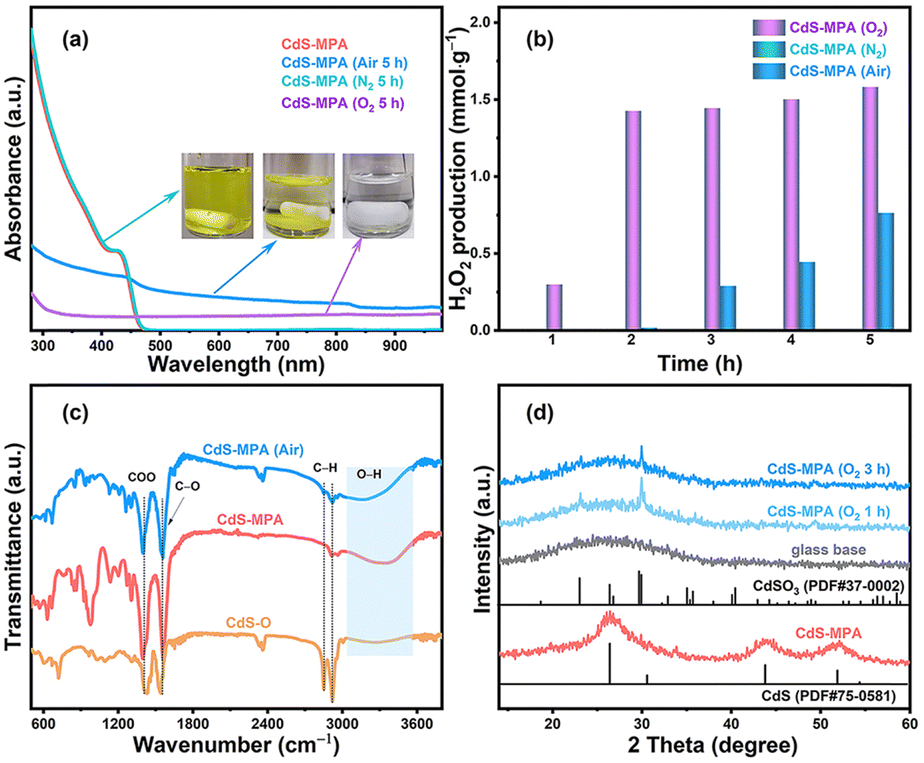 | ||
| Fig. 2 (a) Ultraviolet-visible (UV-vis) spectra of CdS-MPA QDs samples under different atmospheric conditions. The illustration shows changes in the samples after illumination. (b) Photocatalytic production of H2O2 by CdS-MPA QDs in aqueous solutions, (c) FTIR spectra of CdS-O, CdS-MPA, and illuminated CdS-MPA samples under air, and (d) XRD patterns of CdS-MPA QDs samples after different illumination durations in oxygen. | ||
In the air environment, the CdS-MPA QDs solution exhibited minimal change during the initial stage of irradiation (within 1 hour, Fig. S1b, ESI†), and the H2O2 yield was close to zero (Fig. 2b). However, after 2 hours of irradiation, noticeable aggregation of CdS-MPA QDs occurred, and H2O2 concentration increased to approximately 0.02 mmol g−1. As irradiation was extended to 5 hours, the H2O2 yield further increased to 0.8 mmol g−1, and the absorbance of the solution gradually decreased (Fig. 2a). This change indicates that the presence of oxygen significantly accelerates both the aggregation and oxidation of CdS-MPA QDs, resulting in a decline in the stability of the catalyst. Fourier transform infrared (FTIR) spectroscopy results (Fig. 2c) further confirmed these changes, with absorption peaks observed at 2850 and 2921 cm−1, attributed to C–H vibrations in the CdS-O QDs (oil-soluble CdS quantum dots) bond.8 Additionally, the broad O–H peak at 3400 cm−1, typically associated with the vibration of MPA's hydroxyl group,9 showed a decrease in intensity, manifesting that the hydroxyl (O–H) groups of MPA had undergone some changes during irradiation, potentially contributing to the aggregation of the QDs.
Under oxygen-rich conditions, CdS-MPA QDs exhibited more pronounced photocorrosion (Fig. S1c, ESI†). After 1 h of irradiation, the solution became stratified, with the H2O2 concentration reaching 0.3 mmol g−1 (Fig. 2b). By 5 hours of irradiation, the solution gradually faded to colorless, the absorbance dropped significantly (Fig. 2a), and the H2O2 yield stabilized at about 1.6 mmol g−1. This phenomenon indicates that in an oxygen-rich environment, oxygen accelerates the corrosion of CdS-MPA QDs, leading to notable changes in the catalyst structure. Based on the above experimental results, it can be inferred that O2 acts as an electron acceptor, capturing photogenerated electrons to form superoxide radicals (⋅O2−). The superoxide radicals then undergo a two-step electron transfer to generate H2O2.10 These reactive oxygen species (⋅O2−/H2O2) further react with the surface of CdS, initiating a spontaneous oxidation process that gradually degrades the crystal structure of CdS-MPA QDs, causing the loss of active sites and accelerating the deactivation of the catalyst. Besides, the X-ray diffraction (XRD) pattern shows that the synthesized CdS-MPA QDs gradually oxidize into sulfide-oxide in oxygen environment (Fig. 2d).11
To further investigate the role of H2O2 concentration in the corrosion of CdS-MPA QDs, comparative experiments were conducted under varying concentrations of H2O2 in a nitrogen atmosphere (Fig. S3, ESI†). After 2 hours of irradiation, at a H2O2 concentration of 1.6 mM, minimal changes in the solution were observed. However, when the concentration was increased to 3.2 mM, significant changes in the catalyst structure occurred. Under nitrogen conditions, the lower concentration of H2O2 was insufficient to generate reactive free radicals (e.g., ⋅O2−), thus reducing the oxidation reaction and bond cleavage. This is likely due to the protective role of surface ligands or functional groups on the CdS-MPA QDs, which remain relatively intact at lower H2O2 concentrations, ensuring structural stability. However, at high H2O2 concentrations (3.2 mM), light exposure causes H2O2 to react with photogenerated electron–hole pairs (H2O2 + e− + H+ → ⋅OH + H2O, H2O2 + h+ → ⋅O2− + 2H+),12 producing strong oxidizing agents (such as ⋅O2−, ⋅OH, etc.) that can attack the CdS-MPA surface. This leads to surface oxidation, structural damage, and bond dissociation. The formation of oxidation products alters the surface properties of the CdS-MPA QDs, causing them to lose their inherent hydrophilicity, ultimately leading to aggregation.
To identify the key reactive species responsible for the corrosion of CdS-MPA QDs, we conducted an additional experiment using a H2O2 concentration of 1.6 mM, which corresponds to the H2O2 concentration generated in situ by the CdS-MPA QDs after 5 hours of irradiation under oxygen-rich conditions. Under these conditions, only slight changes in the QD structure were observed. A control experiment was performed by introducing K2Cr2O7 where all conditions remained the same except no H2O2 was added. In this case, no significant structural changes were observed (Fig. S4b, ESI†), suggesting that in the absence of H2O2, photogenerated holes (h+) did not significantly interact with the CdS-MPA QDs. This was confirmed by the addition of an electron scavenger (K2Cr2O7), which effectively prevented the formation of superoxide radicals and preserved the catalyst structure. However, when H2O2 and an electron scavenger were present, aggregation of the catalyst was observed after 1 hour of irradiation (Fig. S4c, ESI†). This indicates that after the electrons are captured, a large number of photogenerated holes can react with H2O2 to produce superoxide radicals (⋅O2−), which have a destructive effect on the structure of CdS-MPA QDs. Furthermore, when the electron scavenger was replaced with a hole or hydroxyl radical scavenger in the experiment, no significant changes in the solution were observed after 2 hours of irradiation (Fig. S5, ESI†), indicating that the hydroxyl radicals were not the primary active species responsible for CdS-MPA QDs corrosion.
