
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePushing the limits of electron donation for cis-chelating ligands via an alliance of phosphonium ylide and anionic abnormal NHC†
Mustapha
El Kadiri
 ab,
Abdelhaq
Cherradi
a,
Oleg A.
Filippov
*c,
Carine
Duhayon
a,
Vincent
César
a,
Elena S.
Shubina
c,
Mohammed
Lahcini
*bd,
Dmitry A.
Valyaev
*a and
Yves
Canac
*a
ab,
Abdelhaq
Cherradi
a,
Oleg A.
Filippov
*c,
Carine
Duhayon
a,
Vincent
César
a,
Elena S.
Shubina
c,
Mohammed
Lahcini
*bd,
Dmitry A.
Valyaev
*a and
Yves
Canac
*a
aLCC-CNRS, Université de Toulouse, CNRS, UPS, 205 route de Narbonne, 31077 Toulouse Cedex 4, France. E-mail: dmitry.valyaev@lcc-toulouse.fr; yves.canac@lcc-toulouse.fr
bIMED-Lab, Faculty of Sciences and Techniques, Cadi Ayyad University, Bd Abdelkrim Al Khattabi, BP 549, 40000, Marrakech, Morocco. E-mail: m.lahcini@uca.ac.ma
cA. N. Nesmeyanov Institute of Organoelement Compounds (INEOS), Russian Academy of Sciences, 28/1 Vavilov Str., GSP-1, B-334, Moscow, 119334, Russia. E-mail: h-bond@ineos.ac.ru
dMohammed VI Polytechnic University, Lot 660, Hay Moulay Rachid, 43150 Ben Guerir, Morocco
First published on 7th January 2025
Abstract
The grafting of a –(CH2)2PR3+ moiety on an NHC ligand backbone in the Mn(I) complex [Cp(CO)2Mn(IMes)] followed by double deprotonation opens a route to bidentate ligands with extreme electron-donating character. Such remarkable electronic properties can even allow intramolecular sp2 C–H functionalization in typically inert square-planar Rh(I) dicarbonyl complexes.
Strongly electron-donating ligands have been widely investigated over the last three decades, leading to decisive advances in many fields such as homogeneous catalysis,1 photophysics2 and materials science.3 The isolation of the first stable 2H-imidazol-2-ylidene (A, Chart 1) by Arduengo in 19914 was a key milestone in this continuing quest, from which N-heterocyclic carbenes (NHCs) became essential for modern chemistry. The successive preparations of imidazol-4-ylidene (B)5 and anionic imidazol-2,4-diylidene (C),6 so-called “abnormal” (aNHC)7 and “ditopic” (dNHC)8 carbenes, were integral parts of the progress made to access increasingly electron-donating carbon ligands based on the imidazolyl core (Chart 1, top).9 Indeed, aNHC ligands B exhibit stronger σ-donor and weaker π-acceptor character than their “normal” NHC counterparts A10 and these electronic effects are even more pronounced in dNHCs metallated at the C2 position,11 due to the presence of an additional delocalized negative charge in the heterocyclic moiety. Beyond different classes of NHCs, phosphonium ylides derived from triarylphosphines (D) and trialkylphosphines (E) (Chart 1, down) constitute an alternative class of electron-rich neutral C-ligands for transition metals12 and main group elements.13 Moreover, a systematic IR spectroscopy investigation of a series of C,C-chelating Rh(I) dicarbonyl complexes with three different combinations of NHC (A) and phosphonium ylide (D) donor extremities revealed a decrease in the average νCO stretching frequencies in the order A–A > A–D > D–D, thus demonstrating the superior electron donation of phosphonium ylide vs. conventional NHC (Chart 1, bottom).14 In addition, it was recently shown for Pd(II) carbonyl complexes that a PCy3-derived phosphonium ylide fragment (E) is an even stronger donor than is its PPh3-containing analogue (D).15 In a logical continuation of our research program focused on the association of classical NHCs with phosphonium ylides in bidentate,14–16 pincer15,17 and tetradentate18 scaffolds, we envisioned to combine for the first time the strongest donors of each family with the aim of approaching the upper limits of the electronic scale in chelating systems.19 In this contribution, we report the preparation, coordination chemistry with rhodium and evaluation of electronic properties of anionic abnormal NHC/phosphonium ylide ligands (Chart 1, right) as well as unusual C–H functionalization in Rh(I) dicarbonyl species.
 | ||
| Chart 1 General trends in electron donation for different types of imidazole-based N-heterocyclic carbenes (top) and phosphonium ylides (bottom) as well as the structure of hybrid bidentate ligands proposed in this work (right). | ||
For the assembly of the anionic abnormal carbene platform, we chose a readily available, on a multi-gram scale, Mn(I) complex 120 (Scheme 1) bearing the common 1,3-bis(2,4,6-trimethylphenyl)-2H-imidazol-2-ylidene (IMes) ligand, in which the organometallic fragment in addition to protecting the C2 carbene position can also serve as an excellent IR11 and electrochemical21 probe. The –CH2CH2OH arm required for grafting the phosphonium moiety was installed on the IMes ligand backbone via a direct functionalization approach22 consisting of the deprotonation of 1 with nBuLi and the trapping of the resulting anionic abnormal carbene [Cp(CO)2Mn(dIMesLi)] with ethylene oxide. The subsequent transformation of complex 2 to the corresponding triflate derivative followed by one-pot quaternization with PPh3 and PCy3 afforded phosphonium pre-ligands [3a](OTf) and [3b](OTf) in good yields (Scheme 1). Both of them were fully characterized and their exact structures were obtained using X-ray diffraction (Fig. 1). Their deprotonation with 2.5 eq. of nBuLi in THF at room temperature led to a nearly quantitative generation of extremely moisture-sensitive aNHC-ylides 4a and 4b as confirmed by IR spectroscopy having revealed the presence of two low-frequency νCO bands (Table 1) characteristic for Mn(I) complexes bearing anionic aNHC ligands.20,22 The coordination of these species with a slight excess of [Rh(CO)2Cl]2 dimer was performed in toluene at low temperature similarly as described earlier for the related MnFe dIMes derivative,11 leading to the zwitterionic complexes 5a and 5b isolated in excellent yields (Scheme 1).
 | ||
| Scheme 1 Synthesis of anionic aNHC-phosphonium ylide ligands 4a–b and their Rh(I) complexes 5a–b. Reaction conditions: (i) 1.2 eq. nBuLi, THF, r.t., 2 h; (ii) 2 eq. ethylene oxide, −40 °C to r.t., 2 h; (iii) 1.1 eq. nBuLi, THF, r.t., 20 min; (iv) 1.1 eq. Tf2O, PhCl, −40 °C to r.t., 1 h; (v) 3.0 eq. PPh3 or 1.5 eq. PCy3, PhCl, 60–80 °C, 16 h. | ||
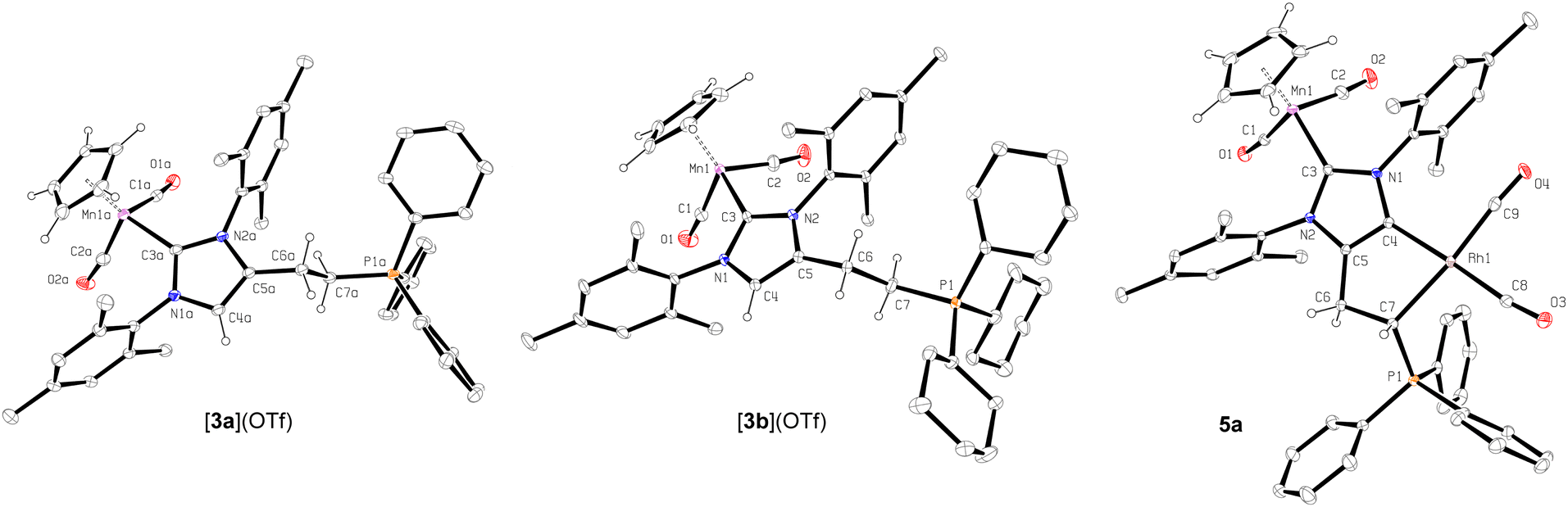 | ||
| Fig. 1 Molecular geometries of complexes [3a](OTf), [3b](OTf) and 5a (25% probability ellipsoids, hydrogen atoms of aryl groups, co-crystallized solvent molecules and counter-anions are omitted for clarity). Selected bond lengths [Å], and angles (°). [3a](OTf): Mn1a–C3a 2.012(3), C4a–C5a 1.344(4), C7a–P1a 1.805(3); [3b](OTf): Mn1–C3 2.012(2), C4–C5 1.337(3), C7–P1 1.806(3); 5a: Mn1–C3 2.011(2), C4–C5 1.349(3), Rh1–C4 2.045(2), Rh1–C7 2.189(2), C7–P1 1.769(2) Rh1–C8 1.890(2), Rh1–C9 1.868(2), C4–C5 1.399(3), C4–Rh1–C7 78.98(8), C4–Rh1–C8 176.67(10), C7–Rh1–C9 174.16(8), C8–Rh1–C9 87.43(10), P1–C7–Rh1 115.69(10), C4–C5–C6–C7 3.9(3). | ||
| No. | Experimental νCO (THF, cm−1) | Calculated νCO (non-scaled, cm−1) | ||
|---|---|---|---|---|
| [Mn(CO)2] | [Rh(CO)2] | [Mn(CO)2] | [Rh(CO)2] | |
| 4a | 1899, 1829 | — | 2028, 1966 | — |
| 4b | 1898, 1828 | — | 2028, 1965 | — |
| 5a | 1906, 1838 | 2035, 1966 | 2032, 1969 | 2147, 2079 |
| 5b | 1906, 1838 | 2030, 1952 | 2031, 1968 | 2139, 2065 |
| 6a | 1912, 1845 | 2034, 1966 | 2040, 1979 | 2143, 2078 |
The identity of heterobimetallic MnRh complexes 5a and 5b was confirmed from spectroscopic methods and HRMS data. IR spectra of both products showed two pairs of νCO bands in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 intensity ratio (Fig. S23, S24, ESI† and Table 1) in agreement with a presence of [Mn(CO)2] and [Rh(CO)2] fragments exhibiting a cis-arrangement of carbonyl groups. The presence of a P-ylide moiety coordinated to the Rh(I) center was confirmed by the observation of 31P NMR signals at δP 41.2 and 43.6 ppm for 5a and 5b, respectively, being deshielded compared to those of their phosphonium precursors ([3a](OTf): δP 22.7 ppm; [3b](OTf): δP 31.3 ppm) as well as characteristic high-field 13C resonances with appropriate multiplicity (5a: δC 13.9 ppm (dd, 1JCRh = 27.5 Hz, 1JCP = 25.5 Hz); 5b: δC 6.4 ppm (dd, 1JCRh = 25.7 Hz, 1JCP = 19.6 Hz)). The characteristic 5a and 5b13C NMR signals resulting from the carbenic atoms of anionic imidazol-2,4-diylidenes were observed as singlets at δC 199.9–200.5 ppm (C2) and doublets at δC 162.2–162.7 ppm with a 1JCRh coupling constant of ca. 42 Hz (C4). Finally, each of the bimetallic complexes yielded four distinct 13C NMR spectral CO signals and a full set of inequivalent resonances of mesityl groups, probably due to blocked rotation across their respective C–N bonds and the existence of a chiral center in the NHC backbone.
:1 intensity ratio (Fig. S23, S24, ESI† and Table 1) in agreement with a presence of [Mn(CO)2] and [Rh(CO)2] fragments exhibiting a cis-arrangement of carbonyl groups. The presence of a P-ylide moiety coordinated to the Rh(I) center was confirmed by the observation of 31P NMR signals at δP 41.2 and 43.6 ppm for 5a and 5b, respectively, being deshielded compared to those of their phosphonium precursors ([3a](OTf): δP 22.7 ppm; [3b](OTf): δP 31.3 ppm) as well as characteristic high-field 13C resonances with appropriate multiplicity (5a: δC 13.9 ppm (dd, 1JCRh = 27.5 Hz, 1JCP = 25.5 Hz); 5b: δC 6.4 ppm (dd, 1JCRh = 25.7 Hz, 1JCP = 19.6 Hz)). The characteristic 5a and 5b13C NMR signals resulting from the carbenic atoms of anionic imidazol-2,4-diylidenes were observed as singlets at δC 199.9–200.5 ppm (C2) and doublets at δC 162.2–162.7 ppm with a 1JCRh coupling constant of ca. 42 Hz (C4). Finally, each of the bimetallic complexes yielded four distinct 13C NMR spectral CO signals and a full set of inequivalent resonances of mesityl groups, probably due to blocked rotation across their respective C–N bonds and the existence of a chiral center in the NHC backbone.
Inspection of the X-ray diffraction structure acquired of complex 5a (Fig. 1) showed the Rh(I) atom in a square-planar environment with the dIMes-ylide behaving as a cis-chelating ligand. While the Rh–carbene bond distance (2.045(2) Å) was measured to be very close to the values found in other Rh(I) complexes bearing IMes-derived aNHCs (2.038(3)–2.061(4) Å),23 the Rh-ylide bond (2.189(2) Å) was found to be slightly longer than those of similar bidentate ylide structures (2.1250(14)–2.155(8)).14,24 The Rh–CO bond in the position trans to the aNHC moiety was also observed to be slightly longer than that opposite to the phosphonium ylide, presumably reflecting higher trans effect of the former ligand. Inspection of the structure also showed, notably, the 5-membered rhodacycle to be virtually coplanar with the NHC fragment. And structural comparisons showed the NHC–Rh-ylide angle in 5a (78.98(8)°) to be significantly more acute that those observed in Rh(I) (94.0(3)°)14 or Pd(II) (91.9(2)°)16a NHC-ylide complexes featuring more flexible 7- and 6-membered chelating cycles, respectively, and to be comparable only with those of related species based on more rigid NHC-bis(ylide) ligands (82.61(6)–85.85(5)°)25 with a similar size of the metallacycle.
Access to the Rh(I) dicarbonyl derivatives 5a–b provided a perfect opportunity to quantify the electronic properties of the novel aNHC-ylide ligands using the frequencies of νCO bands in solution IR spectra, which is applicable for benchmarking both mono-9 and bidentate ligands.19 While νCO values for the [Cp(CO)2Mn] fragment in 5a and 5b were observed to be almost the same (Table 1) and to remain very close to that of the previously reported dIMes complex,11 the average 〈νCO〉 value for the [Rh(CO)2] moiety of 5b was found to be ca. 10 cm−1 lower, consistent with the higher electron-donation ability of PCy3-derived ylide vs. its PPh3 analogue. The same trends were confirmed using cyclic voltammetry studies revealing a reversible Mn(I)/Mn(II) oxidation wave at −0.38 V vs. Fc/Fc+ followed by the occurrence of irreversible one-electron oxidation at +0.61 and +0.51 V vs. Fc/Fc+ for the Rh(I) moiety in complexes 5a and 5b, respectively. Very importantly, the values of the 〈νCO〉 parameter for both 5a (2001 cm−1) and 5b (1991 cm−1) were observed to be far lower than those for known Rh(I) dicarbonyl complexes bearing bis(NHC) (2057 cm−1),14 phosphine-phosphonium ylide (2053 cm−1),24a bis(1,2,3,-triazol-4-ylidene) (2047–2048 cm−1),26 NHC-iminophosphorane (2046 cm−1),27 NHC-phosphonium ylide (2039 cm−1)14 and bis(phosphonium ylide) ligands (2017 cm−1),14 thus highlighting the extreme donation ability of our developed systems.
Intrigued by these outstanding electronic properties, we decided to study the structure and bonding situation in bimetallic species by performing DFT calculations. To our delight, all the trends observed in the experimental IR spectra of 4a–b and their Rh(I) complexes 5a–b were essentially perfectly reproduced in the νCO frequency calculations (Table 1). The calculations yielded rather similar HOMOs localized at the [CpMn(CO)2] moiety in both species (Fig. 2, top), but markedly different LUMOs for 5a and 5b, revealing the main contribution of PPh3+ and [Rh(CO)2] fragments, respectively (Fig. 2, bottom). The global AIM charge of the [Rh(CO)2] fragment in complex 5b was found to be slightly lower than that in the case of 5a (0.171 vs. 0.177), consistent with a stronger electron donation of the PCy3-derived ylide moiety. Interestingly, the AIM charge distribution in carbonyl ligands coordinating the Rh(I) center in bimetallic complexes was found to be quite asymmetrical, accumulating more electron density on the CO in the position trans to the aNHC ligand (−0.207 and −0.225 for 5a and 5b, respectively) than on those having the cis arrangement (ca. −0.160). An ETS-NOCV analysis of the NHC-ylide bonding (Fig. S4, see the ESI† for details)—beyond showing the major primary channel responsible for σ-donation from both C-ligands to the [Rh(CO)2] unit—also revealed an additional channel for the aNHC moiety having ca. 40% weaker associated energy, with the charge flowing only to the metal atom and opposite CO ligand (Fig. S4, ESI†). These results seemed to indicate the anionic aNHC ligand to be a stronger σ-donor than neutral phosphonium ylide regardless of the occurrence of some π-retrodonation for the metal–carbene bond (Fig. S5, ESI†).
 | ||
| Fig. 2 Frontier molecular orbitals of complexes 5a and 5b (isosurface set at 0.05 a.u.). | ||
Unlike complex 5b, which was observed to be stable in solution over long periods of time, its PPh3+ analogue 5a slowly transformed even at room temperature to another species, namely 6a, isolated in 95% yield after being heated at 50 °C (Scheme 2). While IR and cyclic voltammetry data for [Rh(CO)2] remained virtually unchanged (Table 1), both νCO frequencies (1912, 1845 cm−1)11 and oxidation potential (−0.32 V vs. Fc/Fc+)21 of the Mn(I) center clearly indicated its coordination with a regular imidazol-2-ylidene ligand, which was consistent with the re-appearance of the characteristic 13C NMR resonance of the CH moiety belonging to the NHC backbone (δC 122.9 ppm). Finally, the presence of the coordinated ylide moiety (δC 22.5 ppm, dd, 1JCP = 32.2 Hz, 1JCRh = 24.1 Hz) and downfield signal at δC 181.6 (pseudo t, 1JCRh = 2JCP = 33.7 Hz) characteristic for cyclometallated phosphonium fragment,17 unambiguously confirmed the structure of 6a as a zwitterionic heterobimetallic MnRh species bearing a bidentate aryl/phosphonium ylide ligand. A preliminary hypothesis concerning the formation of complex 6a might include the intramolecular activation of the ortho-C–H bond of a P+–Ph substituent by the electron-rich metal site followed by a reductive elimination of the aNHC ligand in a putative Rh(III) hydride intermediate. Despite an extensive use of Rh(I) derivatives in catalytic C–H functionalization,28 this transformation represented the first example, to the best of our knowledge, of intramolecular sp2 C–H functionalization in typically inert square-planar Rh(I) dicarbonyl complexes.29
 | ||
| Scheme 2 Transformation of complex 5a into its isomer 6avia cyclometallation with the phenyl ring of the phosphonium moiety. | ||
In summary, we have combined, for the first time, anionic abnormal carbene and phosphonium ylide to obtain structurally rigid cis-chelating ligands with extreme electron-donating properties. The incorporation of alkyl substituents at the phosphorus atom in such scaffolds improved both ligand donicity and stability of the resulting transition metal complexes. The application of these ligands in homogeneous catalysis as well as the studies on the comparison of electronic donation between aNHC and phosphonium ylide is currently ongoing in our group.
We thank the CNRS and Russian Science Foundation (grant no. 24-13-00283, theoretical studies) for financial support. M. E. K. is grateful to French Embassy in Morocco for a PhD mobility grant and the Centre National de la Recherche Scientifique et Technique (CNRST) of Morocco. Computational studies were performed using HPC resources from CALMIP (no. P18038).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981 CrossRef CAS PubMed; (b) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233 RSC; (c) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493 CrossRef PubMed; (d) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed.
- (a) T.-Y. Li, S.-J. Zheng, P. I. Djurovich and M. E. Thompson, Chem. Rev., 2024, 124, 4332 CrossRef CAS PubMed; (b) H. Amouri, Chem. Rev., 2023, 123, 230 CrossRef CAS PubMed; (c) N. Sinha and O. S. Wenger, J. Am. Chem. Soc., 2023, 145, 4903 CrossRef CAS PubMed.
- (a) Y. Wang, J.-P. Chang, R. Xu, S. Bai, D. Wang, G.-P. Yang, L.-Y. Sun, P. Li and Y.-F. Han, Chem. Soc. Rev., 2021, 50, 13559 RSC; (b) M. Koy, P. Bellotti, M. Das and F. Glorius, Nat. Catal., 2021, 4, 352 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- E. Aldeco-Perez, A. J. Rosenthal, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Science, 2009, 326, 556 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, M. Y. Abraham, P. Wei, H. F. Schaefer, P. V. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2010, 132, 14370 CrossRef CAS PubMed.
- The term abnormal NHC (aNHC) was first coined by Crabtree et al., see: S. Gründemann, A. Kovacevic, M. Albrecht, J. W. Faller Robert and H. Crabtree, Chem. Commun., 2001, 2274 RSC.
- For a general review on ditopic NHC (dNHC), see: J. B. Waters and J. M. Goicoechea, Coord. Chem. Rev., 2015, 293–294, 80 CrossRef CAS.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457 CrossRef CAS PubMed.
- J. C. Bernhammer, G. Frison and H. V. Huynh, Chem. – Eur. J., 2013, 19, 12892 CrossRef CAS PubMed.
- A. A. Grineva, O. A. Filippov, Y. Canac, J. B. Sortais, S. E. Nefedov, N. Lugan, V. César and D. A. Valyaev, Inorg. Chem., 2021, 60, 4015 CrossRef CAS PubMed.
- (a) D. A. Valyaev and Y. Canac, Dalton Trans., 2021, 50, 16434 RSC; (b) J. Handelmann, C. N. Babu, H. Steinert, C. Schwarz, T. Scherpf, A. Kroll and V. H. Gessner, Chem. Sci., 2021, 12, 4329 RSC; (c) E. P. Urriolabeitia, Top. Organomet. Chem., 2010, 30, 15 CrossRef CAS PubMed.
- (a) C. Mohapatra, L. Scharf, T. Scherpf, B. Mallick, K.-S. Feichtner, C. Schwarz and V. H. Gessner, Angew. Chem., Int. Ed., 2019, 58, 7459 CrossRef CAS PubMed; (b) T. Scherpf, K. S. Feichtner and V. H. Gessner, Angew. Chem., Int. Ed., 2017, 56, 3275 CrossRef CAS PubMed; (c) L. T. Scharf and V. H. Gessner, Inorg. Chem., 2017, 56, 8599 CrossRef CAS PubMed.
- Y. Canac, C. Lepetit, M. Abdalilah, C. Duhayon and R. Chauvin, J. Am. Chem. Soc., 2008, 130, 8406 CrossRef CAS PubMed.
- M. El Kadiri, A. Chihab, R. Taakili, C. Duhayon, D. A. Valyaev and Y. Canac, Organometallics, 2022, 41, 456 CrossRef CAS.
- (a) I. Benaissa, R. Taakili, N. Lugan and Y. Canac, Dalton Trans., 2017, 46, 12293 RSC; (b) Y. Canac, C. Duhayon and R. Chauvin, Angew. Chem., Int. Ed., 2007, 46, 6313 CrossRef CAS PubMed.
- (a) M. Ameskal, R. Taakili, E. S. Gulyaeva, C. Duhayon, J. Willot, N. Lugan, C. Lepetit, D. A. Valyaev and Y. Canac, Inorg. Chem., 2023, 62, 20129 CrossRef CAS PubMed; (b) R. Taakili, C. Barthes, C. Lepetit, C. Duhayon, D. A. Valyaev and Y. Canac, Inorg. Chem., 2021, 60, 12116 CrossRef CAS PubMed.
- C. Barthes, C. Bijani, N. Lugan and Y. Canac, Organometallics, 2018, 37, 673 CrossRef CAS.
- Y. Canac and C. Lepetit, Inorg. Chem., 2017, 56, 667 CrossRef CAS PubMed.
- A. A. Grineva, D. A. Valyaev, V. César, O. A. Filippov, V. N. Khrustalev, S. E. Nefedov and N. Lugan, Angew. Chem., Int. Ed., 2018, 57, 7986 CrossRef CAS PubMed.
- R. Brousses, V. Maurel, J.-M. Mouesca, V. César, N. Lugan and D. A. Valyaev, Dalton Trans., 2021, 50, 14264 RSC.
- (a) A. A. Grineva, O. A. Filippov, S. E. Nefedov, N. Lugan, V. César and D. A. Valyaev, Organometallics, 2019, 38, 2330 CrossRef CAS; (b) D. A. Valyaev, M. A. Uvarova, A. A. Grineva, V. César, S. N. Nefedov and N. Lugan, Dalton Trans., 2016, 45, 11953 RSC.
- (a) F. M. Chadwick, B. F. E. Curchod, R. Scopelliti, F. F. Tirani, E. Solari and K. Severin, Angew. Chem., Int. Ed., 2019, 58, 1764 CrossRef CAS PubMed; (b) M. Brill, D. Marrwitz, F. Rominger and P. Hofmann, J. Organomet. Chem., 2014, 775, 137 CrossRef.
- (a) I. Popovici, C. Barthes, T. Tannoux, C. Duhayon, N. Casaretto, A. Monari, A. Auffrant and Y. Canac, Inorg. Chem., 2023, 62, 2376 CrossRef CAS PubMed; (b) R. Zurawinski, B. Donnadieu, M. Mikolajczyk and R. Chauvin, Organometallics, 2003, 22, 4810 CrossRef CAS.
- M. Asay, B. Donnadieu, A. Baceiredo, M. Soleilhavoup and G. Bertrand, Inorg. Chem., 2008, 47, 3949 CrossRef CAS PubMed.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Organometallics, 2011, 30, 6017 CrossRef CAS PubMed.
- K. Azouzi, C. Duhayon, I. Benaissa, N. Lugan, Y. Canac, S. Bastin and V. César, Organometallics, 2018, 37, 4726 CrossRef CAS.
- (a) Y. Zhang, J.-J. Zhang, L. Lou, R. Lin, N. Cramer, S.-G. Wang and Z. Chen, Chem. Soc. Rev., 2024, 53, 3457 RSC; (b) C.-X. Liu, S.-Y. Yin, F. Zhao, H. Yang, Z. Feng, Q. Gu and S.-L. You, Chem. Rev., 2023, 123, 10079 CrossRef CAS PubMed.
- Upon UV irradiation complex [Tp*Rh(CO)2] was shown to activate the C–H bond in arenes and alkanes, see: C. K. Ghosh and W. A. G. Graham, J. Am. Chem. Soc., 1987, 109, 4726 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, spectroscopic, crystallographic and DFT details. CCDC 2393836–2393838. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06177b |
| This journal is © The Royal Society of Chemistry 2025 |