
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilisation of iron-oxo dimers in a natural layered clay for efficient photocatalysts comparable to TiO2†
Hamza
El-Hosainy‡
ab,
Ezz-Elregal M.
Ezz-Elregal‡
acd,
Shinichiro
Takano‡
e,
Akio
Iwanade
f,
Takumi
Miyakage
g,
Duotian
Chen
g,
Chenxi
He
g,
Takashi
Toyao
 g,
Ken-ichi
Shimizu
g,
Yusuke
Ide
*ac and
Tomohiko
Okada
*e
g,
Ken-ichi
Shimizu
g,
Yusuke
Ide
*ac and
Tomohiko
Okada
*e
aResearch Centre for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
bInstitute of Nanoscience and Nanotechnology, Kafrelsheikh University, Kafrelsheikh 33511, Egypt
cGraduate School of Engineering Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
dChemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo 11566, Egypt
eDepartment of Materials Chemistry, Faculty of Engineering, Shinshu University, 4-17-1, Wakasato, Nagano 380-8553, Japan
fResearch Network and Facility Services Division, National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
gInstitute for Catalysis Hokkaido University, N-21 W-10, Sapporo, Hokkaido 001-0021, Japan
First published on 5th March 2025
Abstract
We report the stabilisation of otherwise fleeting iron(III)-oxo dimers in the interlayer space of a natural layered clay. The material shows a good photocatalytic activity toward the oxidation of formic acid in water and formaldehyde in air comparable to a benchmark TiO2 photocatalyst.
Because solid photocatalysis is a (potentially) feasible means of tackling environmental and energy issues, research into a pioneering solid photocatalyst, TiO2, is still growing. In addition, the development of solid photocatalysts alternative to TiO2 has been burgeoning due to its cost-ineffectiveness, environmental and health concerns.1–3 An ultimate alternative to TiO2 is naturally-occurring photocatalysts;4 however, few reports have demonstrated natural materials showing photocatalytic activities higher than or comparable to that of TiO2.
Metal-(hydr)oxo-clusters are of great interest due to their unique structural, electronic and magnetic properties, which distinguish them from bulk metal (hydr)oxides.5–8 Some metal cations, such as Al3+, Ga3+ and U6+, and d0 transition ones, including V5+, Nb5+, Ta5+, Mo6+ and W6+, can form oxo-clusters that remain stable without the need for organic or inorganic ligands.9–12 In contrast, it is difficult for Fe3+ to form oxo-clusters. Fe3+-aqua complexes (e.g., [FeIII(H2O)6]3+, partly hydrolysed [FeIII(OH)(H2O)5]2+ and their dimers) are particularly prone to instability in water. [FeIII(H2O)6]3+ is stable only in highly acidic conditions, whereas it rapidly forms colored iron oxide crystals/particles through hydrolysis and condensation as the solution pH increases.13–16 To maintain their oligomeric structure or to form their clusters, Fe3+ requires stabilisation through organic or inorganic ligands but this limits their broader applicability.17–20
In our previous research, we successfully addressed this issue by using synthetic silica matrices. Ferric oxide nanoparticles were immobilised on the pore surface of a mesoporous silica SBA-15 (pore diameter of approx. 9 nm), after which Fe and O sites were etched to leave Fe3+-oxo dimers immobilised on the pore surface.13 The material absorbed UV light and was durable to work as a good UV-induced photocatalyst toward the synthesis of nylon precursors significantly more active than a benchmark TiO2 photocatalyst.13 On the other hand, similar-structure-Fe3+-oxo dimers were encapsulated within the microchannels (pore diameter of approx. 0.6 nm) of a microporous layered silicate.14,21–23 Because the dimer so fitted the micropore that the photocatalytic activity was suppressed due to the limited access of reactants, the material could be used as a UV absorber for sunscreen products.14 These results demonstrated the stabilising methodology of Fe3+-oxo clusters to control their properties while showing drawbacks for their practical applications, including the use of relatively high-cost silica matrices and requirement of organic solvents and complicated procedures for the synthesis. Here, we present a simple and organic solvent-free method for stabilising Fe3+-oxo dimers by utilising a natural layered clay matrix. We propose the local structure of the dimer via characterisations technique including X-ray absorption spectroscopy (XAS) and evaluate the photocatalytic performance of the material toward organic compound removal by comparing a benchmark TiO2 photocatalyst.
The intercalation of functional units (e.g., molecules, clusters and nanoparticles) in the interlayer space of layered solids like layered clays is a promising methodology for stabilising otherwise fleeting functional units and then functionalising them.24 In this study, as a silica matrix for Fe3+-oxo dimers, we used a natural sodium-type smectite layered clay, Kunipia F (named Na–Sm), purchased from Kunimine Industries, Japan. As shown in Fig. 1 (left), smectite is a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1-type phyllosilicate characterised by one sheet composed of two-dimensionally connected AlO6 octahedra (Al3+ is apparently substituted with Mg2+etc.) sandwiched by two sheets composed of two-dimensionally connected SiO4 tetrahedra (Si4+ is partially substituted with Al3+etc.). To compensate the negative charge due to the isomorphous (partial) substitution, Na+ is located in the interlayer space. Thus, by mixing a highly acidic aqueous solution containing [FeIII(H2O)6]3+ with Na–Sm, we expected that the interlayer Na+ would be replaced with [FeIII(H2O)6]3+ and/or the dimers without losing the layered framework, as reported for the stabilisation of Fe3+-oxo dimers via mixing the [FeIII(H2O)6]3+ solution with a microporous layered silicate.14 The natural smectite layered clay would be suitable because a synthetic smectite was readily decomposed in the acidic condition, due to its fine crystals. We prepared aqueous solutions of Fe(NO3)3 by dissolving controlled amounts of Fe(NO3)3·9H2O in water (5.5 mL) followed by adding HNO3 (1.5 mL) to adjust the solution pH (<0.13). To the solution, the Na–Sm powder (0.1 g) was added and the mixture was stirred at room temperature for 24 h. After the separation of the solid by vacuum filtration, the cake was dried in air for 6 days. The product was named xFe–Sm, where x is actual Fe content (wt%) per sample determined based on inductively coupled plasma optical emission spectroscopy analysis of the dissolved products (Table S1 in ESI†) and increased depending on the amount of the added Fe(NO3)3·9H2O (1.5, 2.4, 3.9 and 5.3 wt% for 50, 610, 1220 and 2440 mg, respectively).
:1-type phyllosilicate characterised by one sheet composed of two-dimensionally connected AlO6 octahedra (Al3+ is apparently substituted with Mg2+etc.) sandwiched by two sheets composed of two-dimensionally connected SiO4 tetrahedra (Si4+ is partially substituted with Al3+etc.). To compensate the negative charge due to the isomorphous (partial) substitution, Na+ is located in the interlayer space. Thus, by mixing a highly acidic aqueous solution containing [FeIII(H2O)6]3+ with Na–Sm, we expected that the interlayer Na+ would be replaced with [FeIII(H2O)6]3+ and/or the dimers without losing the layered framework, as reported for the stabilisation of Fe3+-oxo dimers via mixing the [FeIII(H2O)6]3+ solution with a microporous layered silicate.14 The natural smectite layered clay would be suitable because a synthetic smectite was readily decomposed in the acidic condition, due to its fine crystals. We prepared aqueous solutions of Fe(NO3)3 by dissolving controlled amounts of Fe(NO3)3·9H2O in water (5.5 mL) followed by adding HNO3 (1.5 mL) to adjust the solution pH (<0.13). To the solution, the Na–Sm powder (0.1 g) was added and the mixture was stirred at room temperature for 24 h. After the separation of the solid by vacuum filtration, the cake was dried in air for 6 days. The product was named xFe–Sm, where x is actual Fe content (wt%) per sample determined based on inductively coupled plasma optical emission spectroscopy analysis of the dissolved products (Table S1 in ESI†) and increased depending on the amount of the added Fe(NO3)3·9H2O (1.5, 2.4, 3.9 and 5.3 wt% for 50, 610, 1220 and 2440 mg, respectively).
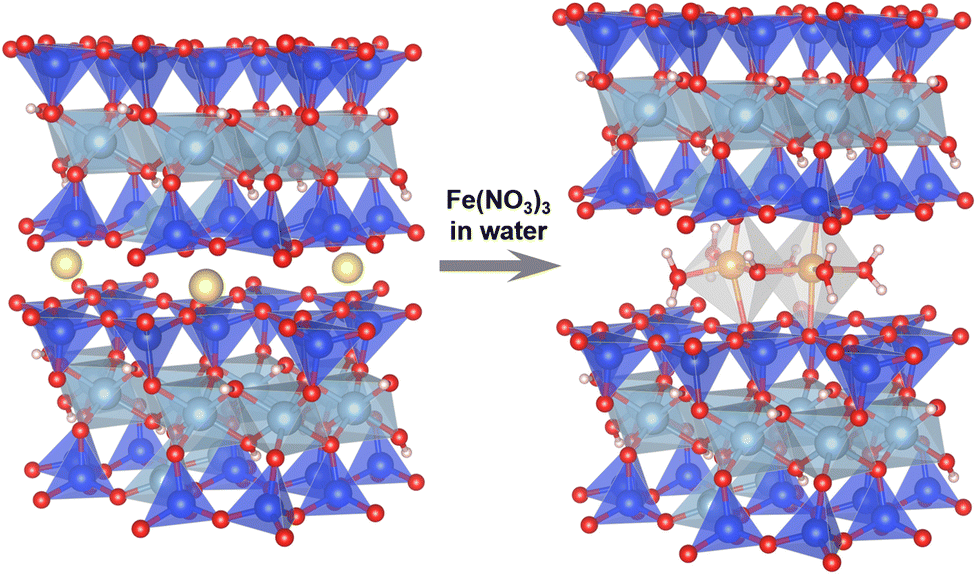 | ||
| Fig. 1 Schematic illustration for material design. Color code: yellow = Na, blue = Si, pale blue = Al, red = O, pink = H, brown = Fe. | ||
As shown in Table S1 (ESI†), the pristine Na–Sm has Na (2.8 wt%) and Fe (1.8 wt%) contents which come from the interlayer cations and framework ones, respectively. On the other hand, xFe–Sm samples have a negligible Na content while having Fe contents ranging from 1.5 to 5.3 wt%. Even after treating with highly acidic solutions, xFe–Sm samples preserved the original layer framework, which was confirmed by a negligible change in wide-angle powder X-ray diffraction (XRD) patterns (Fig. S1, ESI†) and specific surface areas (N2 BET from 5 to 3 m2 g−1, Fig. S2, ESI†). Considering that Na–Sm just treated with HNO3 (without adding any Fe(NO3)3·9H2O) has a negligible Na content but has the Fe content of 1.4 wt% that must come from the framework Fe species, the detected Fe contents for the xFe–Sm samples are thought to come from the intercalated Fe3+-oxo species. For lower Fe content-products like 1.5Fe–Sm, oxonium cations (H3O+) are thought to co-exist in order to compensate the negative layer charge.
The interlayer structure of the products was examined using small-angle XRD, as shown in Fig. 2a. The pristine Na–Sm had a diffraction peak corresponding to the basal spacing of 1.21 nm, reflecting hydrated Na cations in the interlayer space. 1.5Fe–Sm showed a diffraction peak corresponding to the basal spacing of 1.26 nm, which was slightly larger than that of Na–Sm. 2.4Fe–Sm and 3.9Fe–Sm had a broader diffraction peak composed of the diffraction peak seen in the XRD pattern of 1.5Fe–Sm and new one corresponding to the basal spacing of 1.53 nm. 5.3Fe–Sm had a relatively sharp diffraction peak corresponding to the basal spacing of 1.53 nm. These results suggest the intercalation of Fe species to form an homoionic intercalation compound with the interlayer space of 0.57 nm obtained by subtracting the thickness of a silicate layer (0.96 nm) from the basal spacing.
