
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming kinetic barriers of remote electrochemiluminescence on boron-doped diamond via catalytic coreactant oxidation†
Alessandro
Fracassa
 a,
Chiara
Mariani
a,
Andrea
Fiorani
b,
Yasuaki
Einaga
b,
Conor F.
Hogan
c,
Francesco
Paolucci
ad,
Neso
Sojic
e,
Paul S.
Francis
*f and
Giovanni
Valenti
*a
a,
Chiara
Mariani
a,
Andrea
Fiorani
b,
Yasuaki
Einaga
b,
Conor F.
Hogan
c,
Francesco
Paolucci
ad,
Neso
Sojic
e,
Paul S.
Francis
*f and
Giovanni
Valenti
*a
aDepartment of Chemistry “Giacomo Ciamician”, Alma Mater Studiorum – University of Bologna, Via Gobetti 85, 40129 Bologna, Italy. E-mail: g.valenti@unibo.it
bDepartment of Chemistry, Keio University, 3-14-1 Hiyoshi, Yokohama 223-8522, Japan
cThe Biomedical and Environmental Sensor Technology (BEST) Research Centre, Biosensors Program, La Trobe Institute for Molecular Science (LIMS), La Trobe University, Melbourne, 3086, VIC, Australia
dICMATE-CNR, Corso Stati Uniti 4, 35127 Padova, Italy
eUniversity Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, ENSMAC, 33607, Pessac, France
fCentre for Sustainable Bioproducts, Faculty of Science, Engineering and Built Environment, Deakin University, Geelong, Victoria 3220, Australia
First published on 3rd February 2025
Abstract
The effectiveness of boron-doped diamond (BDD) as a platform for electrochemiluminescence (ECL) bead-based bioassays is hindered by the sluggish rate of heterogeneous tri-n-propylamine (TPrA) oxidation. To address this, we investigate the ECL of Ru(II)-coated microbeads in the presence of a redox mediator, exploring the effect of applied potential and electrode surface terminations. Using a redox mediator, the ECL signal on BDD is enhanced by up to 46-fold.
Electrochemiluminescence (ECL) is a phenomenon that involves a cascade of chemical reactions that ultimately convert an electrical input into an optical output.1 The ECL process starts with either the oxidation or reduction of a sacrificial molecule known as a coreactant, producing strongly reducing or oxidizing radicals on the electrode surface.2 These species initiate homogeneous electron transfer reactions that culminate in luminophore excitation, followed by light emission. In this context, the signal efficiency strongly depends on the electrode material, particularly due to the typically slow rate of the coreactant heterogeneous electron transfer.3,4
Without an optical excitation source, the photodetector encounters a negligible background,5,6 making ECL one of the most sensitive analytical tools. Moreover, the absence of interferences associated with traditional imaging such as autofluorescence, photobleaching and light scattering makes ECL an ideal transduction technique for microscopy.7 Empowered with the spatial resolution of a microscope, ECL microscopy (ECLM) enables imaging of single objects and two-dimensional mapping of reaction dynamics by capturing the ECL signal generated during potential sweep.8–14
The most commercially successful application of ECL to date is its use as a transduction vector in automated analyzers for early detection of biomarkers.15 The core strategy employed in these analyzers is the use of 2.8 μm magnetic beads as platforms to perform immunoassays in the vicinity of the electrode surface. The detection antibody, labeled with tris(2,2′-bipyridine)ruthenium(II) ([Ru(bpy)3]2+) units, converts the amount of analyte in the sample into a proportionally intense luminescent signal. The electronically excited state responsible for the emission is generated through reactions with the radical intermediates of an electrochemically oxidized coreactant, namely tri-n-propylamine (TPrA),16 as shown in Fig. 1a.
 | ||
| Fig. 1 Schematics of (a) the conventional remote ECL pathway on BDD and (b) the ECL-enhancing catalytic pathway in a bead-based immunoassay. The magnetic microbead is represented by the orange sphere while the immobilized [Ru(bpy)3]2+ luminophore and the freely-diffusing [Ir(sppy)3]3− redox mediator are labelled as Ru2+ and Ir3−, respectively. | ||
The ECL process is typically carried out on electrodes such as platinum, gold or glassy carbon (GC).3 While Pt and Au working electrodes provide chemical inertness, remarkable cycling stability, and ease of in situ electrochemical cleaning, they are expensive materials that form non-conductive oxide layers during ECL measurements.17 In contrast, GC electrodes are cheaper alternatives, which also exhibit much faster oxidation rate constants for TPrA. However, they are not suitable for long-term use as their surface tends to passivate over time and the cleaning procedure requires tedious mechanical effort.4,18
Boron-doped diamond (BDD) represents the most attractive alternative as it offers superior mechanical and electrochemical properties compared to the aforementioned electrodes: exceptionally wide electrochemical stability window, low capacitive current, remarkable chemical and physical durability, and ease of in situ cleaning.19,20 So far, BDD has also been successfully adapted for ECL generation, as our group demonstrated a 70% enhancement in the signal of [Ru(bpy)3]2+-labeled beads.21 However, we also found that the kinetics of TPrA oxidation at BDD is particularly sluggish. This issue arises from the inner-sphere nature of TPrA heterogeneous electron transfer. The low rate constant of this electron transfer (k0ET) on BDD, measured between 7–75 × 10−4 cm s−1, suggests considerable scope for further improvement.21
Recently, Ir(III)-complex-based redox mediators have garnered significant attention within the ECL community.22 This interest was ignited when Kerr et al. demonstrated that the addition of tris(2-(2-pyridinyl-κN)-4-sulfonatophenyl-κC)iridium(III) ([Ir(sppy)3]3−) to a TPrA solution could significantly amplify the ECL signal of [Ru(bpy)3]2+.23 Since then, we extensively studied such redox-mediated ECL systems to elucidate the underlying electrochemical pathways leading to signal enhancement.24–28 Our findings suggest that, parallel to the sluggish heterogeneous oxidation of TPrA, the simultaneous rapid oxidation of [Ir(sppy)3]3− provides a vehicle to catalyze the coreactant oxidation. This process, particularly effective at low overpotentials due to reduced TPrA reactivity at the electrode, enhances the generation of radicals, thereby producing stronger ECL from [Ru(bpy)3]2+ labels (Fig. 1b and Scheme S1, ESI†).
Herein, we address the modest k0ET of TPrA on BDD,21 using [Ir(sppy)3]3− as a redox mediator. By accelerating the coreactant oxidation rate, this approach boosts the ECL signal of [Ru(bpy)3]2+-coated beads, positioning BDD as an appealing material for commercial ECL bioanalysis. The impact of the enhancing Ir(III) complex on the ECL intensity of [Ru(bpy)3]2+-functionalized beads was explored through ECLM. This invaluable tool provides novel insights into ECL reaction mechanisms in bead-based systems, as it allows for imaging down to the level of single beads. As demonstrated in a previous study of ours on heterogeneous ECL systems mediated by emitting Ir(III) complexes, ECL microscopy also enables the straightforward subtraction of the background ECL signal from [Ir(sppy)3]3−*, distinguishing it from that of [Ru(bpy)3]2+*.25,29
The widespread use of the [Ru(bpy)3]2+/TPrA pair in bead-based immunoassays encouraged extensive studies to understand the mechanism underlying ECL emission (remote pathway, Fig. 1a).16,30–32 Once the electrode potential is pushed sufficiently anodic to oxidize the coreactant, TPrA (E0 = 0.83 V vs. Ag/AgCl) is converted into TPrA˙+. The latter radical is relatively short-lived (τ1/2 = 200 μs) and readily loses a proton in an aqueous environment to yield TPrA˙. This α-aminoalkyl radical (Eox = −1.7 V vs. Ag/AgCl) reduces the Ru(II) labels to the [Ru(bpy)3]+ state. Subsequent exergonic oxidation of [Ru(bpy)3]+ by TPrA˙+ produces the excited luminophore [Ru(bpy)3]2+* that relaxes to the ground state by emitting a photon.33,34
TPrA heterogeneous oxidation is a surface-sensitive process, thus precise control over BDD surface conditions is crucial for tuning the amine oxidation kinetics and, in turn, the intensity of the ECL signal. Different strategies have been adopted to electrochemically modify BDD surface terminations, namely anodic oxidation (AO) and cathodic reduction (CR): AO is an anodic pretreatment that exposes oxygen functionalities, while CR is a pretreatment that develops hydrogen terminations.35,36
The impact of the different surface chemistries was assessed by recording the ECL emission of Ru(II)-covalently functionalized 2.8 μm beads in a 180 mM TPrA solution, either in the absence (Ru@Beads) or presence of 100 μM [Ir(sppy)3]3− (Ru@Beads/[Ir(sppy)3]3−), during cyclic voltammetry (CV-ECL) on AO-BDD (Fig. 2) or CR-BDD (Fig. S1, ESI†) surfaces. Previous studies on similar redox-mediated systems have shown that, when using 180 mM TPrA, the greatest enhancement is achieved by introducing a [Ir(sppy)3]3− concentration of the order of magnitude of 100 μM.24–26 Despite Ru(II) labels being covalently attached to the surface of microbeads, this system closely mimics the behavior of a conventional bead-based immunoassay while improving the signal-to-noise ratio by virtue of the superior surface density of the luminophore.21,30,31,34,37 Concerning the electrochemistry of Ru@Beads, CR pretreatment produces overall higher currents and lowers the onset potential for TPrA oxidation (Fig. S2, ESI†). This behavior arises from a stronger affinity of the coreactant towards the hydrogen-terminated electrode surface.38,39 The latter, displaying enhanced hydrophobicity compared to the surface upon AO treatment, promotes the adsorption and, in turn, a faster oxidation rate for TPrA. This hypothesis is supported by the k0ET values reported in the literature for TPrA oxidation at 0.92 V (vs. Ag/AgCl, KCl sat.): the rate constant on AO-BDD is 7 × 10−4 cm s−1 while that on CR-BDD is 75 × 10−4 cm s−1.21 This difference is expected to translate to the ECL behavior of Ru@Beads, as the coreactant oxidation represents a key bottleneck in the ECL heterogeneous mechanism. However, counterintuitively, the Ru@Beads signal is stronger on AO-BDD than on CR-BDD, consistent with previous studies.21
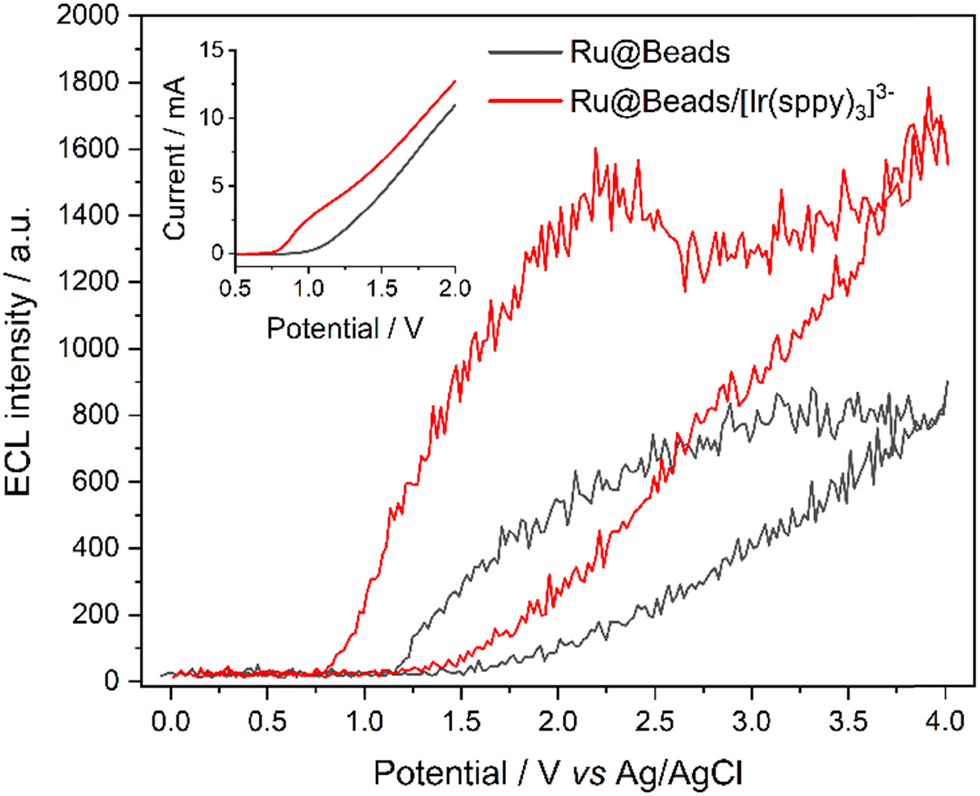 | ||
| Fig. 2 CV-ECL of Ru@Beads (grey line) and Ru@Beads/[Ir(sppy)3]3− (red line) on AO-BDD. The potential was scanned at 100 mV s−1 from open circuit potential (OCP) up to 4 V, back to 0 V, and eventually terminating the cycle at OCP. ECL data points were determined as reported in the ESI† by elaborating ECL images captured every 200 ms during potential scan. Inset: LSV i–V curves of Ru@Beads (grey line) and Ru@Beads/[Ir(sppy)3]3− (red line) on AO-BDD. | ||
The introduction of a redox mediator displaying specific electrochemical properties,25 such as [Ir(sppy)3]3−, significantly alters the electrochemical landscape as the oxidation current onset potential is anticipated to be ∼0.79 V on both AO-BDD and CR-BDD. This shift aligns with the oxidation of [Ir(sppy)3]3− to [Ir(sppy)3]2−, as evidenced by the onset potential matching the complex half-wave oxidation potential. This proves the outer-sphere nature of [Ir(sppy)3]3− electron transfer, which remains unaffected by the state of the BDD electrode surface.
Although TPrA oxidation on BDD requires high overpotentials, the early oxidation of [Ir(sppy)3]3− provides a tool to catalyze the reaction, allowing it to occur at the redox mediator formal potential. The catalytic effect of [Ir(sppy)3]2− is the most evident on AO-BDD, where the current intensifies upon the addition of the redox mediator due to the faster generation of TPrA˙, which readily oxidizes at the electrode surface (Fig. S3, ESI†).
Consistent with the proposed catalytic pathway for TPrA oxidation, the ECL onset potential is shifted to 0.83 V on both AO-BDD and CR-BDD, with enhanced signal intensity across the entire explored potential range. This enhancement, however, is especially pronounced at lower overpotentials, showing 20.7- and 7.8-fold enhancement at 1.1 V, and 2.7- and 6.2-fold enhancement at 2.2 V, on AO-BDD (Fig. 2) and CR-BDD (Fig. S1, ESI†), respectively. This result shows that the catalytic oxidation of the coreactant by [Ir(sppy)3]2− is a far more effective mechanism than the redox-mediated oxidation of [Ru(bpy)3]+ labels on the beads. The latter hypothesis is further supported by the signal gain at 1.1 V: compared to the hydrogen-terminated surface, the oxygen-terminated one is characterized by a smaller k0ET that translates into greater hindering of TPrA oxidation, promoting an enhanced rate of interaction between [Ir(sppy)3]2− and the coreactant. Instead, at 2.2 V, where the k0ET on the two surfaces tends to converge,21 the magnitude of signal gain aligns as TPrA oxidation approaches diffusion control.
Due to the superior performance of AO-BDD compared to CR-BDD, the former surface was chosen as the platform for ECL double-step chronoamperometric (CA) measurements.
At 1.1 V, no ECL emission is observed on Ru@Beads, as the applied overpotential is insufficient to oxidize TPrA at the electrode surface. Under the same conditions, as expected, Ru@Beads/[Ir(sppy)3]3− generates a strong luminescent signal due to the catalytic effect of oxidized [Ir(sppy)3]2−, resulting in a 46.3-fold enhancement of the ECL profile peak (Fig. 3).
 | ||
| Fig. 3 ECL images of 2.8 μm beads covalently labelled with [Ru(bpy)3]2+ in a 0.3 M PB solution (pH 6.8) with 180 mM TPrA (a) without (Ru@Beads) and (b) with 100 μM [Ir(sppy)3]3− (Ru@Beads/[Ir(sppy)3]3−) on AO-BDD. The images were captured with an EM-CCD camera by integrating the ECL signal for 7 s during a two-step chronoamperometry measurement: 2 s at OCP and 5 s at 1.1 V vs. Ag/AgCl. Magnification, 100×; objective numerical aperture, 1.1; gain, 1; sensitivity, 255; contrast scale, 3000 to 5000; scale bar, 3 μm. The yellow regions of interest (14.28 × 0.63 μm) centered on the beads were used to compute the ECL profiles of single beads (see ESI†). The bright field image of (a) is reported in the ESI† (Fig. S4). (c) Statistical analysis of ECL profile peak values obtained from Ru@Beads (left, n = 18) and Ru@Beads/[Ir(sppy)3]3− (right, n = 13). | ||
On the other hand, upon applying 2.2 V, the addition of [Ir(sppy)3]3− yields a 1.14-fold ECL signal enhancement (Fig. S5, ESI†). This increase is notably more modest than that observed in CV-ECL at the same potential, likely due to differences in how the voltage is controlled, with CV involving a sweep while CA a step. As a result, at the same applied potential, the concentration profiles of TPrA and [Ir(sppy)3]3− in close proximity to the electrode surface may differ, potentially affecting the resolution between the two reaction mechanisms. Nonetheless, the ECL trend observed in CA aligns well with that in CV.
Although the ECL intensity of Ru@Beads at 2.2 V surpasses that of Ru@Beads/[Ir(sppy)3]3− at 1.1 V, one must consider that electrochemical biosensors should be designed to work at the lowest possible overpotential to minimize undesired side reactions that could harm biomolecules, cells, or DNA.
In conclusion, we introduced a simple yet powerful approach to tackle the slow TPrA oxidation on BDD. Just by incorporating an Ir(III) complex as an additive in the coreactant solution, we achieved significant enhancements in the ECL signal—up to 20.7-fold in CV and 46.3-fold in CA—within a safe potential range for most biological components. These findings could finally promote BDD, already recognized for its superior mechanical and electrochemical properties compared to other electrodes more commonly employed for ECL, as a reliable platform for commercial bioanalysis.
This work was supported by the ECLectic project that has received funding from the European Union's MSCA Doctoral network Horizon Europe programme Grant Agreement Number 101119951 and PRIN 20225P4EJC.
Data availability
Experimental data are available at AMS Acta at https://amsacta.unibo.it/id/eprint/8119.Conflicts of interest
The authors declare no competing financial interest.Notes and references
- G. Giagu, A. Fracassa, A. Fiorani, E. Villani, F. Paolucci, G. Valenti and A. Zanut, Microchim. Acta, 2024, 191, 359 CrossRef CAS
.
- Y. Yuan, S. Han, L. Hu, S. Parveen and G. Xu, Electrochim. Acta, 2012, 82, 484–492 CrossRef CAS
- G. Valenti, A. Fiorani, H. Li, N. Sojic and F. Paolucci, ChemElectroChem, 2016, 3, 1990–1997 CrossRef CAS
- C. I. Santo, G. Conejo-Cuevas, F. Paolucci, F. J. Del Campo and G. Valenti, Chem. Biomed. Imaging, 2024, 2, 835–841 CrossRef CAS
- J. Shu, Z. Qiu, Q. Zhou, Y. Lin, M. Lu and D. Tang, Anal. Chem., 2016, 88, 2958–2966 CrossRef CAS PubMed
- Z. Yu, H. Gong, Y. Li, J. Xu, J. Zhang, Y. Zeng, X. Liu and D. Tang, Anal. Chem., 2021, 93, 13389–13397 CrossRef CAS PubMed
- X. Gou, Z. Xing, C. Ma and J.-J. Zhu, Chem. Biomed. Imaging, 2023, 1, 414–433 CrossRef CAS PubMed
- Y. Yan, L. Ding, J. Ding, P. Zhou and B. Su, ChemBioChem, 2024, 25, e202400389 CrossRef CAS
- C. Mariani, S. Bogialli, F. Paolucci, P. Pastore, A. Zanut and G. Valenti, Electrochim. Acta, 2024, 489, 144256 CrossRef CAS
- N. Sojic, S. Knežević, D. Han, B. Liu and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202407588 CrossRef PubMed
- B. R. Layman and J. E. Dick, J. Am. Chem. Soc., 2024, 146, 26216–26222 CrossRef CAS
- S. Knežević, J. Totoricaguena-Gorriño, R. K. R. Gajjala, B. Hermenegildo, L. Ruiz-Rubio, J. L. Vilas-Vilela, S. Lanceros-Méndez, N. Sojic and F. J. Del Campo, J. Am. Chem. Soc., 2024, 146, 22724–22735 CrossRef
- Y. C. Kong, D. Ye, C. H. Xu, Z. Ma, H. Zhao and W. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202318748 CrossRef CAS PubMed
- K. Ino, M. Mashiko, Y. Kanno, Y. Tang, S. Masui, T. Nisisako, K. Hiramoto, H. Abe and H. Shiku, Anal. Chem., 2024, 96, 18967–18976 CrossRef CAS PubMed
-
E. Faatz, A. Finke, H. P. Josel, G. Prencipe, S. Quint and M. Windfuhr, Analytical Electrogenerated Chemiluminescence, Royal Society of Chemistry, 2019, pp. 443–470 Search PubMed
- J. Yu, D. Stankovic, J. Vidic and N. Sojic, Sens. Diagn., 2024, 3, 1887–1898 RSC
-
I. Leka Kottaiveedu Sivakumar, L. Bouffier, N. Sojic and S. Senthil Kumar, Angew. Chem., Int. Ed., 2024, e202421185 Search PubMed
- D. Han, B. Goudeau, D. Jiang, D. Fang and N. Sojic, Anal. Chem., 2021, 93, 1652–1657 Search PubMed
- S. Yu, S. Liu, X. Jiang and N. Yang, Carbon, 2022, 200, 517–542 CrossRef CAS
- A. Fiorani, G. Valenti, F. Paolucci and Y. Einaga, Chem. Commun., 2023, 59, 7900–7910 RSC
- K. Sakanoue, A. Fiorani, C. I. Santo, I. Irkham, G. Valenti, F. Paolucci and Y. Einaga, ACS Sens., 2022, 7, 1145–1155 Search PubMed
- Y. Zhang, Y. Xu, J. Li, R. Chen, W. Chen and H. Peng, TrAC, Trends Anal. Chem., 2024, 178, 117812 CrossRef CAS
- E. Kerr, D. J. Hayne, L. C. Soulsby, J. C. Bawden, S. J. Blom, E. H. Doeven, L. C. Henderson, C. F. Hogan and P. S. Francis, Chem. Sci., 2022, 13, 469–477 RSC
- E. Kerr, S. Knezevic, P. S. Francis, C. F. Hogan, G. Valenti, F. Paolucci, F. Kanoufi and N. Sojic, ACS Sens., 2023, 8, 933–939 CrossRef CAS PubMed
- A. Fracassa, C. I. Santo, E. Kerr, S. Knežević, D. J. Hayne, P. S. Francis, F. Kanoufi, N. Sojic, F. Paolucci and G. Valenti, Chem. Sci., 2024, 15, 1150–1158 RSC
- S. J. Blom, N. S. Adamson, E. Kerr, E. H. Doeven, O. S. Wenger, R. S. Schaer, D. J. Hayne, F. Paolucci, N. Sojic, G. Valenti and P. S. Francis, Electrochim. Acta, 2024, 484, 143957 CrossRef CAS
- S. Knežević, E. Kerr, G. Valenti, F. Paolucci, P. S. Francis, C. F. Hogan, N. Sojic and F. Kanoufi, Electrochim. Acta, 2024, 499, 144677 CrossRef
- N. S. Adamson, S. J. Blom, E. H. Doeven, T. U. Connell, C. Hadden, S. Knežević, N. Sojic, A. Fracassa, G. Valenti, F. Paolucci, Y. Wang, J. Ding, B. Su, C. Hua and P. S. Francis, Angew. Chem., Int. Ed., 2024, 136, e202412097 CrossRef
- S. Knežević, E. Kerr, B. Goudeau, G. Valenti, F. Paolucci, P. S. Francis, F. Kanoufi and N. Sojic, Anal. Chem., 2023, 95, 7372–7378 CrossRef
- D. Han, D. Fang, G. Valenti, F. Paolucci, F. Kanoufi, D. Jiang and N. Sojic, Anal. Chem., 2023, 95, 15700–15706 CrossRef CAS
- D. Han, D. Jiang, G. Valenti, F. Paolucci, F. Kanoufi, P. C. Chaumet, D. Fang and N. Sojic, ACS Sens., 2023, 8, 4782–4791 Search PubMed
- Y. Feng, W. Zhou, X. Wang, J. Zhang, M. Zou, C. Zhang and H. Qi, Chem. Biomed. Imaging, 2023, 1, 648–658 CrossRef CAS PubMed
- W. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS
- A. Zanut, A. Fiorani, S. Canola, T. Saito, N. Ziebart, S. Rapino, S. Rebeccani, A. Barbon, T. Irie, H. P. Josel, F. Negri, M. Marcaccio, M. Windfuhr, K. Imai, G. Valenti and F. Paolucci, Nat. Commun., 2020, 11, 2668 Search PubMed
- S. Kasahara, K. Natsui, T. Watanabe, Y. Yokota, Y. Kim, S. Iizuka, Y. Tateyama and Y. Einaga, Anal. Chem., 2017, 89, 11341–11347 CrossRef CAS PubMed
- S. Kasahara, T. Ogose, N. Ikemiya, T. Yamamoto, K. Natsui, Y. Yokota, R. A. Wong, S. Iizuka, N. Hoshi, Y. Tateyama, Y. Kim, M. Nakamura and Y. Einaga, Anal. Chem., 2019, 91, 4980–4986 Search PubMed
- Y. Wang, J. Ding, P. Zhou, J. Liu, Z. Qiao, K. Yu, J. Jiang and B. Su, Angew. Chem., Int. Ed., 2023, 62, e202216525 CrossRef CAS
- Y. Zu and A. J. Bard, Anal. Chem., 2001, 73, 3960–3964 CrossRef CAS
- K. Sakanoue, A. Fiorani, I. Irkham and Y. Einaga, ACS Appl. Electron. Mater., 2021, 3, 4180–4188 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc00270b |
| This journal is © The Royal Society of Chemistry 2025 |