
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
5,5′-Biindeno[2,1-c]fluorene: importance of C5–C5′ linkage for an indeno[2,1-c]fluorene dimer exhibiting an open-shell ground state†
Neha
Maurya
 a,
Palash
Jana
b,
Himanshu
Sharma
a,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
a,
Palash
Jana
b,
Himanshu
Sharma
a,
Subhajit
Bandyopadhyay
b and
Soumyajit
Das
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar 140001, Punjab, India. E-mail: chmsdas@iitrpr.ac.in
bDepartment of Chemical Sciences, Indian Institute of Science Education and Research (IISER) Kolkata, Mohanpur 741246, West Bengal, India
First published on 21st February 2025
Abstract
Mesityl-protected indeno[2,1-c]fluorene and its C2–C2′-linked dimer 2,2′-biindeno[2,1-c]fluorene are closed-shell molecules. Herein, we report the synthesis and characterization of a mesityl-protected 5,5′-biindeno[2,1-c]fluorene displaying 26.4% diradical and 0.9% tetraradical characters. The C5–C5′ linkage between two [2,1-c]IFs is crucial for its open-shell character, resulting in its far-red UV-vis absorption, redox amphoterism, partially para-quinoidal as-indacene core, and temperature-dependent EPR response due to a small singlet–triplet energy gap.
Formally, antiaromatic parent indeno[2,1-c]fluorene (Fig. 1a) is an open-shell (OS) diradicaloid polycyclic hydrocarbon (PH) with the weakest diradical character index (y0 = 0.021)1 among the five indenofluorene (IF) isomers.2 The 5,8-disubstitution of [2,1-c]IF by bulky aryl (like mesityl for 1) or ethynyl groups is proven to be an effective approach to isolate sterically protected stable [2,1-c]IF derivatives,2d,3 but these are closed-shell (CS) quinoidal PHs. Symmetric2d and unsymmetric3b helical [2,1-c]IF derivatives usually have very small P/M-inversion barriers unless helically π-extended,4 a promising strategy to develop chiral diradicaloids with potential chiroptical properties.4b The curved shape of [2,1-c]IF was utilized to construct unique PHs, such as a helical monoradical,5 a macrocyclic tetraradicaloid,6 indacenodifluorene derivatives,3a,4a and benzo-extended [2,1-c]IFs7 with tunable antiaromaticity. From an application viewpoint, IF 1 was employed in bulk-heterojunctions8 as an electron-acceptor and recently our group found it to be an ambipolar charge carrier with balanced hole and electron mobilities in the order of 10−3 cm2 V−1 s−1.3b Additionally, the potential of IFs for application in non-linear optics9 and a recent report on IF-based PHs in single-molecule conductance10 studies suggested that IF derivatives with an OS ground state could be potential organic spintronics materials, tempting us to explore new IF-based polycyclic antiaromatic hydrocarbons with an OS ground state.
 | ||
| Fig. 1 (a) Antiaromatic indeno[2,1-c]fluorene 1, our reported 2,2′-biindeno[2,1-c]fluorene 2, and 6,6′-biindeno[1,2-b]fluorene 3. (b) Closed-shell structure 4 for the targeted mesityl disubstituted 5,5′-biindeno[2,1-c]fluorene, and its open-shell diradical and tetraradical resonance forms 4-OS1 and 4-OS2, respectively. | ||
Dimerization of 1 by carbons 2 and 2′ resulted in an IF dimer 2,2′-biindeno[2,1-c]fluorene 2 with a smaller energy gap between the highest occupied and lowest unoccupied molecular orbitals (HOMO and LUMO) in comparison to monomer 1.11 Although a small HOMO–LUMO gap (henceforth denoted as HLG) is the usual criterion for displaying OS properties,12 BIF 2 did not show an OS ground state, likely due to a large singlet(S)–triplet(T) energy gap (ΔES–T = −13.25 kcal mol−1). Very recently, we reported the synthesis of 6,6′-[1,2-b]BIF 3 as an OS IF dimer with a y0 of 0.268 and a tetraradical character index (y1) of 0.007.13 Dimerizing two indeno[1,2-b]fluorene monomers by carbons 6 and 6' bearing large HOMO and LUMO distributions resulted in a decrease in HLG and S–T gap (ΔES–T = −4.85 kcal mol−1) for 3 compared to those of known 3,3′-linked [1,2-b]BIF.11 Therefore, connecting two [2,1-c]IF units by the non-bridgehead (apical) carbons 5 and 5′ bearing larger HOMO and LUMO coefficients3b than the C2 and C2′ positions is envisaged to significantly tune the ground-state properties for 5,5′-biindeno[2,1-c]fluorene 4 (Fig. 1b) by further compressing the HLG and ΔES–T compared to those of 2. Herein, we report a facile synthesis for the newly designed BIF 4 that can be represented in three ground-state canonical structures: CS quinoidal 4, diradicaloid 4-OS1, and tetraradicaloid 4-OS2. The ground state of 4 was systematically investigated by various analytical techniques, with the support of density functional theory (DFT) calculations.
As parent [2,1-c]IF possesses the weakest y0 among the IF isomers,1 we performed DFT studies at the (U)CAM-B3LYP/6-31G(d,p) level of theory14 to examine the electronic ground state of BIF 4. Compound 4 was found to have an OS singlet ground state with a small ΔES–T = −4.07 kcal mol−1, including a modest y0 = 0.264 and a minor y1 = 0.009, as estimated from the natural orbital occupancy numbers (NOON). The spin densities are delocalized across the entire molecule, with the larger Mulliken spin density values of 0.53 and 0.47 at the apical carbons 8/8′ and 5/5′, respectively (Fig. 2a). The frontier molecular orbital (FMO) profiles of the singly occupied molecular orbitals (SOMO) reveal a typical disjointed nature of the alpha (SOMO-α) and beta (SOMO-β) spins of 4 (Fig. 2b and c). Based on the computations, BIF 4 is expected to exhibit an OS singlet ground state with a smaller ΔES–T than that of 3, while the OS characters are found to be identical to those of 3.13 A modest y0 and an incredibly low y1 indicate that the two radicals in 4-OS2 at the 5,5′-linkage may couple to form a bond, suggesting the contribution of 4-OS1 is more important.
 | ||
| Fig. 2 (a) Spin density distribution in 4. Isovalue for surfaces: MO = 0.02, density = 0.005; frontier molecular orbitals of the (b) α-spin and (c) β-spin of 4. | ||
The synthesis of BIF 4 is shown in Scheme 1. Dimerization of 4-bromo-9-fluorenone155 by Lawesson's reagent afforded 4,4′-dibromo-9,9′-bifluorenylidene 6 as E/Z-diastereomers. Treating diastereomeric mixture 6 with (2-formylphenyl)boronic acid under Suzuki reaction condition gave dialdehyde 7 in 55% yield as a mixture of stereoisomers. The stereoisomeric mixture 7 was treated with an excess of 2-mesitylmagnesium bromide (2-MesMgBr) under ambient conditions to produce diol 8. Crude 8 was reacted with BF3·Et2O to afford the dihydro precursor 9. As partial formation of 4 was experienced in thin layer chromatography during the conversion of 8 to 9, crude 9 was oxidatively dehydrogenated by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in 1,2-dichloroethane to produce desired BIF 4 as a greenish-black solid in 54% yield over three steps after column chromatographic purification. Nuclear magnetic resonance (NMR), high-resolution mass spectrometry (HRMS), and single crystal X-ray diffraction (SCXRD) analyses unequivocally confirmed the formation of BIF 4.
 | ||
| Scheme 1 Synthesis of [2,1-c]IF dimer 4. | ||
Single crystals of BIF 4 (Fig. 3a), obtained by slow diffusion of acetonitrile into hexane solution, are found to pack in either slightly helical P–P linked or M–M linked homodimeric arrangement (Fig. 3b), presumably due to a low P/M-inversion barrier for [2,1-c]IF.2d,3b Two [2,1-c]IF units of BIF 4 form a torsional angle of ∼37.9°, which is smaller than that observed for BIF 3.13 A reduced torsional angle between two [2,1-c]IF units, which are linked via carbons 5 and 5′ with large orbital coefficients for HOMO and LUMO,3b suggests the possibility of greater π-electronic communication between the IF units as the large C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C bond alternation in para-quinodimethane16 (p-QDM) subunit of as-indacene2d,3b forms a polyene-like13 π-conjugation path (shown in red for 4 in Fig. 1b). As a result, the CC double and C–C single bond lengths of the p-QDM subunit are expected to be increased and decreased, respectively.13
C/C–C bond alternation in para-quinodimethane16 (p-QDM) subunit of as-indacene2d,3b forms a polyene-like13 π-conjugation path (shown in red for 4 in Fig. 1b). As a result, the CC double and C–C single bond lengths of the p-QDM subunit are expected to be increased and decreased, respectively.13
 | ||
| Fig. 3 (a) X-ray crystallographic structure of 4 with the ellipsoids drawn at 30% probability level (hydrogens omitted), including NICS(1)zz (blue) and HOMA (green) values. (b) P–P and M–M linked homodimers of 4 in crystal packing. (c) EPR spectra of powder 4 recorded at variable temperatures (VT-EPR). | ||
The bond lengths (labelled a to j in Fig. 3a) determined from SCXRD and DFT analyses are summarized in Table 1 (see Fig. S12, ESI† for complete bond length analyses, including e.s.d. values), and compared with the SCXRD data of 1.2d Our analyses found that exo-methylene CC double bonds a (1.377 Å; DFT: 1.396 Å) and c (1.393 Å; DFT: 1.419 Å) are longer than those of 1 (Table 1 and Fig. S4, ESI†); in particular, the length of c is similar to 3, but bonds b and d are identical in length to those of 1 by SCXRD analysis. C–C single bonds e–h of the central benzenoid ring are relatively shorter than those of 1. Furthermore, C–C single bond i that links two IF units is 1.451 Å (DFT: 1.421 Å), which is shorter than C–C single bond j (1.484 Å; DFT: 1.480 Å) linking the mesityl group to the [2,1-c]IF core and identical to that in BIF 3 (1.452 Å),13 suggesting the gain of of a partial double bond character for bond i due to efficient delocalization of π-electrons between two [2,1-c]IF units. Although the X-ray structure of 2 is unavailable, the DFT data also suggest a greater π-bond character for C5–C5′ bond i in 4 than that of the C2–C2′ bond in 2 (1.481 Å, Table S4, ESI†).11 While longer CC bonds c and a, and shorter C–C bond i suggest the contribution of a bifluorenylidene-type13,17 diradical form 4-OS1 in the ground state, the short CC bonds b and d indicate the presence of fairly p-quinoidal CS form 4. We attribute such observation in BIF 4 to a room-temperature (rt)-accessible low-lying CS state, which is computed to be only 1.88 kcal mol−1 (Table S1, ESI†) higher in energy than the OS singlet ground state; a phenomenon that is comparable to the IF derivative exhibiting low OS character.12b
Electron paramagnetic resonance (EPR) spectroscopy confirmed the OS behaviour of 4. As shown in Fig. 3c, the powder sample of 4 displayed a characteristic broad EPR signal at 340 K. As the temperature was lowered, the signal intensity decreased due to reduced population of paramagnetic triplet species at lower temperatures (Fig. 3c), confirming an OS singlet ground state. A careful fitting of the EPR data using the Bleaney–Bowers equation18 gave an experimental ΔES–T = −4.12 ± 0.26 kcal mol−1 (Fig. S14, ESI†), which closely matches the theoretical value. The harmonic oscillator model for aromaticity (HOMA, Fig. 3a)19 analysis of optimized structure 4 indicated aromaticity for rings A (0.96) and E (0.95) as these rings displayed insignificant bond length alternation (BLA).20 Rings B (0.03) and D (0.09) are essentially nonaromatic as they showed large BLA (Fig. 3a), whereas central benzenoid ring C exhibits a relatively small BLA (0.58), suggesting its weak aromaticity. Nucleus independent chemical shift21 [NICS(1)zz, Fig. 3a] values for 4 are in line with the HOMA values, suggesting greater aromaticity for rings A (−17.11) and E (−18.82), while ring C is weakly aromatic (−2.73). Rings B (5.59) and D (8.60) exhibit weak antiaromatic character. The geometrical and magnetic criteria of aromaticity analyses are in agreement with the X-ray analysis, suggesting that the OS form of 4 partly contributes to the singlet ground state.
The electronic absorption spectrum of dark-green-colored 4 (Fig. 4a) in chloroform showed several high-energy absorption bands in the UV-vis region including a broad and intense band in the far-red region (λmax = 720 nm, ε = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 M−1 cm−1) that accompanies a shoulder at 785 nm and extends to 935 nm in the near-IR region. The lowest energy absorption band originates from the HOMO → LUMO and HOMO−1 → LUMO+1 transitions, as per the time-dependent-DFT calculations (TD-DFT: λmax(TD) = 644.9 nm, oscillator strength (f) = 0.6203, Table S2, ESI†). The lowest energy wavelength maximum of BIF 4 was found to be red-shifted by 88 nm compared to that of BIF 2, clearly indicating greater electronic communication in 4, which is also evident from the higher molar absorptivity (ε). The optical HLG of 4 was roughly estimated to be 1.33 eV from the absorption edge, which is approximately 0.17 eV smaller than those of BIF 2 and 3. Like compounds 1–3, BIF 4 was also found to be non-emissive to the naked eye. It has a half-life of around 2.7 days in toluene under ambient light and temperature in air (Fig. S13, ESI†), suggesting relatively less stability than 3. However, 4 was stable as a solid for several months when stored in a freezer.
200 M−1 cm−1) that accompanies a shoulder at 785 nm and extends to 935 nm in the near-IR region. The lowest energy absorption band originates from the HOMO → LUMO and HOMO−1 → LUMO+1 transitions, as per the time-dependent-DFT calculations (TD-DFT: λmax(TD) = 644.9 nm, oscillator strength (f) = 0.6203, Table S2, ESI†). The lowest energy wavelength maximum of BIF 4 was found to be red-shifted by 88 nm compared to that of BIF 2, clearly indicating greater electronic communication in 4, which is also evident from the higher molar absorptivity (ε). The optical HLG of 4 was roughly estimated to be 1.33 eV from the absorption edge, which is approximately 0.17 eV smaller than those of BIF 2 and 3. Like compounds 1–3, BIF 4 was also found to be non-emissive to the naked eye. It has a half-life of around 2.7 days in toluene under ambient light and temperature in air (Fig. S13, ESI†), suggesting relatively less stability than 3. However, 4 was stable as a solid for several months when stored in a freezer.
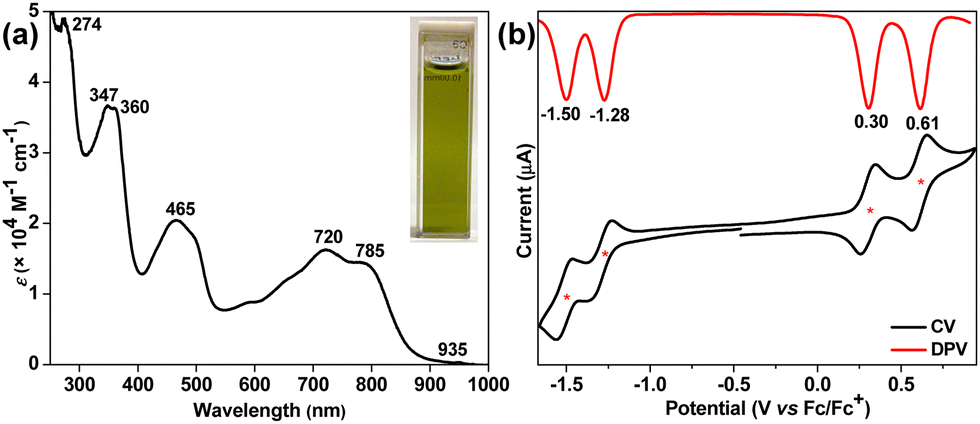 | ||
| Fig. 4 (a) UV-vis-NIR spectrum of 4 in chloroform. (b) CV and DPV of 4 in DCM. | ||
The electrochemical behavior of 4 was studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in dichloromethane using Bu4NPF6 as electrolyte. It displays four-stage redox amphoterism (Fig. 4b) with two reversible reduction waves at half-wave potentials Ered11/2 = −1.28 V and Ered11/2 = −1.50 V, and two reversible oxidation waves at Ered11/2 = 0.30 V and Ered11/2 = 0.61 V (vs. ferrocene/ferrocenium (Fc/Fc+)). The HOMO and LUMO energy levels of 4 were −4.98 eV and −3.65 eV, respectively, as measured from the onset oxidation and reduction potentials. The electrochemical HLG is 1.33 eV for 4, which is 0.29 eV and 0.05 eV smaller than those of 2 and 3, respectively. Notably, both HOMO and LUMO energies of 4 are destabilized in comparison to 3. Importantly, the greater destabilization of the HOMO level for BIF 4 makes it more susceptible to oxidative degradation in air compared to isomeric BIF 3, which is evidenced by the photostability test.
The larger HOMO and LUMO coefficients on the C5/C8 position of 1, relative to C2/C11, suggest that attaching an ambipolar [2,1-c]IF unit3b to the C5 position of another [2,1-c]IF scaffold should influence the HOMO and LUMO energies more than it could do for C2 substitution. Indeed, 4 has more destabilized HOMO and stabilized LUMO levels than those of 2 (Table S6, ESI†).11 Thus, 4 exhibits a smaller HLG than 2, which is crucial12,13 for exhibiting OS properties as a triplet excited state is pushed12b toward the singlet ground state. The SCXRD data of 4 indicate the presence of a moderately quinoidal p-QDM subunit in the IF core, while VT-EPR confirms a singlet OS ground state, which is in line with computations suggesting the OS singlet state of 4 to be slightly more stable than the CS state. Structurally, BIF 2 is CS with a strong p-quinoidal core as it gains no extra aromatic stabilization in its π-extended OS form (Fig. S15, ESI†). However, BIF 4 restores two additional Clar sextets13,22 in diradical form 4-OS1, but has no major driving force toward 4-OS2. Overall, a modest OS character and the presence of a moderately p-quinoidal IF core due to a low-lying CS state suggest the partial contribution of the quinoidal form of 4 to the OS ground state alongside 4-OS1.
In summary, the synthesis and characterization of a novel BIF 4 suggest that linking two [2,1-c]IF units by C5 and C5′ positions, instead of C2 and C2′ positions, can remarkably tune the electronic ground state. A small dihedral angle due to C5–C5′ linkage between two IFs extends the π-delocalization path, resulting in smaller HLG and ΔES–T for 4 than for known BIFs. BIF 4 may be viewed as a desymmetrized [2,1-c]IF due to the attachment of electronically different aryls ([2,1-c]IF/Mes) at C5/C8 positions, resulting in an OS ground state with a low-lying CS state. Though the OS character of [2,1-c]IF is too weak1 to be detected experimentally for 1,2d,3 it is now observable by EPR and SCXRD analyses for 4 and supported by DFT calculations. The fullerene-C60 fragment [2,1-c]IF can act as an electron-accepting scaffold in a bulk-heterojunction device,8 while the high HOMO of [2,1-c]IF dimer 4 suggests its ability as a donor for organic electronics.
S. D. and S. B. gratefully acknowledge the financial support from SERB, India, for the research grants CRG/2022/003012 and CRG/2022/006776, respectively. N. M. and H. S. thank IIT Ropar for junior and senior research fellowships, respectively. P. J. thanks CSIR for a senior research fellowship. The authors thank Kamlesh Satpute (IIT Ropar) for SCXRD data of 4.
Data availability
The data supporting the findings are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Fukuda, T. Nagami, J. Fujiyoshi and M. Nakano, J. Phys. Chem. A, 2015, 119, 10620 CrossRef CAS PubMed.
- (a) D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127 CrossRef CAS PubMed; (b) A. Shimizu and Y. Tobe, Angew. Chem., Int. Ed., 2011, 50, 6906 CrossRef CAS PubMed; (c) A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076 CrossRef CAS PubMed; (d) A. G. Fix, P. E. Deal, C. L. Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362 CrossRef CAS PubMed; (e) J. J. Dressler, Z. Zhou, J. L. Marshall, R. Kishi, S. Takamuku, Z. Wei, S. N. Spisak, M. Nakano, M. A. Petrukhina and M. M. Haley, Angew. Chem., Int. Ed., 2017, 56, 15363 CrossRef CAS PubMed.
- (a) H. Sharma, P. K. Sharma and S. Das, Chem. Commun., 2020, 56, 11319 RSC; (b) H. Sharma, A. Ankita, P. Bhardwaj, U. K. Pandey and S. Das, Org. Mater., 2023, 5, 72 CrossRef CAS.
- (a) Q. Jiang, Y. Han, Y. Zou, H. Phan, L. Yuan, T. S. Herng, J. Ding and C. Chi, Chem. – Eur. J., 2020, 26, 15613 CrossRef CAS PubMed; (b) Á. Martínez-Pinel, L. Lezama, J. M. Cuerva, R. Casares, V. Blanco, C. M. Cruz and A. Millán, Org. Lett., 2024, 26, 6012 CrossRef PubMed; (c) E. Sidler, R. Hein, D. Doellerer and B. L. Feringa, J. Am. Chem. Soc., 2024, 146, 19168 CrossRef CAS PubMed.
- S. Herzog, A. Hinz, F. Breher and J. Podlech, Org. Biomol. Chem., 2022, 20, 2873 RSC.
- S. Nobusue, H. Miyoshi, A. Shimizu, I. Hisaki, K. Fukuda, M. Nakano and Y. Tobe, Angew. Chem., Int. Ed., 2015, 54, 2090 CrossRef CAS PubMed.
- T. Jousselin-Oba, P. E. Deal, A. G. Fix, C. K. Frederickson, C. L. Vonnegut, A. Yassar, L. N. Zakharov, M. Frigoli and M. M. Haley, Chem. – Asian J., 2019, 14, 1737 CrossRef CAS PubMed.
- K. Paudel, B. Johnson, M. Thieme, M. M. Haley, M. M. Payne, J. E. Anthony and O. Ostroverkhova, Appl. Phys. Lett., 2014, 105, 043301 CrossRef.
- S. Thomas and K. S. Kim, Phys. Chem. Chem. Phys., 2014, 16, 24592 RSC.
- (a) R. Casares, Á. Martínez-Pinel, S. Rodríguez-González, I. R. Márquez, L. Lezama, M. T. González, E. Leary, V. Blanco, J. G. Fallaque, C. Díaz, F. Martín, J. M. Cuerva and A. Millán, J. Mater. Chem. C, 2022, 10, 11775 RSC; (b) R. Casares, S. Rodríguez-González, Á. Martínez-Pinel, I. R. Márquez, M. T. González, C. Díaz, F. Martín, J. M. Cuerva, E. Leary and A. Millán, J. Am. Chem. Soc., 2024, 146, 29977 CrossRef CAS PubMed.
- H. Sharma, Ankita, V. Mittal, U. K. Pandey and S. Das, Org. Lett., 2024, 26, 2617 CrossRef CAS PubMed.
- (a) S. Das and J. Wu, Phys. Sci. Rev., 2017, 2, 20160109 Search PubMed; (b) P. K. Sharma, P. Jana, S. Bandyopadhyay and S. Das, Chem. Commun., 2024, 60, 7319 RSC.
- H. Sharma, P. Jana, D. Mallick, S. Bandyopadhyay and S. Das, Chem. Sci., 2024, 15, 20215 RSC.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Molecules, 2020, 25, 711 CrossRef CAS.
- S. Thiery, D. Tondelier, C. Declairieux, B. Geffroy, O. Jeannin, R. Métivier, J. Rault-Berthelot and C. Poriel, J. Phys. Chem. C, 2015, 119, 5790 CrossRef CAS.
- S. Das, S. Lee, M. Son, X. Zhu, W. Zhang, B. Zheng, P. Hu, Z. Zeng, Z. Sun, W. Zeng, R. Li, K. Huang, J. Ding, D. Kim and J. Wu, Chem. – Eur. J., 2014, 20, 11410 CrossRef CAS PubMed.
- Y. Ishigaki, T. Harimoto, T. Shimajiri and T. Suzuki, Chem. Rev., 2023, 123, 13952 CrossRef CAS PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- H. Sharma, N. Bhardwaj and S. Das, Org. Biomol. Chem., 2022, 20, 8071 RSC.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Org. Lett., 2006, 8, 863 CrossRef CAS PubMed.
- (a) P. Yadav, S. Das, H. Phan, T. S. Herng, J. Ding and J. Wu, Org. Lett., 2016, 18, 2886 CrossRef CAS PubMed; (b) Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed syntheses and characterization data, NMR spectra; X-ray crystallographic data of 4. CCDC 2411678. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06643j |
| This journal is © The Royal Society of Chemistry 2025 |