
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination-induced axial chirality controls the metal-centred configuration in a stereogenic-at-iron catalyst†
Benedikt
Buchberger‡
 ,
Nemrud
Demirel‡
,
Xiulan
Xie
,
Sergei I.
Ivlev
and
Eric
Meggers
*
,
Nemrud
Demirel‡
,
Xiulan
Xie
,
Sergei I.
Ivlev
and
Eric
Meggers
*
Fachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Strasse 4, 35043 Marburg, Germany. E-mail: meggers@chemie.uni-marburg.de
First published on 8th January 2025
Abstract
A new approach is introduced to control the metal-centred configuration of stereogenic-at-iron catalysts by utilizing axial ligand chirality, which becomes locked upon metal coordination. This strategy is applied to an iron catalyst containing two chelating N-(2-pyridyl)-substituted triazol-5-ylidene mesoionic carbenes (MICs) resulting in a helical topology with a stereogenic iron centre.
Chiral transition metal complexes play a crucial role in asymmetric catalysis.1 Traditionally, these catalysts are developed by combining chiral ligands with metal precursors, a strategy that has dominated for decades. In contrast, a recently emerging chiral-at-metal approach utilizes metal-centred chirality, with achiral ligands arranged around a stereogenic metal centre.2 This concept has been successfully applied to the noble metals iridium(III),3 rhodium(III),4 and ruthenium(II).5 More recently, our focus has shifted to 3d metals, which are appealing for sustainable catalysis due to their abundance and lower toxicity. We reported the first example of a chiral-at-iron catalyst, in which chirality is generated exclusively by achiral ligands.6 In this catalyst, two bidentate N-(2-pyridyl)-substituted N-heterocyclic carbenes (PyNHC) chelate iron(II) to induce a helical topology with a stereogenic iron centre. Two additional acetonitrile ligands, positioned trans to the σ-donating NHC ligands, create labile coordination sites that facilitate substrate binding and catalysis. While this catalyst demonstrated promising catalytic activity in asymmetric Cannizzaro reactions, a Nazarov cyclisation, and hetero-Diels–Alder reactions, some racemisation occurred over time.6,7 To address this, we have explored various scaffold modifications aimed at improving configurational stability and expanding the reactivity of the iron catalysts (Fig. 1a). Replacing the imidazol-2-ylidene NHC ligand with a more π-accepting benzimidazol-2-ylidene improved configurational stability, but we were unable to demonstrate catalytic activity beyond Lewis acid catalysis.8 To enhance reactivity, we introduced a stronger σ-donor, 1,2,3-triazol-5-ylidene, which enabled nitrene-mediated intramolecular C(sp3)–H amination.9 However, this scaffold suffered from significant configurational lability. For compensation, we incorporated chirality into the ligand framework by using a chiral pinene-modified pyridyl ligand. However, this had the drawback of providing reduced asymmetric induction (Fig. 1b).
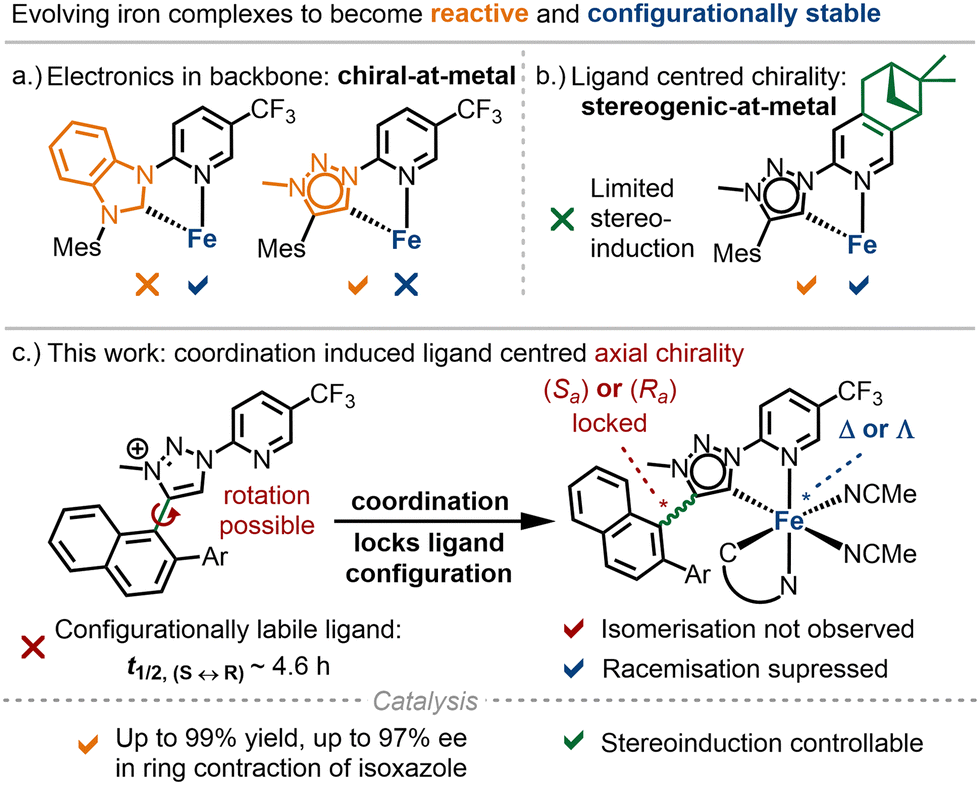 | ||
| Fig. 1 Previous work and this study to control the stereogenic iron centre in chiral iron catalysts. | ||
Herein, we present a new strategy to increase configurational stability of chiral-at-iron catalysts by exploiting a locked axial chirality in the ligands upon metal coordination (Fig. 1c).
We selected the naphthyl triazolium ligands 1 and 2 (Scheme 1), which inherently feature axial chirality but undergo rapid racemisation (half-life of 4.6 hours at room temperature for 1).10 We hypothesised that converting these bidentate ligands into iron-coordinated mesoionic carbenes (MICs),11 specifically by replacing the C–H bond at the 5-position of the triazolium ion with a C–Fe bond, would restrict rotation due to the sterically demanding coordination sphere around the iron. This would effectively freeze the induced axial chirality and promote a matched and stable metal-centred configuration.
 | ||
| Scheme 1 Synthesis of ligands 1 and 2 from the alkyne 3. | ||
The ligand synthesis started with the alkyne 3. A CuAAC reaction with tetrazole 4 at 70 °C in toluene afforded the 1,4-disubstituted triazole 5 (49%).12 This was followed by a Suzuki coupling with an aryl boronic acid to introduce the desired aryl moiety (Ar = Ph or 3,5-(tBu)2Ph), a methylation with MeOTf, and subsequent anion exchange with NH4PF6 or NH4BF4 to provide ligands 1 (Ar = Ph) and 2 (Ar = 3,5-(tBu)2Ph) in 99% yield over three steps.
Next, with the ligands in hand, we synthesised racemic iron(II) complexes by first generating silver carbenes of the triazolium ligands using Ag2O, followed by transmetalation with FeCl2 (Table 1).13 Interestingly, two diastereomers formed, one with C2-symmetry and one with C1-symmetry: rac-Fe1-C2 and rac-Fe1-C1 for Ar = Ph, as well as rac-Fe2-C2 and rac-Fe2-C1 for Ar = 3,5-(tBu)2Ph. We found that the reaction conditions significantly affect the ratio. For example, conducting the transmetalation at room temperature for 2.5 hours, followed by the addition of 10 equiv. of NH4PF6, favoured the formation of the C1-symmetric isomers (entries 1 and 2). In contrast, when the transmetalation was performed in the presence of 2.5 equiv. of AgBF4, the diastereomeric ratio shifted toward the C2-symmetric diastereomer for Ar = 3,5-(tBu)2Ph (entry 3), while for Ar = Ph, both diastereomers were generated in equal amounts (entry 4).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Method | Ar | T (°C) | t (h) | Isomer-ratioc | Yieldd (%) | X | |
| C 2 | C 1 | |||||||
| a The depiction of the complexes only features Δ-enantiomers; a representation of the possible isomers and their relation can be found in the ESI. b Referred to the intermediate monocationic Ag–carbene dimer. c Ratio determined by 1H and 19F NMR after short workup. d Yields determined from crude NMR and absolute weight of the crude after short workup. Free ligand was calculated out by factoring the crude NMR ratios and the respective molecular masses. e See ESI for a detailed gradual warming protocol. f Yield after silica column chromatography to ensure full removal of unreacted traces of the Ag–carbene intermediate. | ||||||||
| 1 | A | Ph | r.t. | 3 | 1 | 2.4 | 91 | PF6 |
| 2 | A | 3,5-(tBu)2Ph | r.t. | 3 | 1 | 2.2 | 66 | PF6 |
| 3 | B | 3,5-(tBu)2Ph | −40 °C → r.t.e | 70e | 7 | 1 | 64f | BF4 |
| 4 | B | Ph | 0 °C → r.t.e | 17e | 1 | 1 | 78f | BF4 |
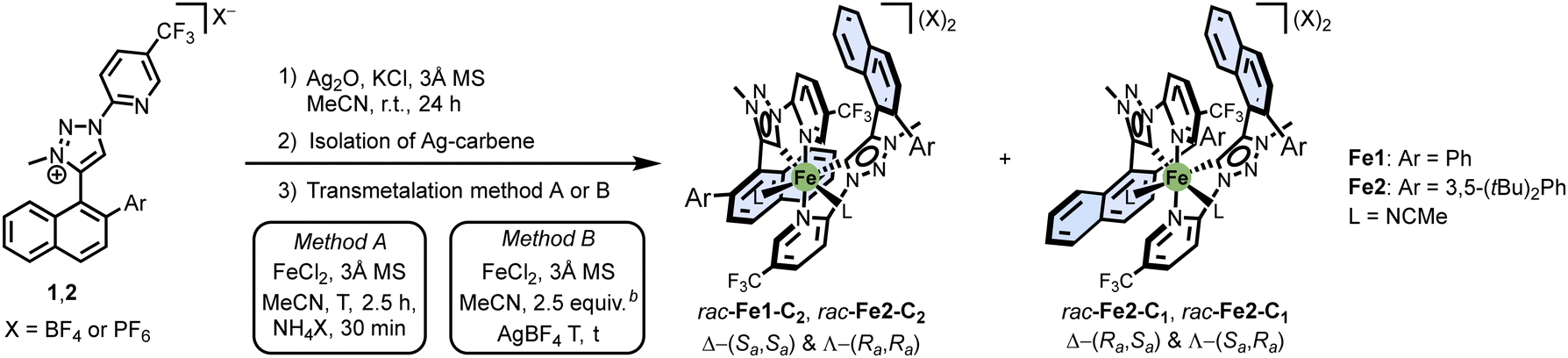
Fig. 2 displays crystal structures of the racemic complexes rac-Fe1-C1 and rac-Fe2-C2 representative for the major isomers described in this work. In the obtained stereogenic-at-iron complexes, iron is coordinated by two chelating 1,2,3-triazol-5-ylidenes in addition to two MeCN ligands, while two PF6− anions complement the dicationic complexes. The structures reveal an interligand π–π interaction between the N-pyridyl moiety of one ligand and the naphthyl ring of the other, while also suggesting a high steric barrier of rotation around the C–C axis between the naphthalene and the triazolyl carbene. This induces the prior configurationally labile ligands to lock their axial chirality as stable atropisomers in the Sa or Ra configuration. In the case of the C2-symmetrical rac-Fe1,2-C2 both naphthyl substituents feature the same axial chirality (Δ-Sa,Sa and Λ-Ra,Ra), while in the C1-symmetrical rac-Fe1,2-C1 the two naphthyl moieties feature differing axial chirality (Δ-Ra,Sa and Λ-Sa,Ra).
 | ||
| Fig. 2 XRD-structures of rac-Fe1-C1 (left) and rac-Fe2-C2 (right). The PF6− anions, the acetonitrile solvent molecule and the hydrogen atoms are not shown. No disorder is shown. Displacement ellipsoids are shown at 50% probability level at 100 K. | ||
In order to investigate the configurational stability of the chiral iron complexes, we attempted the chiral resolution of the racemic complexes rac-Fe1-C1 and rac-Fe1-C2via our previously established auxiliary-ligand mediated route by using a chiral fluoro-salicyloxazoline (Salox) (Scheme 2).14,15 Accordingly, when we coordinated (R)-Salox to rac-Fe1-C1 under basic conditions, we obtained a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two diastereomers in overall 99% yield. The two diastereomers were identified as Δ-(Sa,Ra)-(R)-Fe1Aux-a and Δ-(Sa,Ra)-(R)-Fe1Aux-b by NMR (Scheme 2a) (see ESI,† for further details). Interestingly, both diastereomers contain a metal-centred Δ-configuration. Since the racemic starting material was composed from a 1:1 mixture of Λ and Δ, it can be concluded that the metal-centred configuration in Fe1-C1 is labile. Therefore, the C1-symmetric complex Fe1-C1 was not investigated further.
:1 mixture of two diastereomers in overall 99% yield. The two diastereomers were identified as Δ-(Sa,Ra)-(R)-Fe1Aux-a and Δ-(Sa,Ra)-(R)-Fe1Aux-b by NMR (Scheme 2a) (see ESI,† for further details). Interestingly, both diastereomers contain a metal-centred Δ-configuration. Since the racemic starting material was composed from a 1:1 mixture of Λ and Δ, it can be concluded that the metal-centred configuration in Fe1-C1 is labile. Therefore, the C1-symmetric complex Fe1-C1 was not investigated further.
 | ||
| Scheme 2 Chiral resolution of rac-Fe1-C2 and attempted resolution of rac-Fe1-C1. | ||
In contrast, reacting rac-Fe1-C2 with (R)-Salox furnished Λ-(Ra,Ra)-(R)-Fe1Aux and Δ-(Sa,Sa)-(R)-Fe1Aux with a dr of 1:1. However, only the diastereomer Δ-(Sa,Sa)-(R)-Fe1Aux was stable during column chromatography and could be isolated in 46% yield (Scheme 2b). In analogy, Λ-(Ra,Ra)-(S)-Fe1Aux was obtained via the same route by employing the mirror-imaged chiral auxiliary (S)-Salox. Cleavage of the chiral auxiliary with the weak acid NH4BF4 at 50 °C then afforded the two enantiomers Δ-(Sa,Sa)-Fe1-C2 (60%) and Λ-(Ra,Ra)-Fe1-C2 (54%). The mirror imaged behaviour of the enantiomers was verified by circular dichroism (Fig. 3). Their enantiomeric purity was confirmed through 19F NMR after recoordination of the chiral auxiliary (see ESI,† for details). Thus, it can be concluded that the metal-centred configuration in Fe1-C2 is stable in contrast to a facile isomerisation observed in Fe1-C1.
 | ||
| Fig. 3 Circular dichroism spectra of Λ-(Ra,Ra)-Fe1-C2 (black) and Δ-(Sa,Sa)-Fe1-C2 (red) (0.25 mM in MeCN). | ||
Having established the configurational stability of the stereogenic-at-iron complex Fe1-C2, we next assessed the atropisomerism16 of the ligand 1 employed in this complex. As a result, the rotamers interconvert via zero-order kinetics with a half-life of 280 min, corresponding to a rotational energy barrier of 23 kcal mol−1 (see ESI,† for details). Thus, ligand 1 can be classified as a LaPlante class 2 atropoisomer.17 Axially chiral compounds within this class represent major challenges for further application such as chiral ligands or drug candidates, as they feature rapid interconversion via bond-rotation.
However, this rotation is effectively frozen in the corresponding iron complexes,18,19 which has direct implications for the configurational stability of the stereogenic iron centre. In the C1-symmetric complex Fe1-C1, both the Sa- and Ra-configured ligand 1 is present, meaning that inversion of the metal-centred configuration (Λ vs. Δ) leads to the formation of enantiomers (Fig. 4). As demonstrated, this isomerisation occurs rapidly, consistent with our prior findings on the configurational lability of iron MIC complexes. In contrast, the C2-symmetric complex Fe1-C2 contains axially chiral ligands with identical configurations (either Sa or Ra). Here, inversion of the metal-centred configuration (Λ vs. Δ) results in the formation of diastereomers. For instance, Δ-(Sa,Sa)-Fe1-C2 would isomerize into its diastereomer Λ-(Sa,Sa)-Fe1-C2*. Notably, this diastereomer is not observed experimentally. Structural modelling suggests that steric clashes between the phenyl substituents on the naphthyl moieties prevent this isomerisation. Thus, the configuration of the metal centre in Δ-(Sa,Sa)-Fe1-C2 and its enantiomer Λ-(Ra,Ra)-Fe1-C2 is controlled thermodynamically rather than kinetically by the axial chirality of the ligands.
 | ||
| Fig. 4 Configurational stabilities of the discussed stereogenic-at-iron complexes. | ||
Finally, we investigated the catalytic properties of the stereogenic-at-iron complexes. We found that Δ-(Sa,Sa)-Fe1-C2 exhibited high catalytic activity in the ring contraction of isoxazole 6 to the chiral 2H-azirine 7 (Table 2).20 Initial experiments employing 0.1 mol% of Δ-(Sa,Sa)-Fe1-C2 at room temperature achieved full conversion after only 15 minutes, albeit with a moderate enantiomeric excess (ee) of 72% (entry 1). However, decreasing the temperature to −40 °C improved the ee to 96%, while an increased catalyst loading of 1.0 mol% was necessary to achieve full conversion after 24 hours (entry 2). Further reducing the temperature to −50 °C and extending the reaction time to 40 hours, while using 1.5 mol% of the catalyst, provided the best result with an isolated yield of 93% and 97% ee (entry 3). Although catalytic asymmetric versions of this ring contraction have been reported, the enantioselectivity demonstrated here is unprecedented for iron-catalysed systems and rivals that of ruthenium catalysts.20
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst loading | T (°C) | t (h) | Yieldb | ee (%) |
| a Reaction conditions: Under N2 atmosphere. A solution of Δ-(Sa,Sa)-Fe1-C2 (0.1–1.5 mol%) in CH2Cl2 (0.5 mL) was added to substrate 6 (10 mg, 0.05 mmol) and stirred for the indicated time and temperature. b Yield was determined via1H NMR analysis with 1,3,5-trimethoxybenzene as standard. c Isolated yield. | |||||
| 1 | 0.1 mol% | r.t. | 0.25 | 99 | 72 |
| 2 | 1.0 mol% | −40 | 24 | 98 (95)c | 96 |
| 3 | 1.5 mol% | −50 | 40 | 95 (93)c | 97 |

In summary, we here presented an example in which the metal-centred configuration of a stereogenic-at-iron catalyst is regulated by axially chiral ligands. Notably, the free ligand undergoes rapid atropisomerisation but is frozen into a single configuration only upon coordination to the metal. Future work will investigate the use of related non-racemic ligands with fixed axial chirality.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 883212).
Data availability
The ESI,† contains detailed synthetic procedures and complete characterisation data for all new compounds, as well as CD spectra of all non-racemic iron complexes.Conflicts of interest
There are no conflicts to declare.Notes and references
- P. J. Walsh and M. C. Kozlowski, Fundamentals of Asymmetric Catalysis, University Science Books, Sausalito, CA, 2009 Search PubMed.
- P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764–4794 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Acc. Chem. Res., 2017, 50, 320–330 CrossRef CAS PubMed.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- C.-X. Ye and E. Meggers, Acc. Chem. Res., 2023, 56, 1128–1141 CrossRef CAS PubMed.
- Y. Hong, L. Jarrige, K. Harms and E. Meggers, J. Am. Chem. Soc., 2019, 141, 4569–4572 CrossRef CAS PubMed.
- Y. Hong, T. Cui, S. Ivlev, X. Xie and E. Meggers, Chem. – Eur. J., 2021, 27, 8557–8563 CrossRef CAS PubMed.
- N. Demirel, J. Haber, S. I. Ivlev and E. Meggers, Organometallics, 2022, 41, 3852–3860 CrossRef CAS PubMed.
- N. Demirel, M. Dawor, G. Nadler, S. I. Ivlev and E. Meggers, Chem. Sci., 2024, 15, 15625–15631 RSC.
- F. Meloni, W. D. G. Brittain, L. Male, C. S. L. Duff, B. R. Buckley, A. G. Leach and J. S. Fossey, Tetrahedron Chem., 2022, 1, 100004 CrossRef CAS.
- A. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef CAS PubMed.
- A. Bolje, D. Urankar and J. Košmrlj, Eur. J. Org. Chem., 2014, 8167–8181 CrossRef CAS.
- P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534–13535 CrossRef CAS PubMed.
- L. Gong, M. Wenzel and E. Meggers, Acc. Chem. Res., 2013, 46, 2635–2644 CrossRef CAS PubMed.
- J. Ma, X. Zhang, X. Huang, S. Luo and E. Meggers, Nat. Protoc., 2018, 13, 605–632 CrossRef CAS PubMed.
- C. B. Roos, C.-H. Chiang, L. A. M. Murray, D. Yang, L. Schulert and A. R. H. Narayan, Chem. Rev., 2023, 123, 10641–10727 CrossRef CAS PubMed.
- R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef PubMed.
- X. Luan, R. Mariz, M. Gatti, C. Costabile, A. Poater, L. Cavallo, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 6848–6858 CrossRef CAS PubMed.
- D. Enders, H. Gielen, G. Raabe, J. Runsink and H. H. Teles, Chem. Ber., 1997, 130, 1253–1260 CrossRef CAS.
- (a) K. Okamoto, A. Nanya, A. Eguchi and K. Ohe, Angew. Chem., Int. Ed., 2018, 57, 1039–1043 CrossRef CAS PubMed; (b) L. Li, F. Han, X. Nie, Y. Hong, S. Ivlev and E. Meggers, Angew. Chem., Int. Ed., 2020, 59, 12392–12395 CrossRef CAS PubMed; (c) P. S. Steinlandt, X. Xie, S. Ivlev and E. Meggers, ACS Catal., 2021, 11, 7467–7476 CrossRef CAS; (d) P. S. Steinlandt, M. Hemming, X. Xie, S. I. Ivlev and E. Meggers, Chem. Eur. J., 2023, 29, e202300267 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2400042 and 2400043. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06227b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |