
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of highly fluorescent helical quinolizinium salts by a Rh-catalyzed cyclotrimerization/C–H activation sequence†
Timothée
Cadart
a,
Lucia
Feriancová
a,
Petr
Henke
 b,
Robert
Gyepes
c,
Ivana
Císařová
b,
Květa
Kalíková
d and
Martin
Kotora
*a
b,
Robert
Gyepes
c,
Ivana
Císařová
b,
Květa
Kalíková
d and
Martin
Kotora
*a
aDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Praha 2, Czech Republic. E-mail: martin.kotora@natur.cuni.cz
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Praha 2, Czech Republic
cAcademy of Sciences of the Czech Republic J. Heyrovský Institute of Physical Chemistry, v.v.i. Dolejškova 2155/3, 182 23 Praha 8, Czech Republic
dDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University, Hlavova 8, 128 00 Praha 2, Czech Republic
First published on 28th January 2025
Abstract
A series of helical quinolizinium salts were prepared utilizing Rh-catalyzed [2+2+2]cyclotrimerization and C–H activation processes as the crucial synthetic steps. The cyclotrimerization of appropriately substituted diynes with trimethylsilylethyne under Rh-catalyzed conditions provided the 1-arylisoquinolines in up to 61% isolated yields. Their Rh-catalyzed C–H activation/annulation with various aryl and alkyl disubstituted alkynes gave rise to [7]-helical quinolizinium salts in high isolated yields (up to 93%). Enantioselective C–H activation was also tried with asymmetric induction up to 62% ee. The respective boron and platinum complexes of 1-arylisoquinolines were prepared as well. All prepared compounds exhibit fluorescence in the orange-red light region (606–682 nm) with ΦFs 28–99%.
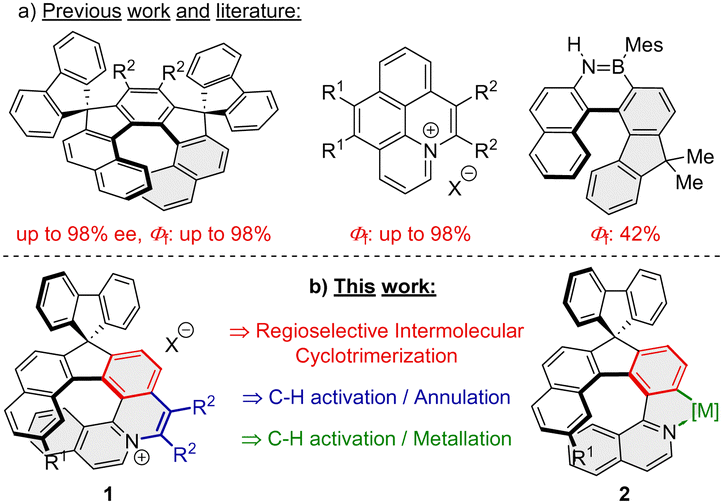
Organic helical compounds are a class of substances possessing a curved scaffold composed of alternating ortho-fused rings.1 They have interesting physical properties for potential applications in material science thanks to their twisted shapes and organization in the solid state.2 Incorporation of heteroatoms into the helicene scaffold has recently emerged as an option to change their structural flexibility and electronic properties, as exemplified by N- and NB-embedded helicenes.3–6 One such potential application of helicenes is CPL light emitting materials. However, [n]helicenes and their respective heteroatom analogues usually exhibit low fluorescence quantum yields (Φf ∼ 4%).7–10 This can be explained by fast intersystem crossing from the singlet excited state to the triplet state, which makes them not so attractive for further application in materials science.
In principle, there are three strategies to overcome this problem and increase the quantum yield of compounds with helical scaffolds: (i) introduction of appropriate substituents and/or heteroatoms,11–13 (ii) lateral extension of the helicene framework,14–16 and (iii) introduction of a 9,9′-spirobifluorene moiety,17 because its presence has proved to avoid excimer formation in the solid state.18 However, it should be kept in mind that the structure–properties relationship between the above-mentioned factors is rather complicated and often combines all the afore-mentioned effects.
We envisioned that the incorporation of an azonia moiety into the helical framework will improve its photophysical properties, because high fluorescence quantum yields (Φf) up to 90% were reported for various planar azonia salts.19–21 Although numerous [n]azoniahelicenes (possessing ≥5 ortho-condensed rings) have been synthesized over the last two decades,22–24 just a handful of data is available regarding photophysical properties.23,25 As far as introduction of a substituted fluorene moiety in the substrate's molecular framework is concerned, its presence increased Φf several fold as can be exemplified by a [7]-helical compound (Φf = 40%), with an improvement by almost one order of magnitude.10 A similar phenomenon was observed for a B,N-embedded [6]-helical compound possessing a 9,9′-dimethylfluorene moiety (Φf = 42%), where the value for the maternal BN-[6]helicene was merely 21%.26 Even higher fluorescence quantum yields (up to 88%) were observed for dispiroindeno[2,1-c]fluorenes possessing [5]-, [7]-, and [9]-helical scaffolds.27–29 Application of the above-mentioned principles may serve design and syntheses of helical compounds having emission maxima in the red or NIR region with reasonably high Φfs, such as an azabuckybowl-helicene hybrid (λem = 770 nm, Φf = 28%),30 contorted superhelicenes (λem = 680 and 697 nm, Φf = 43%),31 and naphthalimide-annulated [n]helicenes (λem = 613 and 655 nm, Φf = 69 and 73%).32
Given our experience with the synthesis of dispiroindenofluorenes27–29 and naphthoquinolizinium salts (Scheme 1a),21 we saw an opportunity in developing a synthetic route towards a new class of compounds that would combine both structural features: the azonia moiety and a helical spirobifluorene fragment. In this report, we would like to demonstrate that the desired helical azonia salts 1 and helical metal complexes 2 (Scheme 1b) can be prepared utilizing two crucial steps relying on transition metal catalyzed reactions: (a) [2+2+2] cyclotrimerization and (b) C–H activation/annulation.
 | ||
| Scheme 1 Previous works (a) and our synthetic proposal (b). | ||
At the outset, we synthesized two diynols 3a and 3b bearing the isoquinoline moiety that are the crucial substrates for catalytic [2+2+2] cyclotrimerization from commercially available starting materials in three step synthesis (for details see Section S2, ESI†). Having 3a and 3b in hand, screening of different transition metal-based catalysts for the [2+2+2] cyclotrimerization of diynols 3 with trimethylsilylacetylene was carried out (Table 1). The intermediate alcohols were not isolated and directly oxidized to ketones 4 to avoid their rather tedious separation and purification. Using an array of common catalytic systems based on Co, Ni, and Ir complexes to induce cyclotrimerization of 3a did not provide encouraging results (entries 1–3). Only catalysis by using Rh complexes33 indicated minor success (entries 4 and 5). According to 1H NMR analysis of the reaction mixtures, the desired product 4a was formed in just 5% yield as a mixture of the ortho4a and meta4a′ regioisomers in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and 79:21 ratios, respectively. Since we assumed that the low yields could be the result of low solubility of the starting material 3a in non-polar solvents, the subsequent cyclotrimerizations were carried out in a 1:1 mixture of THF/MeOH. The change of the solvent had a beneficial effect on the course of the reaction and cyclotrimerization of 3a by using the cationic Rh-complex gave a mixture of 4a and 4′a (95:5) in 26% yield (entry 6). Increase of the catalyst loading from 5 to 10 mol% resulted in an improved isolated yield of 59% (entry 7). Gratifyingly, applying the same reaction conditions as in entry 6 provided 4b/4′b in 61% isolated yield (entry 8). Additionally, isolation and recrystallization of 4a and 4b followed by single crystal X-ray diffraction analyses unequivocally confirmed their structures (see Section S5, ESI†).
:5 and 79:21 ratios, respectively. Since we assumed that the low yields could be the result of low solubility of the starting material 3a in non-polar solvents, the subsequent cyclotrimerizations were carried out in a 1:1 mixture of THF/MeOH. The change of the solvent had a beneficial effect on the course of the reaction and cyclotrimerization of 3a by using the cationic Rh-complex gave a mixture of 4a and 4′a (95:5) in 26% yield (entry 6). Increase of the catalyst loading from 5 to 10 mol% resulted in an improved isolated yield of 59% (entry 7). Gratifyingly, applying the same reaction conditions as in entry 6 provided 4b/4′b in 61% isolated yield (entry 8). Additionally, isolation and recrystallization of 4a and 4b followed by single crystal X-ray diffraction analyses unequivocally confirmed their structures (see Section S5, ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Solvent | Temp. (°C) | Yieldb (%) | 4/4′ |
| a Reaction scale: 0.1 mmol; entries 1–7, 3a: R1 = H; entry 8, 3b: R1 = OMe. b Combined isolated yields. c Performed in a MW reactor. d 0.25 mmol scale. e 1.5 mmol scale. | |||||
| 1 | CpCo(P(EtO)3)dmfu (10) | Toluene | 110 | 0 | |
| 2 | Ni(cod)QD (10), PPh3 (20) | Toluene | 110 | 0 | |
| 3 | [IrCl(cod)]2 (10), PPh3 (20) | Toluene | 110 | 0 | |
| 4 | Rh(cod)2BF4 (5), dppb (6) | DCE | 100 | 5 | 95/5 |
| 5c | RhCl(PPh3)3 (10) | THF | 170 | 5 | 79/21 |
| 6 | Rh(cod)2BF4 (5), dppb (6) | THF/MeOH | 100 | 26 | 95/5 |
| 7d | Rh(cod)2BF4 (10), dppb (12) | THF/MeOH | 100 | 59 (57)e | 95/5 |
| 8 | Rh(cod)2BF4 (5), dppb (6) | THF/MeOH | 100 | 61 | 98/2 |
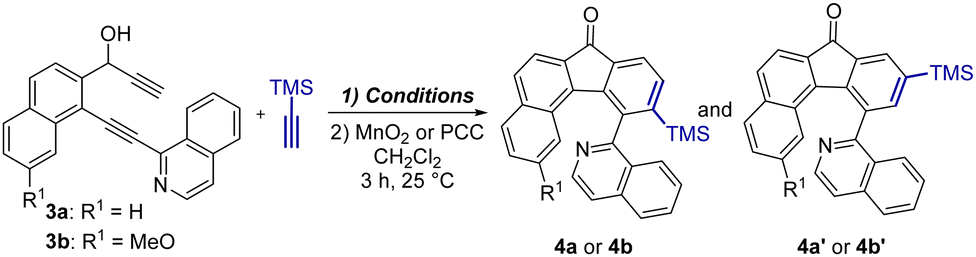
Since it has been previously shown that a proper choice of ligands around the central metal atom can considerably influence the regioselectivity and yields of catalytic cyclotrimerization,33–36 the use of different bidentate phosphines was screened (see Section S2.3 and Table S3, ESI†). In general, none of these catalytic systems could match the result obtained with dppb in terms of the yield, albeit the use of BINAP and dppf gave higher preferential selectivity for the formation of 4a (4:4′ = 97:3 in both cases).
Next, mixtures of regioisomeric ketones were converted to spirofluorenes 5a and 5b in a reaction sequence comprising 1,2-addition of lithiobiphenyl (formed in situ) to the carbonyl group followed by intramolecular Friedel–Crafts reaction induced by trifluoroacetic acid under reflux (Scheme 2, the first step). It is worth mentioning that acidic conditions favour not only the intramolecular Friedel–Crafts reaction but also the TMS-desilylation to give only one product 5a or 5b. Both spirofluorenes 5a or 5b were successfully obtained in high 86% and 82% isolated yields over two steps, respectively.
 | ||
| Scheme 2 Spirocyclization and synthesis of helical quinolizinium salts 1. | ||
Then, we proceeded with a rhodium-catalyzed C–H activation/annulation of spirosubstances 5a and 5b towards the helical quinolizinium salts (Scheme 2, the second step). Based on our experience, the annulation was performed by using the dimeric rhodium catalyst [CpRhCl2]2 in the presence of Cu(OAc)2 and silver salt (AgBF4) in dichloroethane at 100 °C.21 Spirocompound 5a reacted with three symmetric diarylethynes bearing electron-donating or electron-withdrawing groups (pMeO-C6H4, Ph, pCF3-C6H4), and 4-octyne. The corresponding helical azonia salts 1aa–1ad were formed in high yields (up to 93%). Regarding compound 5b, the same reaction conditions and reactants furnished products 1ba–1bd in high yields as well (up to 90%).
Then, we attempted conversion of 1-arylisoquinoline 5a to B,N- and Pt-embedded helical compounds (Scheme 3), because it has been demonstrated that such substances37–39 show high appreciation for their potential applications in OLED devices.40
 | ||
| Scheme 3 Synthesis of helical azabora- and platinum compounds 2. | ||
As for the former, 5a was treated with BBr3 in the presence of i-Pr2NEt and the subsequent methylation with trimethylaluminum in toluene provided azabora[7]-helical compound 2a in a satisfactory yield of 56% (over 2 steps). As for the latter, a two-step reaction sequence starting with 5a in the presence of K2PtCl4 in a mixture of ethoxyethanol and water under reflux furnished a platinum dimer, which upon treatment with acetylacetone in the presence of sodium carbonate yielded the expected platinum complex 2b. Unfortunately, despite all our efforts, the platinum complex 2b was isolated in only 8% yield so far.
UV/Vis absorption and fluorescence spectra of our [7]-helical quinolizinium salts 1 and complexes 2 in dichloromethane are shown in Fig. 1, the spectral data are summarized in Table 2 (for all details, see Section S4, ESI†). Only small differences in emission maxima in the series of 1aa–1ad and 1ba–1bd were recorded. The emission maxima for the methoxy substituted series 1ba–1bd are bathochromically shifted by 40–50 nm with respect to the unsubstituted series of 1aa–1ad. As far as the substituent effect is concerned, the presence of methoxyphenyl substituents slightly shifts the emissions towards the blue light region (610 nm for 1aa and 647 nm for 1ba) with respect to the phenyl substituted derivatives 1ab and 1bb (611 and 653 nm, respectively), on the other hand the presence of (trifluoromethyl)phenyl substituents shifts the emission maxima towards the red-light region (630 nm for 1ac and 682 nm for 1bc). It is worth mentioning that the trend, albeit small, is opposite to the one observed for cationic 11-azapyrenes,21 cationic 12-azapyrene,19,41 and substituted quinolizinium compounds.42 Concerning the quantum yields, higher Φf in the range of 86–99% were recorded for 1aa–1ad, whereas for the series possessing the methoxy substituent 1ba–1bd they dropped to 28–55%. All of the compounds also possess a large Stokes shift with values up to 0.491 eV. HOMO → LUMO transitions for 1ba, 1bb, and 1bc were calculated (see the ESI†).
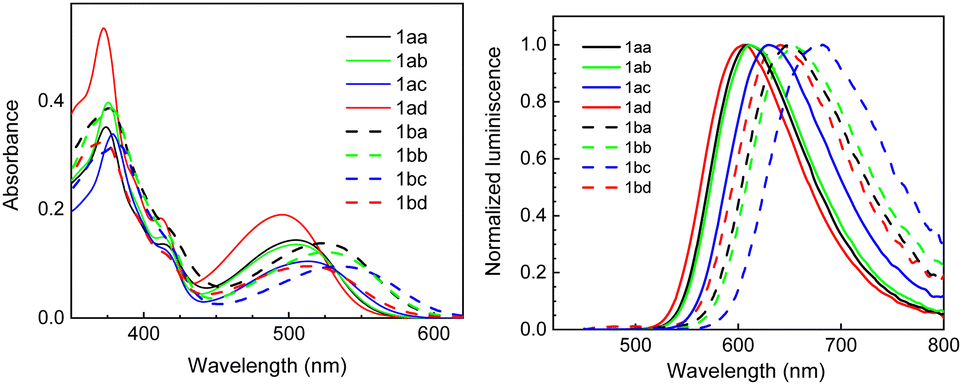 | ||
| Fig. 1 Normalized absorption and emission spectra of 1 recorded in CH2Cl2. | ||
| 1 | λ Amax (nm) | ε | λ Fmax (nm) | Φ f (%) |
|---|---|---|---|---|
| a 103 M−1 cm−1. b In parentheses are reported λFmax (nm) in the solid state. c In parentheses are reported Φf (%) in the solid state. | ||||
| 1aa | 374, 506 | 18, 7 | 610 (608) | >99 (62) |
| 1ab | 376, 506 | 19, 6 | 611 (622) | 95 (34) |
| 1ac | 379, 514 | 19, 6 | 630 (610) | 86 (66) |
| 1ad | 372, 496 | 23, 8 | 606 (614) | 91 (21) |
| 1ba | 376, 523 | 21, 7 | 647 (657) | 55 (34) |
| 1bb | 378, 525 | 19, 6 | 653 (636) | 47 (21) |
| 1bc | 382, 537 | 18, 6 | 682 (676) | 28 (24) |
| 1bd | 372, 514 | 15, 4 | 641 (670) | 48 (4) |
Concerning the metal complexes, azabora compound 2a shows intense blue-green fluorescence with an emission maximum at 497 nm in CH2Cl2 and 505 nm in the solid state with quantum yields (Φf) up to 34% (see Section 4, Fig. S3 and Table S6, ESI†). Emission maxima for structurally similar azabora-helical compounds were reported to be in the range of 459–477 nm.37c On the other hand, the emission maximum of 2b is significantly shifted by almost 200 nm to the red-light region (690 nm in CH2Cl2 and 676 nm in the solid state) similarly to the [7]-helical quinolizinium salts 1 (Table 1), but with rather low quantum yield typical for the previously reported Pt(II)-helicene complexes (1–10%).39
Compounds 5a constitutes also a suitable substrate to attempt an enantioselective C–H activation/annulation reaction sequence towards helical azonia salts, which have been shown to proceed with high asymmetric induction as reported by You et al.24 Preliminary experiments of reactions of 5a and 1,2-bis(4-(trifluoromethyl)phenyl)acetylene as model substrates showed that enantioenriched 1ac could by obtained in 62% ee by using a catalytic system composed of C1 (10 mol%), AgBF4 (1 eq.), and Cu(OAc)2 (1 eq.), but in a low yield of 10% (Section S2.4, ESI†). The level of asymmetric induction is close to the value obtained in the synthesis of a [7]-azoniahelicene, where it reached 73% ee.24 For full account of preliminary experiments see the ESI,† Section 2.4.
In conclusion, a short synthesis of [7]-helical quinolizinium salts possessing a spirofluorene motif was developed by using a Rh-catalyzed cyclotrimerization and a late stage C–H activation/annulation reaction. In this respect, an appropriately substituted 1-arylisoquinoline motif was a suitable choice for post functionalization and gave us access to a library of helical quinolizinium salts, a platinum complex, and an azabora-compound. Photo-physical properties of the prepared compounds were studied, and these showed high fluorescence (Φf up to 99% in solution and up to 66% in solid state) with an emission maximum range of 610–690 nm (orange-red light), bathochromically shifted in comparison with the maternal spirofluorenes and indenofluorenes. Furthermore, preliminary attempts on enantioselective annulation were successful with enantioselectivity up to 62% ee. Although these results indicate that reasonable asymmetric induction could be achieved, it is obvious that further extensive fine tuning of the catalyst design will be needed to improve the results.
The authors acknowledge the financial support from the Czech Science Foundation (21-29124S and 21-39639L), the Charles University Research Centre program No. UNCE/SCI/014, and European Structural and Investment Funds (No. CZ.02.2.69/0.0/0.0/18_053/0016976). We thank Dr Jan Ulč for his help in the synthesis of chiral Cp-complexes.
Data availability
The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- M. Gingras, Chem. Soc. Rev., 2013, 42, 1051–1095 RSC.
- For a review on heterohelicenes, see: K. Dhaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef PubMed.
- W.-W. Yang and J.-J. Shen, Chem. – Eur. J., 2022, 28, e202202069 CrossRef CAS PubMed.
- A. V. Gulevskaya and D. I. Tonkoglazova, Adv. Synth. Catal., 2022, 364, 2502–2539 CrossRef CAS.
- A. Nowak-Król, P. T. Geppert and K. R. Naveen, Chem. Sci., 2024, 15, 7408–7440 RSC.
- A.-C. Bédard, A. Vlassova, A. C. Hernadez-Perez, A. Besette, G. S. Hanan and M. A. Heuft, Chem. – Eur. J., 2013, 19, 16295–16302 Search PubMed.
- H. Kubo, T. Hirose and K. Matsuda, Org. Lett., 2017, 19, 1776–1779 CAS.
- J. Malinčík, S. Gaikwad, J. P. Mora-Fuentes, M.-A. Boillat, A. Prescimone, D. Häussinger, A. G. Campaña and T. Šolomek, Angew. Chem., Int. Ed., 2022, 61, e202208591 Search PubMed.
- H. Oyama, M. Akiyama, K. Nakanmo, M. Naito, K. Nobusawa and K. Nozaki, Org. Lett., 2016, 19, 3654–3657 Search PubMed.
- M. K. Kubitz,W. Haselbach, D. Sretenovič,M. Bracker, M. Kleinschmidt, R. Kühnemuth, C. A. M. Seidel, C. Gilch and C. Czekelius, ChemPhotoChem, 2023, 7, e20220334 Search PubMed.
- X. Lv, C. Gao, T. Han, H. Shi and W. Guo, Chem. Commun., 2020, 56, 715–718 Search PubMed.
- T. Sakurai, M. Kobayashi, H. Yoshida and M. Shimizu, Crystals, 2021, 11, 1105 CrossRef CAS.
- Z. Qiu, C.-W. Ju, L. Frédéric, Y. Hu, D. Schollmeyer, G. Pieters, K. Müllen and A. Narita, J. Am. Chem. Soc., 2021, 143, 4661–4667 CrossRef CAS PubMed.
- P. Izquierdo-Garcia, J. M. Fernandez-Garcia, S. Medina Rivero, M. Šámal, J. Rybáček, L. Bednárová, S. Ramirez-Barroso, F. J. Ramirez, R. Rodriguez, J. Perles, D. Garcia-Fresnadillo, J. Crassous, J. Casado, I. G. Stará and N. Martin, J. Am. Chem. Soc., 2023, 145, 11599–11610 Search PubMed.
- F. Full, Q. Wölflick, K. Radacki, H. Braunschweig and A. Nowak-Król, Chem. Eur. J., 2022, 28, e20220228 CrossRef PubMed.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- C. Poriel, C. Quinton, F. Lucas, J. Rault-Berthelot, Z.-Q. Jiang and O. Jeannin, Adv. Funct. Mater., 2021, 31, 2104980 CrossRef CAS.
- B. Feng, D. Wan, L. Yan, V. D. Kadam, J. You and G. Gao, RSC Adv., 2016, 6, 66407–66411 RSC.
- For a review, see: P. Karak, S. S. Rana and J. Choudhury, Chem. Commun., 2022, 58, 133–154 RSC.
- J. Ulč, J. Jacko, I. Císařová, L. Pospíšil, D. Nečas and M. Kotora, Eur. J. Org. Chem., 2023, e2023001 Search PubMed.
- K. Xu, Y. Fu, Y. Zhou, F. Hennersdorf, P. Machata, I. Vincon, J. J. Weigand, A. A. Popov, R. Berger and X. Feng, Angew. Chem., Int. Ed., 2017, 56, 15876–15881 CrossRef CAS PubMed.
- Z. Wang, L. Jiang, J. Ji, F. Zhou, J. Lan and J. You, Angew. Chem., Int. Ed., 2020, 59, 23532–23536 CrossRef CAS PubMed.
- Q. Wang, W.-W. Zhang, C. Zheng, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 114–120 CrossRef CAS PubMed.
- R. Passeri, G. G. Aloisi, F. Elisei, L. Latterini, T. Caronna, F. Fontana and I. S. Sora, Photochem. Photobiol. Sci., 2009, 8, 1574–1582 Search PubMed.
- K. Yuan, D. Volland, S. Kirschner, M. Uzelac, G. S. Nichol, A. Nowak-Król and M. J. Ingleson, Chem. Sci., 2022, 13, 1136–1145 RSC.
- R. P. Kaiser, D. Nečas, T. Cadart, R. Gyepes, I. Císařová, J. Mosinger, L. Pospíšil and M. Kotora, Angew. Chem., Int. Ed., 2019, 58, 17169–17174 CrossRef CAS PubMed.
- T. Cadart, D. Nečas, R. P. Kaiser, L. Favereau, I. Císařová, R. Gyepes, J. Hodačová, K. Kalíková, L. Bednárová, J. Crassous and M. Kotora, Chem. – Eur. J., 2021, 27, 11279–11284 CrossRef CAS PubMed.
- T. Cadart, T. Gläsel, I. Císařová, R. Gyepes, M. Hapke, D. Nečas and M. Kotora, M. Chem. Eur. J., 2023, 29, e202301491 Search PubMed.
- Y.-F. Wu, S.-W. Ying, L.-Y. Su, J.-J. Du, L. Zhang, B.-W. Chen, H.-R. Tian, H. Xu, M.-L. Zhang, X. Yan, Q. Zhang, S.-Y. Xie and L.-S. Zheng, J. Am. Chem. Soc., 2022, 144, 10736–10742 CrossRef CAS PubMed.
- S. Míguez-Lago, I. F. A. Mariz, M. A. Medel, J. M. Cuerva, E. Maçôas, C. M. Cruz and A. G. Campaña, Chem. Sci., 2022, 13, 10267–10272 RSC.
- X. Tian, K. Shoyama, B. Mahlmeister, F. Brust, M. Stolte and F. Würther, J. Am. Chem. Soc., 2023, 145, 9886–9894 CrossRef CAS PubMed.
- E. Matoušová, R. Gyepes, I. Císařová and M. Kotora, Adv. Synth. Catal., 2016, 358, 254–267 CrossRef.
- I. Caivano, R. P. Kaiser, F. Schnurrer, J. Mosinger, I. Císařová, D. Nečas and M. Kotora, Catalysts, 2019, 9, 942–950 CrossRef.
- J. M. Halford, A. M. Z. Slawin and A. J. B. Watson, ACS Catal., 2023, 13, 3463–3470 CrossRef.
- R. Abe, Y. Nagashima, J. Tanaka and K. Tanaka, ACS Catal., 2023, 13, 1604–1613 Search PubMed.
- (a) C. S. Shen, M. Srebro-Hooper, M. Jean, N. Vanthuyne, L. Toupet, J. A. G. Williams, A. R. Torres, A. J. Riives, G. Muller, J. Autschbach and J. Crassous, Chem. – Eur. J., 2017, 23, 407–418 CrossRef CAS PubMed; (b) Z. Domínguez, R. López-Rodríguez, E. Álvarez, S. Abbate, G. Longhi, U. Pischel and A. Ros, Chem. – Eur. J., 2018, 24, 12660–12668 CrossRef PubMed; (c) J. Full, S. P. Panchal, J. Götz, A.-M. Krause and A. Nowak-Król, Angew. Chem., Int. Ed., 2021, 60, 4350–4357 CrossRef CAS PubMed.
- F. Full, M. J. Wildervanck, D. Volland and A. Nowak-Król, Synlett, 2023, 477–482 CAS.
- E. Anger, M. Rudolph, L. Norel, S. Zrig, C. Shen, N. Vanthuyne, L. Toupet, J. A. G. Williams, C. Roussel, J. Autschbach, J. Crassous and R. Réau, Chem. – Eur. J., 2011, 17, 14178–14198 CrossRef CAS PubMed.
- (a) D. Li, H. Y. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 Search PubMed; (b) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat’ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed.
- Q. Ge, Y. Hu, B. Li and B. Wang, Org. Lett., 2016, 18, 2483–2486 CrossRef CAS PubMed.
- W.-M. Yip, Q. Yu, A. Tantipanjapotn, W.-C. Chan, J.-R. Deng and B. C. Ko Wong, Org. Biomol. Chem., 2021, 19, 8507–8515 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2373208–2373212. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06512c |
| This journal is © The Royal Society of Chemistry 2025 |