 | ||
| Fig. 3 EPR spectra of CdS-MPA QDs for (a) ⋅O2− in methanol and (b) ⋅OH in water. | ||
Moreover, to investigate the key role of ⋅O2− radicals in the corrosion process of CdS-MPA QDs further, electron paramagnetic resonance (EPR) testing was conducted at room temperature (Fig. 3). The analysis results disclosed that, under air conditions (Fig. 3a), the signal for ⋅O2− was prominent, with a stronger intensity in O2 (Fig. S6a, ESI†), while the signal for ⋅OH was weak.10f,13 Under nitrogen, only a weak ⋅OH signal was detected. Interestingly, a strong quadruple peak signal was observed in methanol (Fig. 3a), which is attributed to the signal of MPA itself (Fig. S6b, ESI†). These signals do not affect the corrosion process under nitrogen conditions. Subsequently, when H2O2 was introduced under nitrogen conditions, and upon irradiation in the absence of a catalyst, both ⋅O2− and ⋅OH signals were detected (Fig. S7a, ESI†). Upon irradiation in the presence of CdS-MPA QDs, a stronger ⋅O2− radical signal was observed (Fig. S7b, ESI†),10f while ⋅OH radical signal intensity was consistent with that in pure water. Therefore, based on the results of the capture experiment, it can be concluded that ⋅O2− radicals are the primary cause of the CdS-MPA QDs corrosion, while ⋅OH radicals contribute minimally to the degradation.
To confirm these findings, X-ray photoelectron spectroscopy (XPS) was used to analyze the surface elemental composition and chemical valence states of the CdS-MPA QDs after irradiation. The wide-scan XPS spectrum (Fig. 4a) detected typical signals for Cd, S, C, and O.6,14 For CdS-O QDs, the signals of Cd and C were the strongest, while those of O and S were relatively weak, with C and O primarily originating from surface ligands (e.g., oleylamine or oleic acid). In the CdS-MPA QDs, the C content increased due to the introduction of MPA, while the Cd and S content were lower due to the shielding effect of organic surface groups.
 | ||
| Fig. 4 (a) The XPS spectra, (b) Cd 3d, (c) S 2p, and (d) O 1s spectra for CdS-O, CdS-MPA, CdS-MPA (air) and CdS-MPA (O2). | ||
Interestingly, after exposing CdS-MPA QDs to air and irradiating for 2 hours, the signal of both Cd and S intensify, suggesting that the hydrophilic MPA groups may have been oxidized and removed from the CdS QDs surface, leading to changes to the surface structure and a decrease in stability. These alterations likely contributed to the aggregation of the catalyst. Further analysis revealed additional Cd peaks at 406.1 eV and 412.9 eV (Fig. 4b), indicating the presence of Cd in different chemical environments, possibly due to the partial transformation of CdS into CdSO3 or CdSO4. After prolonged irradiation in an oxygen-rich environment, Cd 3d signals shifted to 405.8 eV and 412.5 eV, corresponding to the binding energies of Cd 3d5/2 and Cd 3d3/2, respectively. The S signal at 159.7 eV completely disappeared, replaced by a high-valence S signal at 168.2 eV, indicative of the oxidation of S2− to a higher oxidation state (such as sulfates and sulfites).5d,15 These shifts in binding energy indicate a transition on the CdS surface from sulfide to oxidized sulfur, with the resulting oxidation products disrupting the electronic structure and reactivity of the surface, thereby degrading photocatalytic performance. This further corroborates the role of reactive oxygen species in the photo-corrosion process.
In summary, this study reveals that O2 acts as a critical electron acceptor in the corrosion of CdS-MPA QDs, generating superoxide radicals that significantly degrade the material. These findings enhance our understanding of the photo-corrosion mechanisms in CdS-based photocatalysts and underscore the role of radical-mediated corrosion in limiting performance. By elucidating these mechanisms, this research provides a foundation for designing more durable and efficient photocatalysts, addressing photo-corrosion, and enabling the development of catalysts with improved stability and longer lifespans.
M. Gu: methodology, investigation, data curation, writing – original draft, visualization. X. Xiang: methodology. B. Cheng, J. Yu and L. Zhang: conceptualization, resources, writing – review & editing, supervision, funding acquisition.
The authors acknowledge the financial support from the National Key Research and Development Program of China (2022YFB3803600); the National Natural Science Foundation of China (52322214, 22278383, 22261142666, 22361132529 and 22361142704); the National Science Foundation of Hubei Province of China (2022CFA001 and 2023AFA088) and Key R&D Program Projects in Hubei Province (2023BAB113).
Data availability
The data supporting this article are included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) B. Zhu, C. Jiang, J. Xu, Z. Zhang, J. Fu and J. Yu, Mater. Today, 2025, 82, 251–273 CAS; (b) X. Xiang, L. Wang, J. Zhang, B. Cheng, J. Yu and W. Macyk, Adv. Photonics Res., 2022, 3, 2200065 Search PubMed; (c) Y. Yang, J. Wu, B. Cheng, L. Zhang, A. A. Al-Ghamdi, S. Wageh and Y. Li, Chin. J. Struct. Chem., 2022, 41, 2206006 CAS.
- (a) C. Bie, B. Zhu, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2019, 31, 1902868 CAS; (b) A. Agosti, Y. Nakibli, L. Amirav and G. Bergamini, Nano Energy, 2020, 70, 104510 Search PubMed; (c) A. Gautam, S. Sk, A. Tiwari, B. M. Abraham, V. Perupogu and U. Pal, Chem. Commun., 2021, 57, 8660–8663 Search PubMed; (d) B. Zhu, J. Liu, J. Sun, F. Xie, H. Tan, B. Cheng and J. Zhang, J. Mater. Sci. Technol., 2023, 162, 90–98 CAS; (e) R. Liu, Y. Chen, H. Yu, M. Položij, Y. Guo, T. C. Sum, T. Heine and D. Jiang, Nat. Catal., 2024, 7, 195–206 Search PubMed.
- (a) G. Han, Y. H. Jin, R. A. Burgess, N. E. Dickenson, X. M. Cao and Y. Sun, J. Am. Chem. Soc., 2017, 139, 15584–15587 CAS; (b) Q. Sun, N. Wang, J. Yu and J. C. Yu, Adv. Mater., 2018, 30, 1804368 Search PubMed; (c) D. Gogoi, A. K. Shah, P. Rambabu, M. Qureshi, A. K. Golder and N. R. Peela, ACS Appl. Mater. Interfaces, 2021, 13, 45475–45487 Search PubMed; (d) Y. Li, S. Wan, W. Liang, B. Cheng, W. Wang, Y. Xiang, J. Yu and S. Cao, Small, 2024, 20, 2312104 CAS; (e) W. Zhong, Y. Huang, X. Wang, J. Fan and H. Yu, Chem. Commun., 2020, 56, 9316–9319 RSC; (f) Z. Zhang, M. Wang, H. Zhou and F. Wang, J. Am. Chem. Soc., 2021, 143, 6533–6541 CrossRef CAS PubMed.
- C. M. Wolff, P. D. Frischmann, M. Schulze, B. J. Bohn, R. Wein, P. Livadas, M. T. Carlson, F. Jäckel, J. Feldmann, F. Würthner and J. K. Stolarczyk, Nat. Energy, 2018, 3, 862–869 CrossRef CAS.
- (a) S. Zhang, X. Ou, Q. Xiang, S. A. C. Carabineiro, J. Fan and K. Lv, Chemosphere, 2022, 303, 135085 CrossRef CAS PubMed; (b) C. Wang, L. Wang, J. Jin, J. Liu, Y. Li, M. Wu, L. Chen, B. Wang, X. Yang and B.-L. Su, Appl. Catal., B, 2016, 188, 351–359 CrossRef CAS; (c) F. Ma, Y. Wu, Y. Shao, Y. Zhong, J. Lv and X. Hao, Nano Energy, 2016, 27, 466–474 CrossRef CAS; (d) M. Wang, L. Cai, Y. Wang, F. Zhou, K. Xu, X. Tao and Y. Chai, J. Am. Chem. Soc., 2017, 139, 4144–4151 CAS; (e) B. Weng, M.-Y. Qi, C. Han, Z.-R. Tang and Y.-J. Xu, ACS Catal., 2019, 9, 4642–4687 CAS.
- X. Xiang, L. Zhang, C. Luo, J. Zhang, B. Cheng, G. Liang, Z. Zhang and J. Yu, Appl. Catal., B, 2024, 340, 123196 CAS.
- W. Zhong, X. Wu, P. Wang, J. Fan and H. Yu, ACS Sustainable Chem. Eng., 2019, 8, 543–551 Search PubMed.
- X. Huang, Y. Chen, X. Feng, X. Hu, Y. Zhang and L. Liu, J. Membr. Sci., 2020, 602, 117956 CAS.
- (a) X. Xiang, B. Zhu, J. Zhang, C. Jiang, T. Chen, H. Yu, J. Yu and L. Wang, Appl. Catal., B, 2023, 324, 122301 CAS; (b) M. N. Kalasad, M. K. Rabinal and B. G. Mulimani, Langmuir, 2009, 25, 12729–12735 CAS.
- (a) M. Sun, X. Wang, Y. Li, H. Pan, M. Murugananthan, Y. Han, J. Wu, M. Zhang, Y. Zhang and Z. Kang, ACS Catal., 2022, 12, 2138–2149 CAS; (b) B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, 2203225 CrossRef CAS PubMed; (c) K. Zhang, L. Tian, J. Yang, F. Wu, L. Wang, H. Tang and Z. Q. Liu, Angew. Chem., Int. Ed., 2023, 63, e202317816 CrossRef PubMed; (d) Y. Wu, Y. Yang, M. Gu, C. Bie, H. Tan, B. Cheng and J. Xu, Chin. J. Catal., 2023, 53, 123–133 CrossRef CAS; (e) L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS PubMed; (f) K. Meng, J. Zhang, B. Cheng, X. Ren, Z. Xia, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2024, 36, 2406460 CrossRef CAS PubMed.
- X. Xiang, B. Zhu, B. Cheng, J. Yu and H. Lv, Small, 2020, 16, 2001024 CrossRef CAS PubMed.
- (a) T. Liu, Z. Pan, J. J. M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu and K. Domen, Nat. Commun., 2022, 13, 1034 CrossRef CAS PubMed; (b) X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- (a) J. Cheng, W. Wang, J. Zhang, S. Wan, B. Cheng, J. Yu and S. Cao, Angew. Chem., Int. Ed., 2024, 63, e202406310 CrossRef CAS PubMed; (b) Y. Yang, B. Zhu, L. Wang, B. Cheng, L. Zhang and J. Yu, Appl. Catal., B, 2022, 317, 121788 CrossRef CAS; (c) L. Xu, Y. Liu, L. Li, Z. Hu and J. C. Yu, ACS Catal., 2021, 11, 14480–14488 CrossRef CAS.
- (a) R.-B. Wei, Z.-L. Huang, G.-H. Gu, Z. Wang, L. Zeng, Y. Chen and Z.-Q. Liu, Appl. Catal., B, 2018, 231, 101–107 CrossRef CAS; (b) Y. Z. Lin, K. Wang, Y. Zhang, Y. C. Dou, Y. J. Yang, M. L. Xu, Y. Wang, F. T. Liu and K. Li, J. Mater. Sci. C, 2020, 8, 10071–10077 CAS.
- P. Cai, R. Momen, Y. Tian, L. Yang, K. Zou, A. Massoudi, W. Deng, H. Hou, G. Zou and X. Ji, Adv. Energy Mater., 2021, 12, 2103221 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00433k |
| This journal is © The Royal Society of Chemistry 2025 |