
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoredox capacity expanded by the Cu site of CuFe-Mabiq†
Matthias
Huber
ab,
Ajeet
Kumar
b,
Jürgen
Hauer
 b,
Erling
Thyrhaug
*b and
Corinna R.
Hess
*ab
b,
Erling
Thyrhaug
*b and
Corinna R.
Hess
*ab
aFaculty of Chemistry and Pharmacy, University of Regensburg, 93053 Regensburg, Germany. E-mail: corinna.hess@ur.de
bTUM School of Natural Sciences, Department of Chemistry and Catalysis Research Center, Technical University of Munich, 85748 Garching, Germany. E-mail: erling.thyrhaug@tum.de
First published on 25th February 2025
Abstract
The monometallic Fe-Mabiq and bimetallic CuFe-Mabiq photoredox catalysts feature similar optical spectra and short excited-state lifetimes. Nevertheless, they exhibit markedly different photochemistry. Photoreduction proceeds significantly faster for the bimetallic complex, and uniquely generates the three-electron reduced form. These characteristics underpin the self-sensitized photocatalytic behaviour of the bimetallic complex.
The development of catalysts for CO2 reduction is largely motivated by the prospect of using this greenhouse gas as a C1-feedstock for valuable chemicals and fuels.1,2 Light-driven CO2 conversion is particularly appealing, since this route could harness a natural renewable energy source, sunlight. Despite the challenges associated with CO2 reduction, several molecular catalysts have been developed that operate with reasonable overpotential requirements and with high turnover numbers (TONs), faradaic efficiencies, and selectivity.3,4 However, in photocatalytic CO2 reduction, very few of these can function in the absence of an added photosensitizer (PS)5 a role commonly served by Ru and Re complexes.6
We recently showed that our monometallic Fe-Mabiq (Mabiq = 2–4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6–8-bis(3,3,4,4-tetramethyldihydropyrrolo)-10–15-(2,2′-biquinazolino)-[15]-1,3,5,8,10,14-hexaene-1,3,7,9,11,14-N6) complex ([Fe(Mabiq)]+, 1, Fig. 1a) acts as both an electro- and photocatalyst for CO2 reduction, selectively generating CO.7,8 The photocatalytic reaction requires [Ru(bpy)3]2+ as PS. A key intermediate in the CO2 reduction, IPhOH (structure in Fig. S17, ESI†), is generated upon protonation of the formal Fe0 form of 1. Consequently, the Mabiq ligand may offer a proton relay site and support hydrogen bonding, which both favor CO formation.7 Our related bimetallic CuFe-Mabiq complex ([Cu(Xantphos)Fe(Mabiq)(MeCN)(OTf)]OTf, 2, Fig. 1a) likewise is an effective and selective CO2 reduction catalyst.8 However, the neighbouring Cu-Xantphos unit of 2 alters both the redox properties and reactivity of the complex, where, in contrast to 1, formation of an IPhOH analogue does not occur in the CO2 reduction pathway. Moreover, photocatalytic CO2 to CO conversion by 2 proceeds even in the absence of added PS. The bimetallic 2 is one of the most efficient self-sensitized catalysts for this reaction, with a TON of 100. Thus, we set out to examine the photochemical properties of the mono- and bimetallic compounds to determine the factors that underpin the enhanced reactivity on association with Cu-Xantphos.
:6–8-bis(3,3,4,4-tetramethyldihydropyrrolo)-10–15-(2,2′-biquinazolino)-[15]-1,3,5,8,10,14-hexaene-1,3,7,9,11,14-N6) complex ([Fe(Mabiq)]+, 1, Fig. 1a) acts as both an electro- and photocatalyst for CO2 reduction, selectively generating CO.7,8 The photocatalytic reaction requires [Ru(bpy)3]2+ as PS. A key intermediate in the CO2 reduction, IPhOH (structure in Fig. S17, ESI†), is generated upon protonation of the formal Fe0 form of 1. Consequently, the Mabiq ligand may offer a proton relay site and support hydrogen bonding, which both favor CO formation.7 Our related bimetallic CuFe-Mabiq complex ([Cu(Xantphos)Fe(Mabiq)(MeCN)(OTf)]OTf, 2, Fig. 1a) likewise is an effective and selective CO2 reduction catalyst.8 However, the neighbouring Cu-Xantphos unit of 2 alters both the redox properties and reactivity of the complex, where, in contrast to 1, formation of an IPhOH analogue does not occur in the CO2 reduction pathway. Moreover, photocatalytic CO2 to CO conversion by 2 proceeds even in the absence of added PS. The bimetallic 2 is one of the most efficient self-sensitized catalysts for this reaction, with a TON of 100. