
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis and structural aspects of the perbromo-functionalised thiaboranes closo-SBnBrn (n = 5, 9, 11): the solid-state structure of the octahedral closo-SB5Br5, governed by strong dihalogen contacts†
Willi
Keller
*a,
Joachim
Ballmann
 b,
Jindřich
Fanfrlík
c and
Drahomír
Hnyk
*d
b,
Jindřich
Fanfrlík
c and
Drahomír
Hnyk
*d
aInstitut für Chemie, Universität Hohenheim, Garbenstrasse 30, 70599 Stuttgart, Germany. E-mail: willi.keller@uni-hohenheim.de
bAnorganisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
cInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, Flemingovo nám. 2, 166 10 Praha 6, Czech Republic
dInstitute of Inorganic Chemistry, Czech Academy of Sciences, 250 68 Husinec-Řež, Czech Republic. E-mail: hnyk@iic.cas.cz
First published on 6th January 2025
Abstract
Co-pyrolysis reactions of B2Br4 with S2Br2 at 350 °C in vacuo yielded the brominated thiaboranes closo-SB5Br5 (1), closo-1-SB9Br9 (2) and closo-SB11Br11 (3), confirmed by high-resolution mass spectrometry, experimental and computational 11B NMR spectroscopy. The strong Brδ+(σ-hole)⋯Brδ−(ring) attraction has been the decisive energy contribution in the crystal of 1.
Small polyhedral borane and heteroborane clusters of octahedral shape have so far been limited to the borate dianions [B6X6]2− (X = H, Cl or Br),1 the carboranes2 1,2- or 1,6-C2B4H6 and the perhalogenated heteroboranes P2B4X4 (X = Cl3a or Br3b), As2B4X4 (X = Cl3c or Br3d), SB5Cl5,3e SeB5Cl53f,g and TeB5Cl5.3h In particular, recent studies of the thermal reactions of B2Cl4 with Se2Cl2 or TeCl4, producing the octahedral SeB5Cl53f,g and TeB5Cl53h have shown surprising results: chalcogens heavier than sulphur apparently induce stronger perturbations to the corresponding cluster frameworks, as manifested by the long body diagonal in chlorinated octahedral selena- and telluraboranes, which may prevent their accommodation in octahedral moieties. There are two known pnictogenaboranes, P2B4Br4 and As2B4Br4, but no brominated octahedral chalcogenaborane as yet, which has prompted us to carry out the thiaborane reaction3e with S2Br2. On that basis, we wish to report the synthesis and characterisation of closo-SB5Br5 (1) and other new brominated thiaboranes.
We have studied the co-pyrolysis reaction of B2Br4 with S2Br2 at temperatures above 350 °C. In comparison with chlorinated thiaboranes, prepared at 280 °C, this reaction required significantly higher temperatures because the bromo compounds are less volatile than the Cl-synthons.3e
The procedure described yielded three new brominated thiaboranes, closo-SB5Br5 (1), closo-1-SB9Br9 (2) and closo-SB11Br11 (3) (Scheme 1), of which we were able to isolate only compound 1 by repeated vacuum-fractionation and crystallisation processes. Compound 1 was obtained in 5% yield based on the idealised eqn (1). The non-separable compounds 2 and 3 (eqn (2) and (3)) were formed in approximately 1% and 3% yields, respectively.
| 11B2Br4 + S2Br2 → 2SB5Br5 (1) + 12BBr3 | (1) |
| 19B2Br4 + S2Br2 → 2SB9Br9 (2) + 20BBr3 | (2) |
| 23B2Br4 + S2Br2 → 2SB11Br11 (3) + 24BBr3 | (3) |
 | ||
| Scheme 1 Molecular diagrams of closo-SB5Br5 (1), closo-1-SB9Br9 (2) and closo-SB11Br11 (3). | ||
The new thiaboranes 1–3 are thermally stable under inert atmosphere at least up to their formation in two experiments at 350 °C and 430 °C. In the very high vacuum applied in EI-MS (10−5 mbar), 1 starts to sublime already at 25 °C, 2 at 50 °C and 3 at 85 °C. They are soluble in common aprotic solvents such as benzene, chloroform and methylene chloride.
The corresponding reaction of B2Cl4 with S2Cl2 has resulted in a much higher number of individual chlorinated thiaboranes closo-SBnCln (with n = 4, 5, 6, 9, 10, 11, 12) than in the present study. This may be attributed to the fact that the isodesmic heats of formation of the bromo-products are less negative than those of the chloro-products.3e However, computational studies have confirmed in both cases that octahedral, bicapped square-antiprismatic and icosahedral cages are the most stable in the entire series. The computed isodesmic heats of formation in the origination of closo-SBnBrn from closo-SBn−1Brn−1 (n = 5, 9, 11) at the B3LYP/def2-QZVP//BP86/DZVP-DFT level (in kcal mol−1) are as follows:
| closo-1-SB4Br4 + B2Br4 → closo-SB5Br5 (1) + BBr3 −15.6 |
| closo-4-SB8Br8 + B2Br4 → closo-1-SB9Br9 (2) + BBr3 −19.6 |
| closo-2-SB10Br10 + B2Br4 → closo-SB11Br11 (3) + BBr3 −34.3 |
The new thiaboranes have been characterised by 11B NMR spectroscopy, high- and low-resolution mass spectrometry, X-ray structure determination for 1 and SO-DFT/ZORA/NMR computations (taking spin–orbit coupling (SO) into account), which have confirmed the experimental 11B NMR chemical shifts. With SO not included in 11B NMR chemical-shift computations, the values have shifted upfield by about 5 ppm4 with respect to the SO-DFT/ZORA/NMR results. Based on these results, we have derived the molecular structures of 1, 2 and 3 in their solutions (see also Table 1).
| 1 Octahedral motif | ||
|---|---|---|
| B(2–5) | B(6) | |
| a SOC-PBE0/T2ZP//B3LYP/6-311 + G**. | ||
| Comp. | −1.2 | 20.5 |
| Exp. | −2.3 | 19.4 |
| 2 Bicapped square-antiprismatic motif | |||
|---|---|---|---|
| B(2–5) | B(6–9) | B(10) | |
| Comp. | 8.9 | −4.7 | 53.5 |
| Exp. | 7.5 | −3.7 | 49.6 |
| 3 Icosahedral motif | |||
|---|---|---|---|
| B(2–6) | B(7–11) | B(12) | |
| Comp. | −1.7 | −6.0 | 14.6 |
| Exp. | −1.4 | −3.7 | 14.2 |
The 11B chemical shifts of 1, 2 and 3 are within the normal range for halogenated heteroborane clusters3a–h and closely follow the trends established for non-halogenated heteroboranes.5a They also reflect the symmetries of C4v, C4v and C5v, respectively.
As expected, 1 shows two broad signals in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at −2.3 ppm (h1/2 ≈ 106 Hz, B(2–5)) and 19.4 ppm (h1/2 ≈ 113 Hz, B(6)), which are in agreement with the signals for closo-SB5Cl5 at −0.3 ppm and 19.4 ppm.3e With the application of the Lorentz–Gaussian transformation with a 1J(11B–11B) of 25 Hz, the signals for B(2–5) are split into a quartet, indicating the coupling with B(6) and showing the expected cross-peak in the COSY 11B11B NMR with the low-field signal.
:1 ratio at −2.3 ppm (h1/2 ≈ 106 Hz, B(2–5)) and 19.4 ppm (h1/2 ≈ 113 Hz, B(6)), which are in agreement with the signals for closo-SB5Cl5 at −0.3 ppm and 19.4 ppm.3e With the application of the Lorentz–Gaussian transformation with a 1J(11B–11B) of 25 Hz, the signals for B(2–5) are split into a quartet, indicating the coupling with B(6) and showing the expected cross-peak in the COSY 11B11B NMR with the low-field signal.
The 11B NMR of 2 consists of three signals in a 4:4:1 ratio at −3.7 ppm (B6–9), 7.5 ppm (B2–6) and 49.6 ppm (B10), which are in good agreement with the signals for closo-SB9Cl9 at −0.3 ppm, 13.1 ppm and 48.8 ppm.3e The signal at −3.7 ppm shows 11B11B cross-peak correlation with the other two signals and thus can be assigned to B(6–9), the signal at 7.5 ppm to B(2–5), and the poorly-resolved intensity-one signal at 49.6 ppm can be connected with the antipodal downfield shift5 of B(10).
The 11B NMR of 3 consists of three signals in a 5:5:1 ratio at −3.7 ppm (B7–11), −1.4 ppm (B2–6) and 14.2 ppm (B12), with expected cross-peaks between B(7–11)/B(2–6) and B(7–11)/B(12). The signals are in agreement with the chemical shifts established for closo-SB11Cl11 at −3.9 ppm, 1.0 ppm and 14.4 ppm.3e
The 70 eV EI mass spectra of all compounds show a strong molecular ion. In the case of thiaborane 1, the major cut-off indicates the abstraction of a SBBr edge from SB5Br5 to leave a [B4Br4]+ fragment. The formation of such a [B4Hal4]+ fragment has been analogously found in the mass spectra of closo-SB5Cl53e and closo-1,2-P2B4Cl4.3a The MS of thiaborane 2 shows the stepwise abstraction of S, Br, BBr2 and BBr3 fragments, whereas 3 exhibits only minimal fragmentation with the abstraction of a single Br atom, which indicates the high stability of the icosahedral cluster framework.
All mass-spectroscopy patterns are consistent with the computed spectra based on natural isotopic abundances. On the basis of simple skeletal electron-counting rules,61, 2 and 3 should adopt octahedral, bicapped square-antiprismatic and icosahedral geometries, respectively, with sulphur contributing four electrons and each BBr unit two electrons to the cluster bonding. The employment of intrinsic-bond orbitals (IBOs)3e,g,h has revealed the nature of bonding in 1, 2 and 3. According to the expansion coefficients (ECs: contributions from individual atoms to a particular IBO to reveal the nature of such orbitals), the sulphur involvement in 1 consists of two IBOs with the ECs (the contributing atom in parentheses) of 1.15 (S), 0.51 (B) and 0.25 (B) and one IBO with the ECs of 1.20 (S) and 0.53 (B). Whereas the first pair of IBOs can be assigned to 3c–2e bonding, the second IBO is of 2c–2e type. The rest of the octahedral cage is assembled through 3c–2e IBOs (Fig. 1). The bonding schemes in 2 and 3 are shown in ESI† (Fig. S2).
 | ||
| Fig. 1 Visualised IBOs for SB5Br5 (1). The colour coding is as follows: blue classical 2c–2e bonding, pink 3c–2e bonding. | ||
The ESP (electrostatic potential molecular surface) of 1 is similar to that of its chlorine analogue,3e but the bromine atoms of 1 have more positive σ-holes than the chlorines of SB5Cl5 (see Fig. 2 and Table 2).
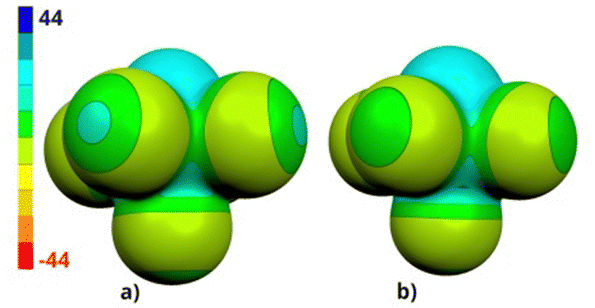 | ||
| Fig. 2 The ESP molecular surfaces of 1 (a) and SB5Cl5 (b) computed at the HF/cc-pVDZ level. | ||
| Compound | V s,max [kcal mol−1] | V s,min [kcal mol−1] | |
|---|---|---|---|
| Sulphur atom | Halogen atoms | Halogen atoms | |
| SB5Br5(1) | 24.5 | 4 × 11.6; 10.2 | 4 × −6.0; −5.8 |
| SB5Cl5 | 24.83e | 4 × 6.6; 5.7 | 4 × −5.6; −5.1 |
To confirm the octahedral structure of 1, single crystals were grown via vacuum sublimation and subjected to X-ray diffraction (see Fig. 3). Compound 1 crystallises in P21/c with three crystallographically independent closo-SB5Br5 octahedrons present in the corresponding asymmetric unit. As a consequence of symmetry, a total of twelve molecules are present in the centrosymmetric monoclinic cell (see ESI,† for details). Crystal packing is dominated by short contacts between the bromine atoms of neighbouring molecules, indicative of dihalogen bonding (d(Br⋯Br) = 339 to 368 pm, see also Fig. 4). Intermolecular bonding interactions between the sulphur and the bromine atoms, however, seem to be absent because no short contacts (<the sum of vdW radii) have been observed.
 | ||
| Fig. 3 ORTEP plot (thermal ellipsoids drawn at the 50% probability level) of the molecular structure of closo-SB5Br5 (1). The asymmetric unit contains three independent molecules with very similar metrical parameters; only one of these molecules is shown. Selected averaged bond lengths (pm) and angles (°): Beq–Breq 189.4(5), Bax–Brax 190.4(5), S–Beq 196.8(2), Beq–Beq 177.8(7), Beq–Bax 170.1(3), Beq–S–Beq 53.7(2), Beq–Beq–Beq 90.0(3), Beq–Bax–Beq 63.0(3), S–Bax–Brax 179.1(2). | ||
 | ||
| Fig. 4 The short contacts below the sum of vdW radii in the crystal packing of 1 (a) and SB5Cl53e (b) are indicated by dot lines (in pm). An example of Brδ+⋯Brδ− is depicted in term of the corresponding molecular ESP surfaces. The color coding of the ESP is in the range of −26 to 26 kcal mol−1. | ||
All the interaction motifs present in the crystal structure of 1 have been investigated using a cluster model. Two-body interaction energies have been computed for the selected crystallographic molecule using the SAPT method,7 which enables energy decomposition into individual terms. For comparison, the same analysis has also been performed for SB5Cl5. Both crystals are dominated by dispersion as expected. Surprisingly, the contribution of dispersion to crystal packing was slightly less pronounced in 1 than in SB5Cl5 (i.e. the dispersion in them formed 63 and 67%, respectively, of the sum of all the attractive terms in the SAPT decomposition). The explanation is that there are considerably more positive σ-holes on Br atoms in 1 than on Cl atoms in SB5Cl5. Compound 1 thus has a better balance of positive and negative sites for crystal packing – it has six highly positive σ-holes and five negative rings around B–Br bonds, unlike SB5Cl5, which has only one highly positive σ-hole on the S atom to match the five negative rings around B–Cl bonds. Consequently, the electrostatic contribution is more pronounced in the crystal packing of 1, which is dominated by dihalogen bonds, than in SB5Cl5, dominated by chalcogen bonds (see Fig. 4 and Table 3). This enhanced electrostatic contribution may be ascribed to the fact that the crystallisation process of 1 is more straightforward than that of SB5Cl5.
| Compound | ΣSAPT0 | ΣEelec | ΣEexch | ΣEind | ΣEdisp |
|---|---|---|---|---|---|
| SB5Br5 (1) | −41.5 | −38.8 (30%) | 86.4 | −8.7 (7%) | −80.4 (63%) |
| SB5Cl5 | −32.2 | −24.0 (26%) | 59.4 | −6.1 (7%) | −61.5 (67%) |
In a thermodynamically controlled reaction at 350 °C, simple inorganic synthons such as B2Br4 and S2Br2 have provided octahedral, bicapped square-antiprismatic and icosahedral structural motifs referred to as closo-SB5Br5, closo-1-SB9Br9 and closo-SB11Br11, respectively. Structural features have been elucidated through a combination of experimental and computational approaches including NMR spectroscopy together with MS spectrometry and DFT/ZORA/NMR model chemistry. These methods have confirmed that these three structural arrangements are good representations of the molecular geometries in solutions. In addition, crystal-structure determination of the octahedral brominated thiaborane shows that the Br⋯Br contacts are strong enough to be able to overcome the repulsion between the σ-holes located on the bromines and the sulphur. As a consequence, the crystal-packing forces are dominated by strong Brδ+(σ-hole)⋯Brδ−(ring) attraction, i.e. the crystal is formed without chalcogen bonding detected in the chlorine analogue of 1.
We thank Dr Jürgen Conrad and Mr Mario Wolf (Institut für Chemie, Universität Hohenheim) for recording NMR spectra and Dipl.-Ing. (FH) J. Trinkner (Institut für Organische Chemie, Universität Stuttgart) for recording MS. Dr M-B. Sárosi (Universität Würzburg) is thanked for the assistance with the ADF computations. Financial support from the Czech Science Foundation, grant no. 23-05083S, is gratefully acknowledged.
Data availability
The data supporting of this article have been included as part of the ESI.† The crystallographic data for compound 1 (closo-SB5Br5) were deposited with the Cambridge Crystallographic Data Centre with CCDC no. 2403282.†Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected surveys on polyhedral boranes, see: (a) W. N. Lipscomb, Boron Hydrides, Benjamin, New York, 1963 Search PubMed; (b) E. L. Muetterties, Boron Hydride Chemistry, Academic Press, New York, 1975 Search PubMed; (c) J. F. Liebman, A. Greenberg and R. E. Williams, Advances in Boron and the Boranes, VCH Verlagsgesellschaft, Weinheim New York, 1988 Search PubMed; (d) R. B. King, Chem. Rev., 2001, 101, 1119–1152 CrossRef CAS.
- For selected surveys on carboranes, see: (a) Electron Deficient Boron and Carbon Clusters, ed. G. A. Olah, K. Wade and R. E. Williams, John Wiley and Sons, New York, 1991 Search PubMed; (b) T. Onak, Polyhedral Carbaboranes, Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, UK, 1995, Vol. 1, 217–255 Search PubMed; (c) M. A. Fox, Polyhedral Carboranes, Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, 2007, Vol. 3, 49–112 Search PubMed; (d) R. N. Grimes, Carboranes, ed., Elsevier, Amsterdam, The Netherlands, 2016 Search PubMed.
- (a) W. Haubold, W. Keller and G. Sawitzki, Angew. Chem., Int. Ed. Engl., 1988, 27, 925–926 CrossRef; (b) W. Keller, L. G. Sneddon, W. Einholz and A. Gemmler, Chem. Ber., 1992, 125, 2343–2346 CrossRef CAS; (c) R. Schäfer, W. Einholz, W. Keller, G. Eulenberger and W. Haubold, Chem. Ber., 1995, 128, 735–736 CrossRef; (d) W. Keller, W. Einholz, D. Rudolph and T. Schleid, Z. Anorg. Allg. Chem., 2017, 643, 664–668 CrossRef CAS; (e) W. Keller, F. Lissner, J. Ballmann, J. Fanfrlík, D. Hnyk and T. Schleid, Angew. Chem., Int. Ed., 2024, 63, e202406751 Search PubMed; (f) W. Keller and M. Hofmann, Z. Anorg. Allg. Chem., 2017, 643, 729–731 CrossRef CAS; (g) W. Keller, M. Hofmann, H. Wadepohl, M. Enders, J. Fanfrlík and D. Hnyk, Dalton Trans., 2023, 52, 16886–16893 RSC; (h) W. Keller, J. Ballmann, M. B. Sárosi, J. Fanfrlík and D. Hnyk, Angew. Chem., Int. Ed., 2023, 62, e202219018 CrossRef CAS PubMed.
- See e.g. (a) W. Keller, M. Hofmann, M. B. Sárosi, J. Fanfrlík and D. Hnyk, Inorg. Chem., 2022, 61, 16565–16572 CrossRef CAS PubMed; (b) W. Keller, M. B. Sárosi, J. Fanfrlík, M. Straka, J. Holub, M. L. McKee and D. Hnyk, RSC Adv., 2023, 13, 19627–19637 RSC.
- (a) S. Heřmánek, Chem. Rev., 1992, 92, 325–362 CrossRef; (b) F. Teixidor, C. Vinas and R. W. Rudolph, Inorg. Chem., 1986, 25, 3339–3346 CrossRef CAS; (c) T. P. Fehlner, P. T. Czech and R. F. Fenske, Inorg. Chem., 1990, 29, 3103–3109 CrossRef CAS; (d) S. Heřmánek, D. Hnyk and Z. Havlas, J. Chem. Soc., Chem. Commun., 1989, 23, 1859–1861 RSC; (e) M. Bühl, P. V. R. Schleyer, Z. Havlas, D. Hnyk and S. Heřmánek, Inorg. Chem., 1991, 30, 3107–3111 CrossRef.
- (a) R. E. Williams, Inorg. Chem., 1971, 10, 210–214 CrossRef CAS; (b) K. Wade, Adv. Inorg. Chem. Radiochem., 1976, 18, 1–66 CrossRef CAS; (c) R. E. Williams, Adv. Inorg. Chem. Radiochem., 1976, 18, 67 CrossRef CAS; (d) R. W. Rudolph, Acc. Chem. Res., 1976, 9, 446–452 CrossRef CAS; (e) R. E. Williams in Electron Deficient Boron and Carbon Clusters, ed. G. A. Olah, K. Wade and R. E. Williams, Wiley and Sons, New York, 1991, 11–93 Search PubMed; (f) R. E. Williams, Chem. Rev., 1992, 92, 177–207 CrossRef CAS.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2403282. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06334a |
| This journal is © The Royal Society of Chemistry 2025 |