
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ru(II)-catalyzed oxidative (4+3) C–H/C–H annulation of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones with allyl alcohol†
Tarun
Jangir‡
a,
Dhananjay S.
Nipate‡
a,
Krishnan
Rangan
 b and
Anil
Kumar
*a
b and
Anil
Kumar
*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Pilani Campus, Vidya Vihar, Pilani, Rajasthan, 333031, India. E-mail: anilkumar@pilani.bits-pilani.ac.in
bDepartment of Chemistry, Birla Institute of Technology and Science, Pilani, Hyderabad Campus, Jawahar Nagar, Kapra Mandal, Medchal District, Telangana 500078, India
First published on 13th March 2025
Abstract
A ruthenium(II)-catalyzed direct oxidative (4+3) C–H/C–H annulation of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones with allyl alcohol has been described. The developed method yielded the corresponding annulated products in moderate to good yields. In addition, based on the control experimental results, a possible reaction mechanism has been proposed for the (4+3) C–H/C–H annulation reaction.
Over the past few decades, transition metal-catalyzed C–H bond functionalization of inert C–H bonds has become an attractive and reliable alternative strategy to conventional cross-coupling methods in organic synthesis.1 This approach is step- and atom-economic, avoids pre-functionalization of C–H bonds, and reduces the generation of salt wastes. In particular, a great deal of effort has been devoted to the direct functionalization of C(sp2)–H bonds with various transition metal catalysts by utilizing different coupling partners.2 Among a large number of coupling partners such as alkynes, maleimides, alkenes, α-diazoketones, sulfonyl ylides, and strained rings, the use of allyl alcohols as coupling partners has attracted significant attention due to their relatively easy availability, unique reactivity, and stability.3 Several groups have reported transition metal-catalyzed (4+1),4 (4+2),5 (3+2),6 and (3+3)7-annulation reactions involving sequential C–H activation/intramolecular cyclization using allyl alcohol as a coupling partner for the synthesis of five- and six-membered rings. Synthesis of seven-membered ring motifs employing allyl alcohol as a coupling partner is limited and challenging. In this direction, a few groups have described Rh(III)-catalyzed (4+3) intermolecular C–H/N–H annulation to construct an azepine ring using allyl alcohol as a coupling partner (Scheme 1a).8
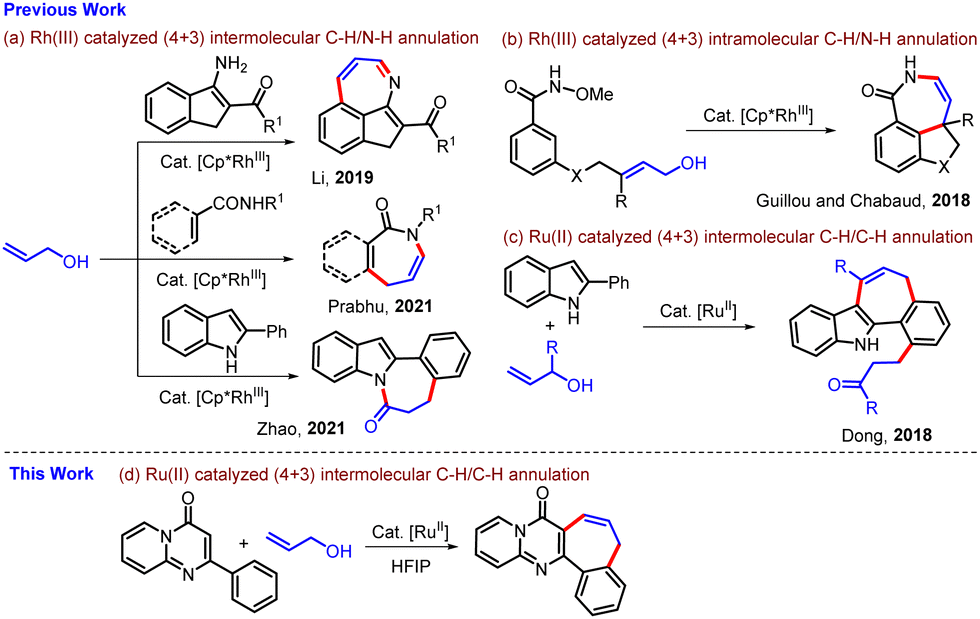 | ||
| Scheme 1 Transition metal-catalyzed (4+3) annulation using allyl alcohol leading to seven-membered rings. | ||
Li's group, in their work on Rh(III)-catalyzed reaction of arylimidates with allyl alcohols, observed that 3-aminoindenes, which were obtained as isolable intermediates on subsequent reaction with monosubstituted allyl alcohol, afforded densely fused azepines in moderate to good yields (Scheme 1a).8a The reaction involves multiple oxidative C–H Heck reactions using allylic alcohols. The Prabhu group8b synthesized benzazepinones and azepinones by the Rh(III)-catalyzed oxidative (4+3) C–H/N–H annulation of benzamides and cinnamamides with allyl alcohol while the Zhao group8c prepared indolo[2,1-a]benzazepinones by Rh(III)-catalyzed (4+3) oxidative C–H/N–H annulation of 2-arylindoles with allyl alcohols (Scheme 1a). Guillou and Chabaud reported an Rh(III)-catalyzed intramolecular (4+3) oxidative C–H/N–H annulation of benzamide tethered allylic alcohols to prepare a variety of tricyclic benzazepinones (Scheme 1b).8d Most of the reactions leading to seven-membered rings involving allyl alcohol are C–H/N–H annulation, and C–H/C–H annulation using allyl alcohol as a coupling partner is a rare and challenging task. In 2018, Dong and co-workers, in their work on Ru(II)-catalyzed coupling of allyl alcohols with indoles, showed a few examples of (4+3) C–H/C–H annulation reactions of allylic alcohols with 2-arylindoles leading to ortho-alkylated 5,12-dihydrobenzo[6,7]cyclohepta-[1,2-b]indoles in moderate yields (Scheme 1c).8e Learning from these examples and our continuing interest in transition metal-catalyzed functionalization of heterocycles,5b,9 herein, we report Ru(II)-catalyzed oxidative (4+3) C–H/C–H annulation of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones with allyl alcohol to synthesize benzo[6,7]cyclohepta[1,2-d]pyrido[1,2-a]pyrimidin-8(5H)-ones (Scheme 1d).
We started our investigation using 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-one (1a) and allyl alcohol (2a) as the starting materials. To our delight, a (4+3) C–H/C–H annulated product, benzo[6,7]cyclohepta[1,2-d]pyrido[1,2-a]pyrimidin-8(5H)-one (3aa), was obtained in 72% yield when 1a was treated with 2a in the presence of [Ru(p-cymene)Cl2]2 (5 mol%) as a catalyst and Cu(OAc)2·H2O (2 equiv.) as an oxidant in HFIP at 120 °C for 12 h (Table 1, entry 1). The replacement of HFIP by alternative solvents, such as TFE, DCE, DMF, 1,4-dioxane, and toluene (Table 1, entries 2–4; Table S1, ESI,† entries 8 and 9), did not furnish the desired product except for TFE, which afforded 3aa in 44% yield together with the ortho-C–H alkylated product (4aa) in 20% yield. In the case of DCE and 1,4-dioxane as the solvent, the (4+2) annulated product, 7-oxo-7H-benzo[h]pyrido[2,1-b]quinazoline-6-carbaldehyde, (5aa) was obtained as the major product together with ortho-C–H alkylated product 4aa. Results from solvent screening highlight the unique role of fluorinated alcohols (HFIP and TFE) for this annulative transformation. Lowering of the reaction temperature led to a decrease in the yield of 3aa and an increase in the yield of 4aa (Table 1, entries 5–7). We then scrutinized various other conditions to optimize the reaction conditions for this annulative reaction (see Table S1, ESI†). The best yield for 3aa was obtained in HFIP using [Ru(p-cymene)Cl2]2 as a catalyst and Cu(OAc)2·H2O (2 equiv.) as an oxidant (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) | ||
| 3aa | 4aa | 5aa | ||
| a Reaction conditions: 1a (0.22 mmol), 2a (0.45 mmol, 2.0 equiv.), catalyst (5 mol%), oxidant (2 equiv.), solvent (1.0 mL) in a sealed tube at 120 °C for 12 h. b Isolated yield. c 3-(2-(4-oxo-4H-pyrido[1,2-a]pyrimidin-2-yl)phenyl)acrylaldehyde (6aa) was also obtained in 23% yield. | ||||
| 1 | None | 72 | — | — |
| 2 | TFE was used as solvent | 44 | 20 | — |
| 3 | DCE was used as solvent | — | 14 | 32 |
| 4 | 1,4-Dioxane was used as solvent | — | 10 | 16 |
| 5 | Reaction at 100 °C | 58 | 12 | — |
| 6 | Reaction at 80 °C | 28 | 40 | — |
| 7 | Reaction at 60 °C for 24 h | — | 48c | — |
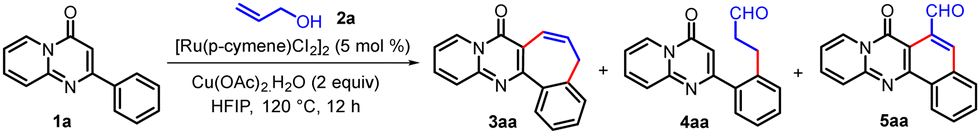
With the optimized reaction conditions in hand, the reactivity of different 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones was investigated (Table 2). A series of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones (1a–h) featuring both electron-donating and electron-withdrawing groups on the para-position of the C2-phenyl ring were converted to the corresponding products (3aa–ha) in moderate to good (54–74%) yields. 2-(3-Methoxyphenyl)-4H-pyrido[1,2-a]pyrimidin-4-one (1i) underwent the reaction to afford two regioisomeric products (3ia and 3i′a) in a 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This may be due to the coordination of the –OMe group with Ru metal in the catalytic cycle. However, 2-(3-(trifluoromethyl)-phenyl)-4H-pyrido[1,2-a]pyrimidin-4-one (1j) produced only one regioisomer of the annulated product (3ja) by oxidative coupling at the less sterically hindered site, i.e. the para-position of the CF3 group. It was observed that substrates with electron-donating groups at the para-position of the C2-phenyl ring produced higher yields of the corresponding annulated product as compared with those having electron-withdrawing groups at the para-position of the C2-phenyl ring. On the contrary, substrates with electron-donating groups at the meta-position of the C2-phenyl ring produced lower yields of the corresponding annulated products as compared with those having electron-withdrawing groups. This was also established by two independent one-pot competitive reactions of 1c/1h with 2a and 1i/1j with 2a, which produced the corresponding products 3ca/3ha in 10:1 and (3ia + 3ia′)/3ja in 1:13 ratios, respectively. Gratifyingly, the oxidative annulation of various 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-ones (1k–o) with substituents at different positions of the pyrido ring afforded the corresponding annulated products (3ka–oa) in good (69–78%) yields.
:1 ratio. This may be due to the coordination of the –OMe group with Ru metal in the catalytic cycle. However, 2-(3-(trifluoromethyl)-phenyl)-4H-pyrido[1,2-a]pyrimidin-4-one (1j) produced only one regioisomer of the annulated product (3ja) by oxidative coupling at the less sterically hindered site, i.e. the para-position of the CF3 group. It was observed that substrates with electron-donating groups at the para-position of the C2-phenyl ring produced higher yields of the corresponding annulated product as compared with those having electron-withdrawing groups at the para-position of the C2-phenyl ring. On the contrary, substrates with electron-donating groups at the meta-position of the C2-phenyl ring produced lower yields of the corresponding annulated products as compared with those having electron-withdrawing groups. This was also established by two independent one-pot competitive reactions of 1c/1h with 2a and 1i/1j with 2a, which produced the corresponding products 3ca/3ha in 10:1 and (3ia + 3ia′)/3ja in 1:13 ratios, respectively. Gratifyingly, the oxidative annulation of various 2-phenyl-4H-pyrido[1,2-a]pyrimidin-4-ones (1k–o) with substituents at different positions of the pyrido ring afforded the corresponding annulated products (3ka–oa) in good (69–78%) yields.
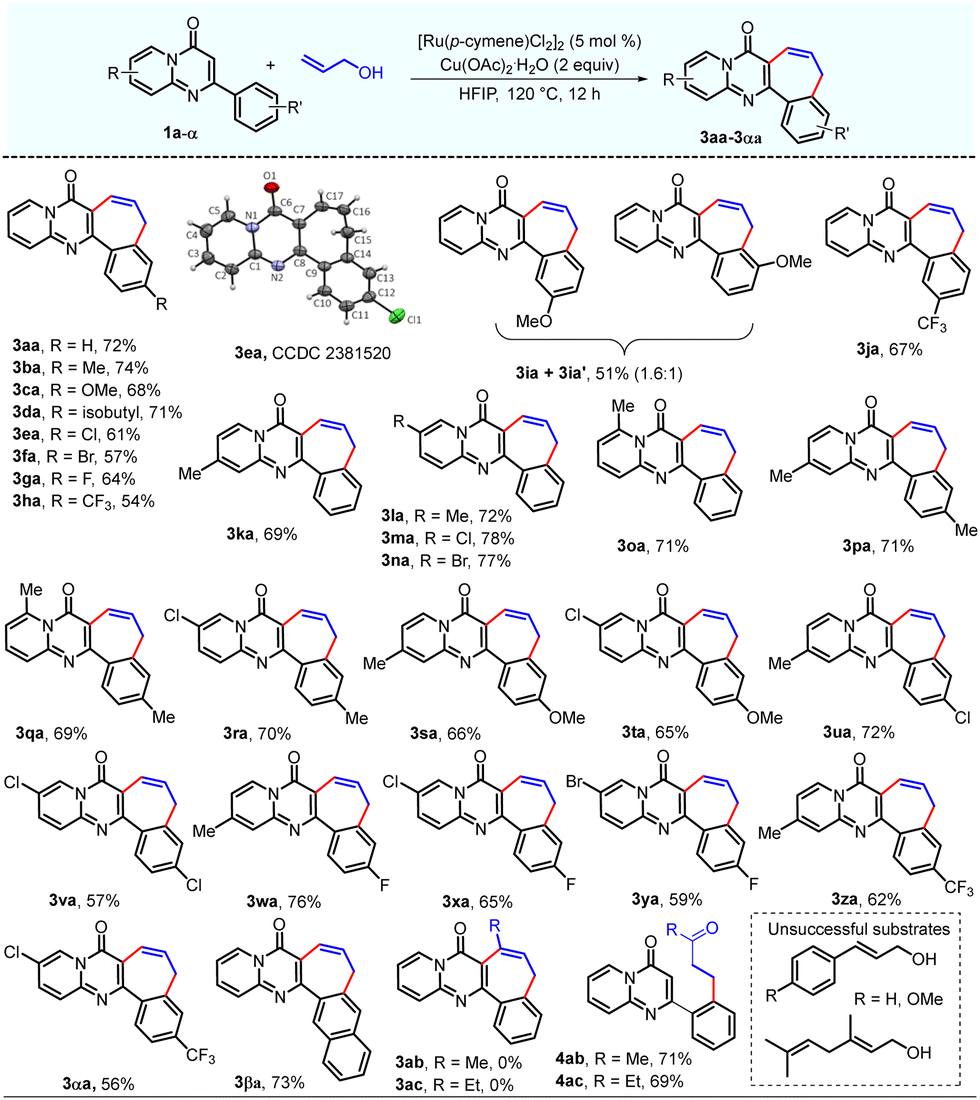
The reaction of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones (1p–α) having substituents both on the C2-phenyl ring and pyrido ring with allyl alcohol (2a) also delivered the desired annulated products (3pa–αa) in moderate to good (56–76%) yields. Finally, the reaction of 2-(naphthalen-2-yl)-4H-pyrido[1,2-a]pyrimidin-4-one (1β) could also participate in this oxidative annulation to produce the corresponding annulated product 3βa in a 73% yield. Reaction of 1a with secondary allyl alcohols, but-3-en-2-ol (b2) and pent-1-en-3-ol (c2), produced the corresponding ortho-alkylated derivatives 4ab and 4ac in 71% and 69% yields, respectively, but failed to afford the desired annulated products 3ab and 3ac under these conditions. The γ-substituted allylic alcohols did not react under standard conditions. Notably, the protocol exhibited excellent compatibility for the halogen (F, Cl, and Br) groups that can be further engaged in additional late-stage functionalization of the products. The structure of all the synthesized products was confirmed by NMR (1H and 13C{1H}) and HRMS data. The structure of 3ea was also unambiguously confirmed by a single crystal X-ray diffraction study (CCDC 2381520†).
To highlight the utility of the developed protocol, we successfully scaled the reaction of 1a to 6 mmol. The reaction of 1a (1.33 g, 6 mmol) with 2a (0.70 g, 12 mmol) in the presence of [Ru(p-cymene)Cl2]2 (5 mol%) and Cu(OAc)2·H2O (2 equiv.) in HFIP (8 mL) at 120 °C after 12 h produced 3aa in 71% (1.11 g) yield (Scheme 2). The potential of 3aa as a valuable scaffold for further synthetic transformations was then considered. The reaction of 3aa with Lawesson's reagent8c and NBS/MeOH8b produced corresponding thio derivative 7 and bromomethoxylated product 8 in 71% and 78% yields, respectively (Scheme 2).
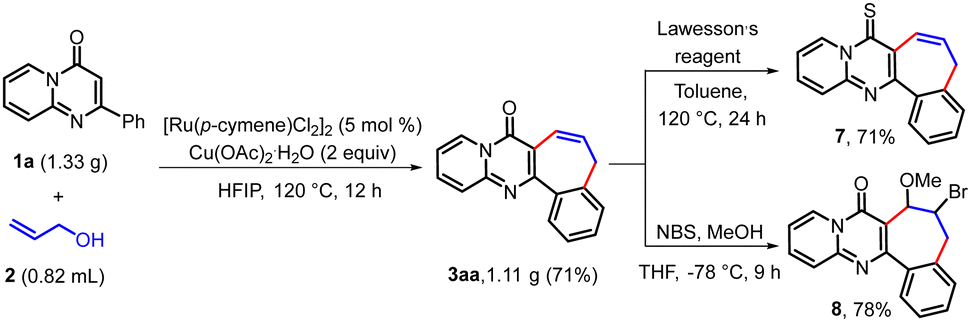 | ||
| Scheme 2 Gram-scale synthesis and chemical transformations of 3aa. | ||
To learn more about the reaction mechanism, a few control experiments were performed (Scheme 3). A 50% hydrogen/deuterium exchange at both ortho-positions of the C2-phenyl ring and at the C3-position of the pyrido[1,2-a]pyrimidin-4-one nucleus was observed when the reaction of 2-(p-tolyl)-4H-pyrido[1,2-a]pyrimidin-4-one (1b) was conducted in HFIP/D2O under standard conditions in the absence of allyl alcohol (Scheme 3a). This result indicated that the ortho C–H bond is involved in the ruthenacycle formation during C–H activation, and this C–H bond activation is of a reversible nature. Intermolecular competition reaction of substrates with electron-donating and electron-withdrawing groups at the meta-position of the C2-phenyl ring 1i and 1j with 2a produced the corresponding annulated products (3ia + 3ia′) and 3ja in 1:13 ratio in favor of C2-phenyl ring with an electron-withdrawing group at the meta-position (Scheme 3b). This result supports that the reaction proceeds via a concerted metalation-deprotonation (CMD)10 pathway rather than a Friedel–Craft-type electrophilic aromatic substitution pathway. The reaction of 1a with 2a in the presence of [Ru(p-cymene)Cl2]2 (5 mol%) and Cu(OAc)2·H2O (2 equiv.) in HFIP at 60 °C for 24 h produced an ortho-alkylated product (4aa) and ortho-alkenylated product (6aa) in 48% and 23% yields, respectively (Scheme 3c(i)). Furthermore, the reaction of 1a with methacrolein produced only alkenylated product 9 in 63% yield (Scheme 3c(i)). The stoichiometric reaction of 1b with [Ru(p-cymene)Cl2]2 in methanol at room temperature produced a five-membered ruthenacycle intermediate Ru-I (Scheme 3c(ii)). Subsequently, the reaction of 1b and 2a under the optimal conditions using Ru-I (5 mol%) as a catalyst instead of [Ru(p-cymene)Cl2]2 afforded 3ba in 69% yield (Scheme 3d), indicating that the developed annulation reaction proceeds through cyclometalated complex Ru–I. Finally, the reaction of intermediate 4aa under standard reaction conditions without an oxidant gave the desired product 3aa in 53% yield, whereas intermediate 6aa failed to produce 3aa (Scheme 3e).
 | ||
| Scheme 3 Control experiments. | ||
Although the mechanism of the (4+3) C–H/C–H annulative reaction still remains to be established, on the basis of preliminary results from control experiments and previous literature reports11 we proposed a plausible mechanistic sequence for the formation of product 3 (Scheme 4). After the generation of active ruthenium complex intermediate A, it reacts with 1a to form five-membered ruthenacycle intermediate Ru–Ivia reversible C–H bond activation through a concerted metalation-deprotonation (CMD) mechanism.10 Coordination of 2a with Ru–I produces complex B which on subsequent 1,2-migratory insertion into the Ru–C bond of Ru–I produces a seven-membered ruthenacycle C. Next, intermediate C undergoes ‘rollover C–H activation’12 to produce a seven-membered ruthenacycle intermediate D, which on β-hydride elimination produces intermediate F. Intermediate F tautomerizes to G (detected in LC-HRMS m/z 513.1121 [M]+) which then either produces intermediate H or ruthenacycle I. Protodemetalation of H or reductive elimination of I generates intermediate J (detected in LC-HRMS m/z 279.1125 [M + H]+), which on dehydration produces the desired product 3aa. The catalytic cycle involving the formation of intermediate 4aa from seven-membered ruthenacycle C by reductive elimination followed by tautomerization cannot be ruled out. Ruthenium and HFIP-assisted cyclodehydration of intermediate 4aa can produce the desired (4+3) annulated product 3aa.
 | ||
| Scheme 4 Proposed reaction mechanism. | ||
In summary, we have developed a Ru(II)-catalyzed (4+3) oxidative C–H/C–H annulation of 2-aryl-4H-pyrido[1,2-a]pyrimidin-4-ones with allyl alcohols leading to the formation of benzo[6,7]cyclohepta[1,2-d]pyrido[1,2-a]pyrimidin-8(5H)-one derivatives in moderate to good yields. The reaction features good substrate scope with respect to pyrido[1,2-a]pyrimidin-4-one with excellent functional group tolerance. The efficacy of the method was demonstrated by gram-scale synthesis of 3aa. In addition, based on a preliminary mechanistic study, a plausible mechanism of (4+3) C–H/C–H annulation reaction is proposed.
The authors gratefully acknowledge BITS Pilani for NMR and single crystal XRD analysis under a central instrumentation facility. T. J. and D. S. N. thank UGC, New Delhi, and BITS Pilani, respectively, for the research fellowship. T. J. and D. S. N. performed the experiments and collected data. A. K., T. J., and D. S. N. discussed and analyzed the data. K. R. analyzed the crystallographic data. A. K. designed and developed this project, and wrote the manuscript with the help of all of the authors.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. A. Shah, T. Sarkar, S. Kar, P. K. Maharana, K. Talukdar and T. Punniyamurthy, Chem. – Asian J., 2024, 19, e202300815 CrossRef CAS PubMed; (b) J. H. Docherty, T. M. Lister, G. McArthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- (a) B. Niu, K. Yang, B. Lawrence and H. Ge, ChemSusChem, 2019, 12, 2955–2969 CrossRef CAS PubMed; (b) K. Ghosh, R. K. Rit, M. Shankar, K. Mukherjee and A. K. Sahoo, Chem. Rec., 2020, 20, 1017–1042 CrossRef CAS; (c) M. Shankar, R. K. Rit, S. Sau, K. Mukherjee, V. Gandon and A. K. Sahoo, Chem. Sci., 2020, 11, 10770–10777 RSC.
- W. Ma, H. Dong, D. Wang and L. Ackermann, Adv. Synt. Catal., 2017, 359, 966–973 Search PubMed.
- (a) R. Manoharan and M. Jeganmohan, Chem. Commun., 2015, 51, 2929–2932 RSC; (b) G. S. Kumar, T. Chand, D. Singh and M. Kapur, Org. Lett., 2018, 20, 4934–4937 CrossRef CAS; (c) J. Jiang, J. Liu, Z. Yang, J. Zheng, X. Tian, L. Zheng and Z.-Q. Liu, Org. Biomol. Chem., 2022, 20, 339–344 RSC; (d) G. Naskar and M. Jeganmohan, Org. Lett., 2024, 26, 6580–6585 CrossRef CAS PubMed; (e) J. Jiang, J. Liu, Z. Yang, J. Zheng, X. Tian, L. Zheng and Z.-Q. Liu, Org. Biomol. Chem., 2022, 20, 339–344 RSC.
- (a) J.-S. Sun, Y.-Y. Wang, M. Liu, J. Zhang, C.-F. Liu, Y.-J. Xu and L. Dong, Org. Chem. Front., 2019, 6, 2713–2717 Search PubMed; (b) D. S. Nipate, N. Meena, P. N. Swami, K. Rangan and A. Kumar, Chem. Commun., 2024, 60, 344–347 Search PubMed; (c) H. Wu, J. Gui, M. Sun, Y. Ma, J. Yang and Z. Wang, J. Org. Chem., 2023, 88, 3871–3882 CrossRef CAS PubMed.
- (a) J. Zhang, J.-S. Sun, Y.-Q. Xia and L. Dong, Adv. Synth. Catal., 2019, 361, 2037–2041 CrossRef CAS; (b) L. Huang, Q. Wang, J. Qi, X. Wu, K. Huang and H. Jiang, Chem. Sci., 2013, 4, 2665–2669 RSC; (c) Z. Shi, M. Boultadakis-Arapinis and F. Glorius, Chem. Commun., 2013, 49, 6489–6491 RSC.
- G. S. Kumar, P. Kumar and M. Kapur, Org. Lett., 2017, 19, 2494–2497 CrossRef CAS PubMed.
- (a) X. Li, J. Rao, W. Ouyang, Q. Chen, N. Cai, Y.-J. Lu and Y. Huo, ACS Catal., 2019, 9, 8749–8756 CrossRef CAS; (b) M. S. Sherikar, R. Devarajappa and K. R. Prabhu, J. Org. Chem., 2021, 86, 4625–4637 CrossRef CAS; (c) Y.-J. Wang, T.-T. Wang, C.-C. Liang, Z.-H. Li and L.-M. Zhao, Org. Lett., 2021, 23, 6272–6277 CrossRef CAS PubMed; (d) A. Peneau, Q. Tricart, C. Guillou and L. Chabaud, Chem. Commun., 2018, 54, 5891–5894 RSC; (e) Y.-Q. Xia, C. Li, M. Liu and L. Dong, Chem. – Eur. J., 2018, 24, 5474–5478 CrossRef CAS.
- (a) N. Meena, V. N. Shinde, Sonam, P. N. Swami, K. Rangan and A. Kumar, J. Org. Chem., 2023, 88, 12902–12913 Search PubMed; (b) Bhawani, V. N. Shinde, Sonam, K. Rangan and A. Kumar, J. Org. Chem., 2022, 87, 5994–6005 CrossRef CAS PubMed.
- (a) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299–2302 Search PubMed; (b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef.
- C. N. Shambhavi and M. Jeganmohan, J. Org. Chem., 2024, 89, 9896–9909 CrossRef CAS.
- (a) J. Kwak, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778–17788 CrossRef CAS; (b) A. Zucca and M. I. Pilo, Molecules, 2021, 26, 328 Search PubMed; (c) B. Butschke and H. Schwarz, Chem. Sci., 2012, 3, 308–326 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Optimization table, experimental procedures, spectral data and copies of 1H and 13C{1H} NMR spectra of products 3aa–6aa and 7, and single crystal X-ray data for 3da. CCDC 2381520. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00318k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |