
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-Monoarylated dihydrophenazines in reduced and oxidized states as efficient organo-photocatalysts†
Maurizio
Prato
 *abc and
Jacopo
Dosso
*a
*abc and
Jacopo
Dosso
*a
aDepartment of Chemical and Pharmaceutical Sciences, CENMAT, Centre of Excellence for Nanostructured Materials, INSTM UdR Trieste, University of Trieste, via Licio Giorgieri 1, 34127 Trieste, Italy. E-mail: Jacopo.dosso@units.it; prato@units.it
bCentre for Cooperative Research in Biomaterials (CIC BiomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, 20014, Donostia San Sebastián, Spain
cBasque Fdn Sci, Ikerbasque, 48013 Bilbao, Spain
First published on 9th January 2025
Abstract
In this work, the synthesis of an N-monoarylated dihydrophenazine is reported together with its interconversion to its oxidized mono-cationic form. While the reduced state was employed for the dechlorination of aromatic substrates, the oxidized mono-cationic one was exploited for the formation of C–N bonds between aryl rings and azoles, which was achieved with high yields and very low catalyst loadings (down to 0.5 mol%).
N,N-Diphenyl-dihydrophenazines belong to a class of aromatic compounds that have undergone a renaissance in the last few years, due to their great potential for sensing,1–3 light emission,4,5 molecular switching6,7 and catalysis.8–11 In particular, dihydrophenazines have been extensively investigated, especially as photo-reducing agents (Fig. 1, I).9,10 In this perspective, we recently demonstrated that through π-extension of the dihydrophenazine scaffolds, it is possible not only to tune the optoelectronic properties, but also to introduce hydroxyl functional groups on the π-extended scaffold, which enables efficient photocatalysis.12 Moreover, π-extension coupled with the design of the aromaticity patterns, resulted in much more accessible and stable dicationic states.7,13 Given their electron-depleted nature and generally intense absorption in the visible region, dicationic oxidized states of dihydrophenazines should present great potential as organo-photo oxidizers, leading to the potential use of both states (neutral and dicationic) as active photocatalysts. This is of course highly interesting since it would enable the use of the same scaffold for different photo-redox reactions, resulting in a great versatility and practicality. Despite this, while many examples of photocatalytic applications of the neutral dihydrophenazine have been reported, the use of dicationic dihydrophenazine derivatives for this application has been hindered by their reactivity14 and general instability towards moisture and nucleophiles which persisted, albeit to a lesser extent, also in π-extended derivatives.7 One possible solution to this, could be the preparation of N-monoarylated dihydrophenazines, which upon oxidation should form mono-cationic phenazine derivatives (1 and 1ox, Fig. 1).
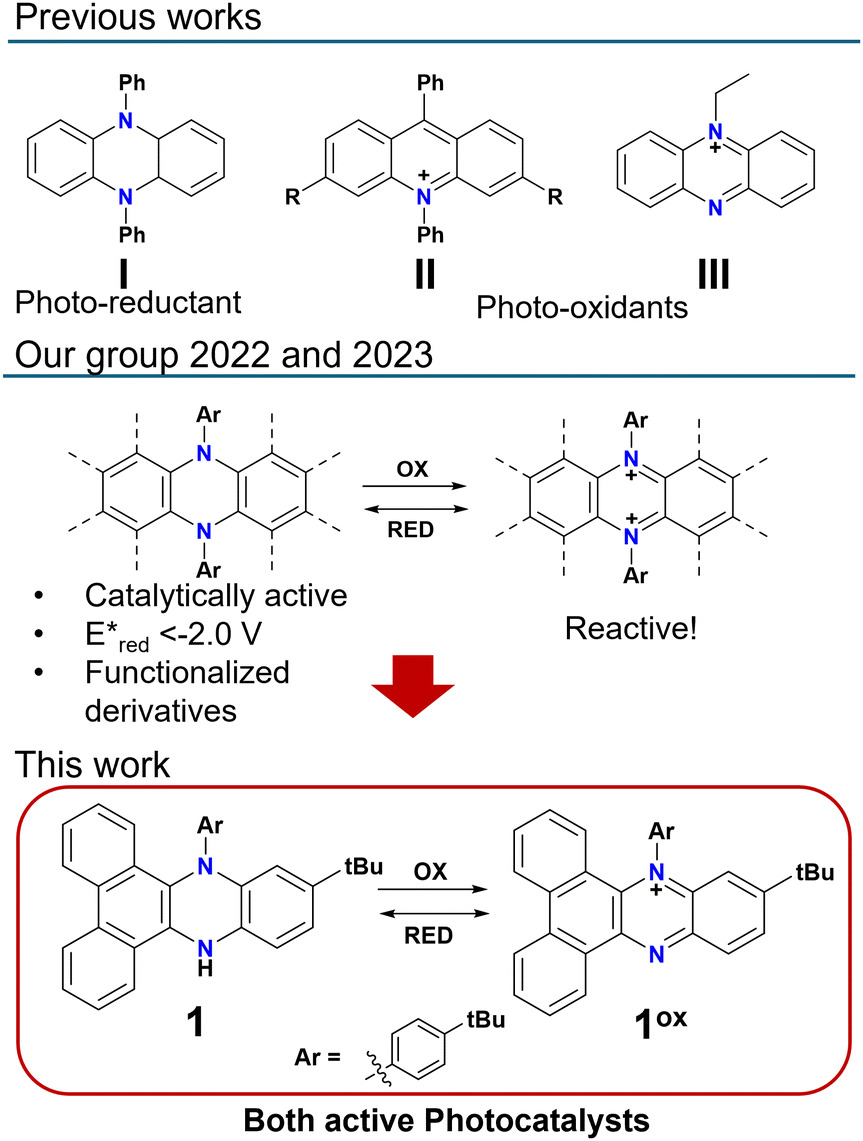 | ||
| Fig. 1 Previously reported photocatalytic systems and recent work on π-extended N,N-diphenyl dihydrophenazine systems. | ||
The latter should present an ideal balance between stability and reactivity aiming at catalytic applications, possibly with activities similar to or better than those of the well-established acridinium catalysts (Fig. 1, II).15–18 From this point of view, a few examples of mono-cationic phenazinium cations have been reported in the literature, and have been generally exploited for DNA binding19 or, in a notable example, photocatalytic oxidation of aldehydes to amides.20 In the former example, π-extended systems were used,19 while in the latter the simpler phenazine ethosulfate (Fig. 1, III) was employed with low (1–2 mol%) catalyst loadings resulting in promising high yields.20 Some more examples of similar mono-cationic systems employed as photocatalysts exist, but are generally based on functionalized safranine dyes presenting excited state oxidation potentials much lower than acridinium systems.21–23
Based on this, to prepare target molecule 1, it was decided to start by synthesising phenanthrene diimine 2, presenting tBu groups for enhancing the solubility of the system. To achieve this, phenanthrenequinone was treated with 4-tBu aniline in the presence of TiCl4 and pyridine, resulting in the formation of 2 in a 40% yield (Scheme 1). As correctly reported by Hoye and co-workers, also diamine 3 was isolated in a modest 9% yield. Subsequently, 2 was refluxed in a THF/EtOH mixture. As a result, after 3 hours, the formation of the desired product could be observed, which proved to be 1 (see Fig. S32–S37, ESI†) in accordance with the expected 6π-electrocyclization.24 Since 1 proved to be partially oxidized during the reaction, this was repeated with the addition of 1 equivalent of NaBH4 resulting in a cleaner outcome with isolation of 1 in a 63% yield. Derivative 1 proved to be stable for weeks in the solid state and could be completely characterised via nuclear magnetic resonance spectroscopy (NMR). Given that the two-electron oxidation of 1 should result in the formation of phenazinium system 1ox, the former was treated with an excess of AgSbF6 (3 equivalents) resulting in an immediate colour change from faint yellow to brown/orange associated with the presence of a new species. After filtration to remove silver salts and reprecipitation from Et2O, the new compound was isolated and characterised by NMR and high-resolution mass spectroscopy (HRMS) confirming the structure of 1ox. Interestingly, the conversion between 1 and 1ox proved to be chemically reversible by treating the latter with NaBH4, as demonstrated by NMR experiments (see Fig. S23–S25, ESI†), thus enabling the interconversion between the two species. To prove the usefulness of derivatives 1 and 1ox as organo-photocatalysts, the optoelectronic properties of both derivatives were analysed. Derivative 1 presents an absorption with a λmax = 354 nm, tailing in the visible region (up to ca. 450 nm), associated with a broad emission λmax = 590 nm (Fig. S1, ESI†). On the other hand, 1ox presents a more red-shifted absorption with λmax = 462 nm in the blue region of the spectra, resembling similar derivatives reported in the literature.19 Also in this case, the emission is broad with λmax = 630 nm associated with a less marked Stokes shift compared to 1 (Fig. S7, ESI†). For both species the quantum yields proved to be modest, with a value <5% for 1 and of 5% for 1ox. Cyclic voltammetry measurements were carried out for both compounds in CH2Cl2. In the case of 1 only a single quasi reversible oxidative event was visible at E1/2 = +0.24 V vs. SCE (Fig. S17, ESI†). On the other hand, for 1ox two reversible reductive events were observed, at E1/2 = −0.21 and −0.84 V vs. SCE (Fig. S18, ESI†) while no oxidations were detected. Based on these values, using the Rehm–Weller equation,25 it was possible to estimate an excited state oxidation potential of −2.25 V for 1 and an excited state reduction potential of +2.13 V for 1ox,26 suggesting that indeed the two derivatives are highly promising in terms of photo-reduction and oxidation of organic substrates. In particular, in the case of 1ox, the observed excited state oxidation potential is close to that of some of the most oxidant acridinium ions reported in the literature.15,16 To evaluate the photocatalytic activity of the 1/1ox couple, it was decided to start with derivative 1 as a photo-reducing agent, by using the dehalogenation of 4-chlorobenzonitrile as the test reaction. As such, 10 mol% of 1 in DMF [0.1 M] was irradiated together with the substrate and 1 equivalent of DIPEA, with two 456 nm kessil lamps for 20 h. As a result, the dehalogenation product could be obtained in a 39% yield (Table S1, ESI†). Increasing DIPEA to 10 equivalents only resulted in a slight increase in the yield. Substituting DIPEA with 10 equivalents of DBU resulted in a marked improvement (84%, entry 3, Table S1, ESI†). While increasing or decreasing the DBU equivalents did not enhance the reaction outcome (entries 4 and 5, Table S1, ESI†), the addition of 1 equivalent of DIPEA (in the presence of 10 equivalents of DBU) resulted in a quantitative conversion (>95%). As a control, a reaction was carried out in the absence of 1, resulting in the complete loss of reactivity. The same happened when the reaction was carried out in the dark, suggesting that the reaction is indeed photocatalytic.
 | ||
| Scheme 1 Synthetic procedure for the preparation of 1 and 1ox. | ||
Finally, performing the reaction in air resulted in only 15% yield, highlighting the radical nature of the process. Based on these results, several substrates were evaluated (Fig. 2), demonstrating that it is possible to dehalogenate systems containing esters (4d), ketones (4b), polyaromatics (4e) and also non-activated biphenyls (4c). To complete the mechanistic investigation, a Stern–Volmer titration was performed with 4a as a quencher on a 1.0 × 10−5 M solution of 1 in DMF resulting in a visible quenching of the emission (Fig. S4, ESI†). Also, no EDA complex formation was observed in different mixtures of 4-chlorobenzonitrile, DBU and 1 in DMF (Fig. S3, ESI†), suggesting that the reaction is based on a SET event from 1 to 4a. Overall, these results and the marked improvement of the reaction outcome in the presence of a base (DBU) led us to hypothesise a mechanism involving a proton-coupled electron-transfer plausibly similar to the one reported by Bortolato et al. (Fig. S14, ESI†).27 This is consistent with the presence of a hydrogen bonding interaction between DBU and the NH proton in 1 as highlighted by an NMR titration performed in DMF (Fig. S20, ESI†). The catalytic behavior of 1ox was then studied in the photo-oxidation of anisole (6a), followed by trapping of the resulting radical with 1H-pyrazole (Table S3, ESI†).17 A first reaction was carried out by irradiating a 0.1 M solution of anisole in dichloroethane (DCE) with 10 mol% of 1ox under air for 18 h. The desired product 7a was formed in a 73% yield as a 4.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of p:o isomers (Table S3, ESI†). Building on this encouraging result, the reaction was repeated twice adding 10 and 20 mol%, respectively, of TEMPO as additive,17 which however did not result in improvement of the reaction yield (entries 2 and 3, Table S3, ESI†). At this point o-dichlorobenzene (ODCB) was used in place of DCE (entry 4, Table S3, ESI†), resulting in a marked increase of the yield up to 92%. Also in this case, performing the reaction in the absence of catalyst resulted in a complete loss of reactivity, as did carrying out the reaction in the absence of light (entries 5 and 6, Table S3, ESI†). Interestingly, decreasing the catalyst content from 10 mol% to 2.5 mol% did not affect significantly the reaction outcome, while moving down to 1.0 and 0.5 mol% only resulted in a moderate decrease in yield (entries 9–11, Table S3, ESI†). Also, irradiating the reaction at less energetic wavelengths (525 nm) resulted in a good reactivity (entry 8, Table S3, ESI†) with a 63% yield of the desired product. Performing the reaction under an inert atmosphere (argon) resulted in a marked yield decrease (entry 12, Table S3, ESI†), highlighting the important role of oxygen in the reaction mechanism. Since 1 is known to be oxidized in air, the reaction was repeated using 2.5 mol% of 1 as the catalyst, resulting in a 24% yield of 7a. This suggests that 1 can be oxidized in the reaction conditions to the active species resulting in reactivity albeit with lower yield than with isolated 1ox. Increasing the amount of 1 resulted in an enhancement in the yield of 7a up to 40% (entry 14, Table S3, ESI†). To understand if 1ox was responsible for this reactivity, a solution of 1 was irradiated for 2 hours at 456 nm and upon UV-Vis analysis the absorption of 1ox was clearly visible (Fig. S13, ESI†). Having defined the best conditions for the reaction, different substrates were tested (Fig. 3). From this point of view, performing the reaction on diphenyl ether resulted in a quantitative conversion to 7b, while using less activated biphenyl as a substrate produced a 64% yield of 7c as a single isomer. Naphthalene derivative 7d was also obtained in a good 52% yield, while halo derivative 7e resulted in a lower 38% yield.
:1 mixture of p:o isomers (Table S3, ESI†). Building on this encouraging result, the reaction was repeated twice adding 10 and 20 mol%, respectively, of TEMPO as additive,17 which however did not result in improvement of the reaction yield (entries 2 and 3, Table S3, ESI†). At this point o-dichlorobenzene (ODCB) was used in place of DCE (entry 4, Table S3, ESI†), resulting in a marked increase of the yield up to 92%. Also in this case, performing the reaction in the absence of catalyst resulted in a complete loss of reactivity, as did carrying out the reaction in the absence of light (entries 5 and 6, Table S3, ESI†). Interestingly, decreasing the catalyst content from 10 mol% to 2.5 mol% did not affect significantly the reaction outcome, while moving down to 1.0 and 0.5 mol% only resulted in a moderate decrease in yield (entries 9–11, Table S3, ESI†). Also, irradiating the reaction at less energetic wavelengths (525 nm) resulted in a good reactivity (entry 8, Table S3, ESI†) with a 63% yield of the desired product. Performing the reaction under an inert atmosphere (argon) resulted in a marked yield decrease (entry 12, Table S3, ESI†), highlighting the important role of oxygen in the reaction mechanism. Since 1 is known to be oxidized in air, the reaction was repeated using 2.5 mol% of 1 as the catalyst, resulting in a 24% yield of 7a. This suggests that 1 can be oxidized in the reaction conditions to the active species resulting in reactivity albeit with lower yield than with isolated 1ox. Increasing the amount of 1 resulted in an enhancement in the yield of 7a up to 40% (entry 14, Table S3, ESI†). To understand if 1ox was responsible for this reactivity, a solution of 1 was irradiated for 2 hours at 456 nm and upon UV-Vis analysis the absorption of 1ox was clearly visible (Fig. S13, ESI†). Having defined the best conditions for the reaction, different substrates were tested (Fig. 3). From this point of view, performing the reaction on diphenyl ether resulted in a quantitative conversion to 7b, while using less activated biphenyl as a substrate produced a 64% yield of 7c as a single isomer. Naphthalene derivative 7d was also obtained in a good 52% yield, while halo derivative 7e resulted in a lower 38% yield.
 | ||
| Fig. 2 Substrate scope for photo-reductive dehalogenation using 1 as a photocatalyst. *NMR yields obtained with trichloroethylene (TCE) as an internal standard. | ||
 | ||
| Fig. 3 Substrate scope for the photo-oxidation reaction using 1ox as a photocatalyst. *NMR yields obtained with TCE as an internal standard. Yields in parentheses are isolated yields. | ||
Finally, different azole derivatives were tested: in the case of methylated 1H-pyrazoles, derivative 7f was obtained with an 89% yield, and when benzotriazole was used instead, 7g was formed quantitatively, whereas the use of the halogenated 5,6-dichloro benzimidazole resulted in the formation of 7h in a 60% yield. To elucidate the reaction mechanism, also in this case UV-Vis experiments were carried out. As for 1 no EDA complexes were visible by adding 100 or even 1000 eq. of 6a to 1ox (Fig. S11, ESI†). Instead, when a Stern–Volmer titration was performed on 8.3 × 10−6 M of 1ox using anisole 6a as a quencher, a clear decrease in the emission was visible, suggesting that the photooxidation is occurring via a SET event from 6a to 1ox (Fig. S9, ESI†). This result, together with the previous experiments suggests that the reaction should occur following a similar mechanism to the one reported by Nicewitz and co-workers for acridinium ions (Fig. S15, ESI†).17 In conclusion, in this work, the synthesis of a novel N-monoaryl dihydrophenazine is reported along with its oxidation to the corresponding monocationic derivative 1ox. The compounds were completely characterised, also demonstrating the reversibility of the oxidation/reduction process. Moreover, both forms proved to behave as effective photocatalysts. In particular, 1ox was shown to promote the oxidative coupling between electron rich aromatics and azoles at low catalyst loadings (from 2.5 to 0.5 mol%) and without the need for additives or pure O2 atmospheres. As such, the present work is a relevant implementation of the recent studies on π-extended dihydrophenazine systems, which are expected to be further developed for catalytic and material applications in both their reduced and oxidized forms.
Maurizio Prato: conceptualization, reviewing and editing of manuscript Jacopo Dosso: synthesis, characterisation, conceptualization, writing of original draft.
J. D. kindly acknowledges FRA2024 funded by the University of Trieste. J. D. acknowledges the RTDa REACT EU-PON ‘ricerca e innovazione’ 2014–2020. M. P. is the AXA Chair for Bionanotechnology (2016–2026). This work was supported by the University of Trieste, INSTM, and the Italian Ministry of Education MIUR (cofin Prot. 20228YFRNL).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- Z. Zhang, Y. S. Wu, K. C. Tang, C. L. Chen, J. W. Ho, J. Su, H. Tian and P. T. Chou, J. Am. Chem. Soc., 2015, 137, 8509–8520 CrossRef CAS PubMed.
- Z. Zhang, W. Song, J. Su and H. Tian, Adv. Funct. Mater., 2020, 30, 1902803 CrossRef CAS.
- Z. Zhang, Q. Wang, X. Zhang, J. Mei and H. Tian, JACS Au, 2024, 4, 1954–1965 CrossRef CAS PubMed.
- J. Lee, K. Shizu, H. Tanaka, H. Nakanotani, T. Yasuda, H. Kaji and C. Adachi, J. Mater. Chem. C, 2015, 3, 2175–2181 RSC.
- G. Sun, J. Pan, Y. Wu, Y. Liu, W. Chen, Z. Zhang and J. Su, ACS Appl. Mater. Interfaces, 2020, 12, 10875–10882 CrossRef CAS PubMed.
- B. Huang, H. Kang, X. L. Zhao, H. B. Yang and X. Shi, Cryst. Growth Des., 2022, 22, 3587–3593 CrossRef CAS.
- J. Dosso, B. Bartolomei, N. Demitri, F. P. Cossío and M. Prato, J. Am. Chem. Soc., 2022, 144, 7295–7301 CrossRef CAS PubMed.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- A. Vega-Peñaloza, J. Mateos, X. Companyó, M. Escudero-Casao and L. Dell’Amico, Angew. Chem., Int. Ed., 2021, 60, 1082–1097 CrossRef PubMed.
- D. A. Corbin, B. G. McCarthy, Z. Van De Lindt and G. M. Miyake, Macromolecules, 2021, 54, 4726–4738 CrossRef CAS PubMed.
- D. A. Corbin, K. O. Puffer, K. A. Chism, J. P. Cole, J. C. Theriot, B. G. McCarthy, B. L. Buss, C. H. Lim, S. R. Lincoln, B. S. Newell and G. M. Miyake, Macromolecules, 2021, 54, 4507–4516 CrossRef CAS PubMed.
- G. Gentile, B. Bartolomei, J. Dosso, N. Demitri, G. Filippini and M. Prato, Chem. Commun., 2024, 60, 602–605 RSC.
- J. Dosso and M. Prato, Chem. – Eur. J., 2023, 29, e202203637 CrossRef CAS PubMed.
- F. Hilgers, W. Kaim, A. Schulz and S. Zalis, J. Chem. Soc., Perkin Trans. 2, 1994, 135–138 RSC.
- N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2021, 122, 1925–2016 CrossRef PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L. C. Campeau, D. Nicewicz and D. A. DiRocco, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS PubMed.
- N. A. Romero, K. A. Margrey, N. E. Tay and D. A. Nicewicz, Science, 2015, 349, 1326–1330 CrossRef CAS PubMed.
- L. Niu, H. Yi, S. Wang, T. Liu, J. Liu and A. Lei, Nat. Commun., 2017, 8, 14226 CrossRef PubMed.
- S. Kundu, M. K. Biswas, A. Banerjee, K. Bhadra, G. S. Kumar, M. G. B. Drew, R. Bhadra and P. Ghosh, RSC Adv., 2013, 3, 3054–3061 RSC.
- D. Leow, Org. Lett., 2014, 16, 5812–5815 CrossRef CAS PubMed.
- O. S. Lizyakina, L. B. Vaganova and D. F. Grishin, Polym. Sci., Ser. B, 2023, 65, 398–408 CrossRef CAS.
- C. H. Liu, J. J. Wang, M. Xu, Q. Luo, Z. Wang, W. Tan, X. Zhao and X. Jia, New J. Chem., 2024, 48, 2367–2370 RSC.
- S. P. Pitre, C. D. McTiernan and J. C. Scaiano, ACS Omega, 2016, 1, 66–76 CrossRef CAS PubMed.
- D. S. Sneddon and T. R. Hoye, Chem. Sci., 2023, 14, 6730–6737 RSC.
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259–271 CrossRef CAS.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- T. Bortolato, G. Simionato, M. Vayer, C. Rosso, L. Paoloni, E. M. Benetti, A. Sartorel, D. Lebœuf and L. Dell’Amico, J. Am. Chem. Soc., 2023, 145, 1835–1846 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06499b |
| This journal is © The Royal Society of Chemistry 2025 |