 | ||
| Fig. 2 (a) XRD patterns and (b) UV-vis spectra of Na–Sm and the products with different Fe contents. | ||
UV-vis spectroscopy was performed to further examine the kind of Fe species contained in xFe–Sm (Fig. 2b). The pristine Na–Sm showed little to no absorption in the UV region. 1.5Fe–Sm had an absorption band centered at around 260 nm. This spectral feature was in good agreement with that reported for mononuclear Fe3+ species in highly acidic solutions and assigned to ligand-to-metal charge transfer (LMCT) transitions between Fe3+ and oxo ligands.25,26 On the other hand, 2.4Fe–Sm, 3.9Fe–Sm and 5.3Fe–Sm, in addition to the absorption band centered at around 260 nm, had those centered at around 360 and 470 nm, which are assignable to LMCT and/or d–d transitions for dimeric Fe3+-oxo species.25–27 No spectral features typical for iron oxide crystals/particles, like absorption onsets from a wavelength of 500–600 nm, were detected. Considering the wide-angle XRD results, where weak diffraction peaks due to iron oxide are detected for 3.9Fe–Sm and 5.3Fe–Sm (Fig. S1, ESI†), 5.3Fe–Sm is thought to be mainly composed of intercalated dimeric Fe3+-oxo species.
To determine the local structure of 5.3Fe–Sm, we performed XAS and density functional theory (DFT) calculations, as shown in Fig. 3. Fig. 3a shows the Fe K-edge X-ray absorption near-edge structure (XANES) spectra of 5.3Fe–Sm and reference compounds. The absorption edge position of the spectrum of 5.3Fe–Sm is close to that of the spectra of α-Fe2O3 and α-FeOOH, which were used as references, indicating that the Fe in 5.3Fe–Sm exists as Fe3+. Fig. 3b shows the Fourier transforms of extended X-ray absorption fine structure (EXAFS) spectra. This result revealed that 5.3Fe–Sm shows only one major peak at ca. 1.8 Å (without phase shift correction), attributed to Fe–O features. Curve-fitting analysis of the EXAFS data, performed for the first coordination shell and summarised in Table S2 (ESI†), indicates the presence of three Fe–O contributions in the spectrum of 5.3Fe–Sm.
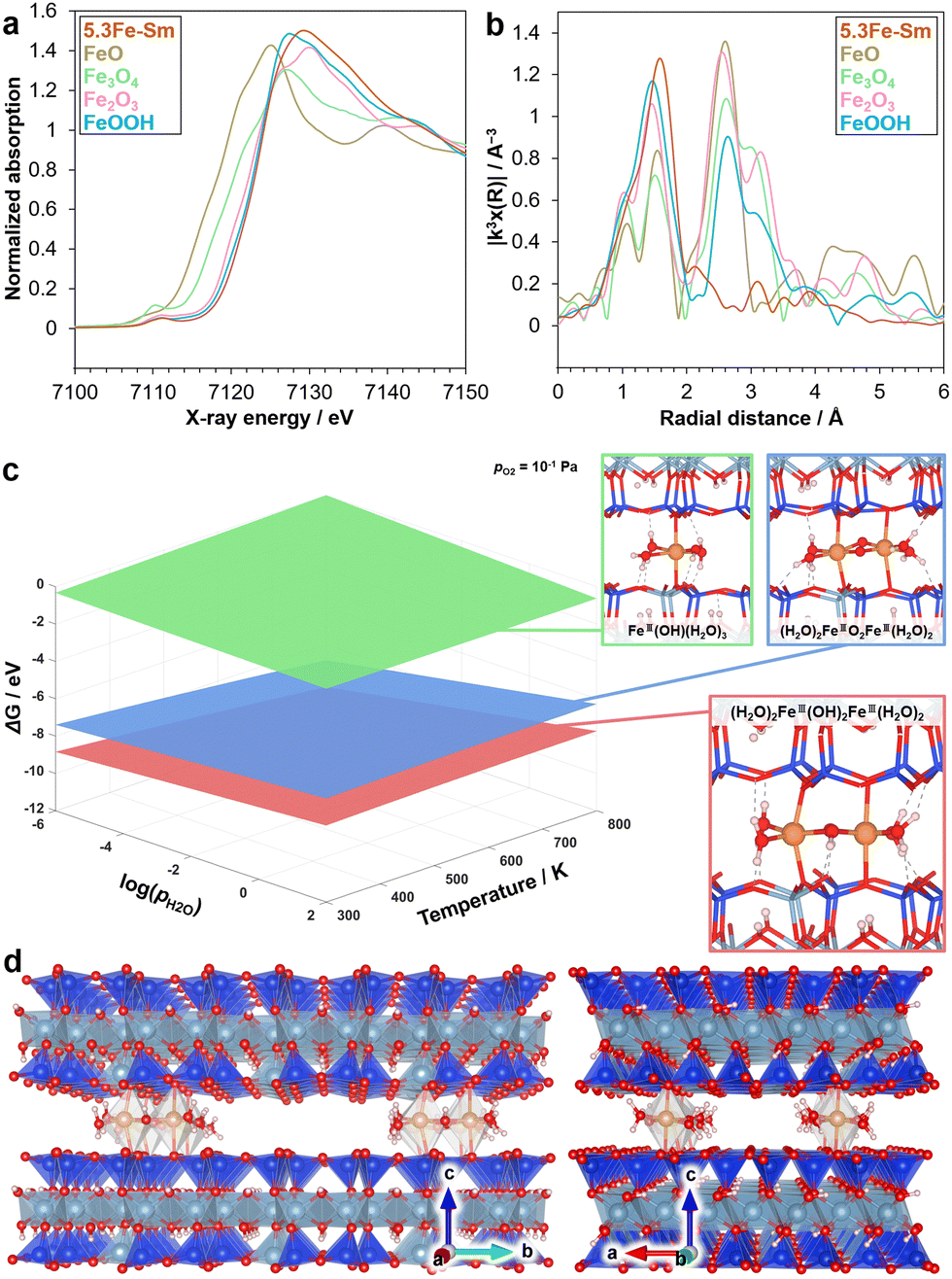 | ||
| Fig. 3 Experimental and theoretical structure analyses. (a) Fe K-edge XANES spectra and (b) Fourier transforms of EXAFS spectra of 5.3Fe–Sm and reference samples. (c) Phase diagram showing the species with the lowest relative Gibbs free energy values as a function of temperature and the partial pressure of H2O. (d) DFT-simulated structure of 5.3Fe–Sm seen from different angles. | ||
To further investigate the plausible structures of Fe species in the 5.3Fe–Sm sample under realistic experimental conditions, ab initio thermodynamic analysis was conducted based on spin-polarised DFT calculations on Fe species intercalated in the silicate layer. The oxidation state of Fe species was set to +3 to be consistent with the aforementioned experimental findings. The interlayer distance was also fixed to the experimentally obtained values from XRD. Fig. 3c shows the phase diagram of Fe species with the lowest relative Gibbs free energy as a function of temperature and partial pressure of H2O. The dimeric (H2O)2Fe3+(OH)2Fe3+(H2O)2 coordinated to oxygen of the silicate layers (Fig. 1 right and 3d) was identified to be the most stable. In contrast, another dimeric species, (H2O)2Fe3+O2Fe3+(H2O)2, and monomeric species, Fe3+OH(H2O)3 were less stable under the conditions considered. Note that other Fe species including self-standing ones without direct coordinations of Fe to the silicate oxygens were found to be unstable, likely due to the narrow interlayer space. These computational findings are consistent with experimental results, including XAS and UV-vis measurements.
We firstly evaluated the photocatalytic activity of xFe–Sm via a well-known reaction for testing the ability of photocatalysts toward organic compound removal, the oxidation of formic acid in water.28–30Fig. 4a shows the time-course CO2 evolution from water containing formic acid and xFe–Sm under solar simulator irradiation (1 SUN). The amount of CO2 evolved almost linearly increased with the increase of irradiation time, indicating the photocatalytic activity of xFe–Sm.28–30 Among all xFe–Sm samples, 5.3Fe–Sm and 3.9Fe–Sm showed the best activity, which was comparable to that of P25 TiO2, that is the highest-active TiO2 photocatalyst among commercially available TiO2 and thus has been used as benchmark TiO2.29
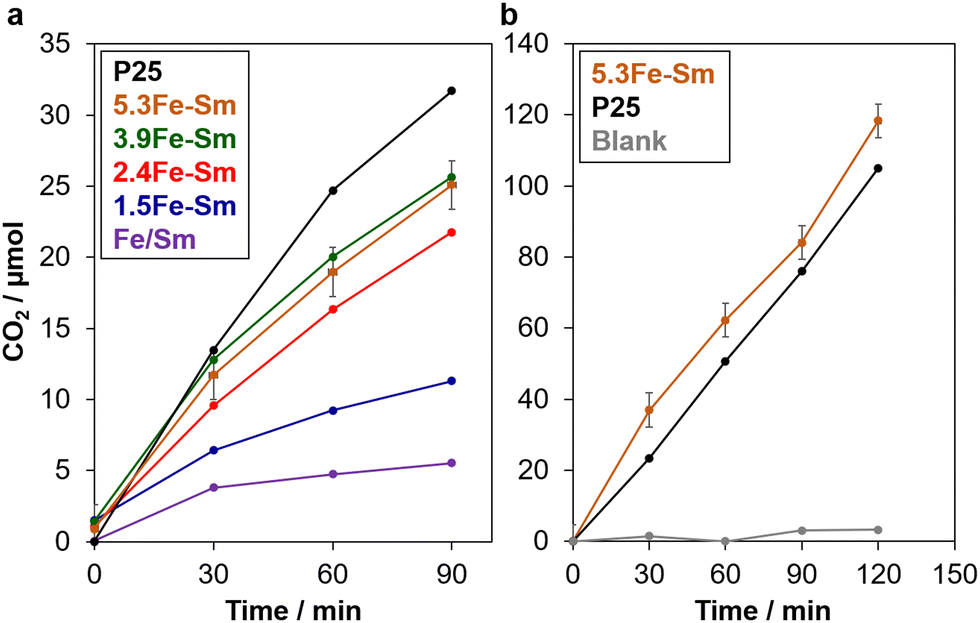 | ||
| Fig. 4 Time course of CO2 evolution on different materials from (a) formic acid in water and (b) formaldehyde in air under solar simulator irradiation. 5.3Fe–Sm samples synthesised in two different batches were tested to investigate the reproducibility of the material and performance. | ||
We then evaluated the photocatalytic ability of the 5.3Fe–Sm sample toward the oxidative decomposition of formaldehyde, a well-known cancer-causing substance in contaminated air. Before the test, we confirmed that formaldehyde gas/vapor gave a negligible amount of CO2 without any photocatalysts under 1 SUN-solar light irradiation, which is shown in Fig. 4b. However, with 5.3Fe–Sm, a significant amount of CO2 evolved linearly with the increase of irradiation time, indicating a good photocatalytic activity of 5.3Fe–Sm also toward this reaction. Remarkably, the photocatalytic activity of 5.3Fe–Sm was comparable to that of P25.
In our previous study,13 it was demonstrated that dimeric Fe3+-oxo species, in contrast to usual iron oxyhydroxide particles having narrower bandgap (little to no photocatalytic oxidation activity), had an expanded electronic structure capable of capturing photoexcited electrons with O2 to produce a relatively stable superoxide radical anion (O2−) and then retard electron–hole recombination. This mechanism can be applied to the present material showing a good photocatalytic oxidation ability comparable to TiO2.
Generally, (photo)catalysts based on layered materials, such as layered titanates and cluster-pillared layered clays, showed activities lower than the corresponding nanoparticulate (photo)catalysts (e.g., TiO2 and cluster-supported TiO2).31–37 The reason for the lower activity is explained by limited access of reactants to narrow interlayer spaces occupied by guest species. In contrast, as shown in Fig. 3d, 5.3Fe–Sm had larger void space in the interlayer space, which originates from the replacement of monovalent Na+ with trivalent Fe3+-oxo dimers (Fig. 1).
Remarkably, xFe–Sm showed a good photocatalytic activity considerably higher than a naturally occurring Fe-containing smectite clay (named Fe/Sm, purple plots in Fig. 4a), where 16.4 wt% (oxide base) of Fe in the framework is thought to be the active site for the photocatalysis.4 Turnover frequency (TOF) on 5.3Fe–Sm was calculated to be 0.06 min−1 by dividing the number of moles of evolved CO2 at 30 min by the number of moles of all Fe. This TOF value is higher than and comparable to those of Fe/Sm (0.004 min−1) and Fe3+-oxo dimers stabilised on the pore surface of SBA-15 (0.1 min−1),13 respectively, tested under identical conditions. These results suggest not only the importance of Fe3+-oxo species in the interlayer space of layered clays for better accessibility of reactants to the active sites but also the merit to use natural clays over synthetic silicas.
In conclusion, we have reported the intercalation of Fe3+-oxo dimers in a natural smectite clay via simple mixing of the clay powder and an aqueous solution containing Fe(NO3)3·9H2O. The obtained material showed a good photocatalytic activity toward the oxidation of formic acid in water and formaldehyde in air comparable to a benchmark TiO2 photocatalyst (P25). Considering the possible structural tunability of clusters in the interlayer space of the layered materials,24,33,34 the present findings hold potential for applications in environmental remediation and sustainable energy, encouraging further exploration of similar stabilisation strategies for various metal-oxo clusters.
This study was supported by the Joint Usage/Research Center for Catalysis. We also acknowledge JSPS KAKENHI (Grant-in-Aid for Scientific Research), Grant numbers of 21H02034 (Y.I.), 24K08057 (T.O.). The XAS measurements were conducted at the BL01B1 beamline of SPring-8 at JASRI (Proposal no. 2023A1931). DFT calculations were performed on supercomputers at ACCMS (Kyoto University). A part of this work was supported by ARIM of MEXT (JPMXP1223NM51).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Riebeling, A. Haase, T. Tralau and A. Luch, Nat. Food, 2020, 1, 523–525 CrossRef PubMed.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- M. D. Hernández-Alonso, F. Fresno, S. Suárez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- A. Phuekphong, T. Hayakawa and M. Ogawa, Chem. Commun., 2022, 58, 12661–12664 RSC.
- D. E. Salazar Marcano, N. D. Savić, K. Declerck, S. A. M. Abdelhameed and T. N. Parac-Vogt, Chem. Soc. Rev., 2024, 53, 84–136 RSC.
- D. Vendrame, G. Bragaggia, M. Carraro and S. Gross, Chem. Mater., 2024, 36, 9259–9278 CrossRef CAS.
- D. Yang, M. Babucci, W. H. Casey and B. C. Gates, ACS Cent. Sci., 2020, 6, 1523–1533 CrossRef CAS PubMed.
- N. Ogiwara and S. Uchida, Chem Catal., 2023, 3, 100607 CrossRef CAS.
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 RSC.
- B. Vlaisavljevich, L. Gagliardi and P. C. Burns, J. Am. Chem. Soc., 2010, 132, 14503–14508 CrossRef CAS PubMed.
- M. Nyman, M. A. Rodriguez and C. F. Campana, Inorg. Chem., 2010, 49, 7748–7755 CrossRef CAS PubMed.
- W. H. Casey, Chem. Rev., 2006, 106, 1–16 CrossRef CAS PubMed.
- Y. Ide, S. Tominaka, Y. Yoneno, K. Komaguchi, T. Takei, H. Nishida, N. Tsunoji, A. Machida and T. Sano, Chem. Sci., 2019, 10, 6604–6611 RSC.
- H. El-Hosainy, S. Mine, T. Toyao, K.-I. Shimizu, N. Tsunoji, M. Esmat, E. Doustkhah, M. El-Kemary and Y. Ide, Mater. Today Nano, 2022, 19, 100227 CrossRef CAS.
- R. Seidel, K. Kraffert, A. Kabelitz, M. N. Pohl, R. Kraehnert, F. Emmerling and B. Winter, Phys. Chem. Chem. Phys., 2017, 19, 32226–32234 RSC.
- S. De and B. Das, ChemPhysChem, 2024, 25, e202400144 CrossRef CAS PubMed.
- A. Kundu, L. Mallick, A. Rajput, Y. Kumar and B. Chakraborty, Mater. Today Chem., 2022, 25, 100927 CrossRef CAS.
- T. Yu, Z. Li, W. Jones, Y. Liu, Q. He, W. Song, P. Du, B. Yang, H. An, D. M. Farmer, C. Qiu, A. Wang, B. M. Weckhuysen, A. M. Beale and W. Luo, Chem. Sci., 2021, 12, 3152–3160 RSC.
- C. Castillo-Blas, I. Romero-Muñiz, A. Mavrandonakis, L. Simonelli and A. E. Platero-Prats, Chem. Commun., 2020, 56, 15615–15618 RSC.
- O. Sadeghi, L. N. Zakharov and M. Nyman, Science, 2015, 347, 1359–1362 CrossRef CAS PubMed.
- M. Esmat, H. El-Hosainy, M. Miyagawa, H. Takaba, N. Tsunoji, S. Ishihara and Y. Ide, ACS Appl. Mater. Interfaces, 2024, 16, 51046–51054 CrossRef CAS.
- Y. Ide, S. Tominaka, H. Kono, R. Ram, A. Machida and N. Tsunoji, Chem. Sci., 2018, 9, 8637–8643 RSC.
- E. Doustkhah and Y. Ide, ACS Appl. Nano Mater., 2019, 2, 7513–7520 CrossRef CAS.
- K. Saito, M. Morita, T. Okada, R. Wijitwongwan and M. Ogawa, Chem. Soc. Rev., 2024, 53, 10523–10574 RSC.
- L. Lopes, J. de Laat and B. Legube, Inorg. Chem., 2002, 41, 2505–2517 CrossRef CAS PubMed.
- C. A. Brown, G. J. Remar, R. L. Musselman and E. I. Solomon, Inorg. Chem., 1995, 34, 688–717 CrossRef CAS.
- J. Stahl and B. König, Green Chem., 2024, 26, 3058–3071 RSC.
- Y. Ide and K. Komaguchi, J. Mater. Chem. A, 2015, 3, 2541–2546 RSC.
- O.-O. Prieto-Mahaney, N. Murakami, R. Abe and B. Ohtani, Chem. Lett., 2009, 38, 238–239 CrossRef CAS.
- H. Kominami, A. Tanaka and K. Hashimoto, Chem. Commun., 2010, 46, 1287–1289 RSC.
- Y. Ide, Y. Nakasato and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 3601–3604 CrossRef CAS PubMed.
- Y. Ide, M. Matsuoka and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 16762–16764 CrossRef CAS PubMed.
- E. Doustkhah, S. Rostamnia, N. Tsunoji, J. Henzie, T. Takei, Y. Yamauchi and Y. Ide, Chem. Commun., 2018, 54, 4402–4405 RSC.
- D. Mani, N. Tsunoji, Y. Yumauchi, M. Arivanandhan, R. Jayavel and Y. Ide, J. Mater. Chem. A, 2018, 6, 5166–5171 RSC.
- R. Tahawy, E. Doustkhah, E.-S. A. Abdel-Aal, M. Esmat, F. E. Farghaly, H. El-Hosainy, N. Tsunoji, F. I. El-Hosiny, Y. Yamauchi, M. H. N. Assadi and Y. Ide, Appl. Catal., B, 2021, 286, 119854 CrossRef CAS.
- A. H. Zaki, N. Tsunoji and Y. Ide, ACS Sustainable Chem. Eng., 2023, 11, 2295–2302 CrossRef CAS.
- E. Doustkhah, N. Tsunoji, M. H. N. Assadi and Y. Ide, Adv. Mater. Interfaces, 2023, 10, 2202368 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00333d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |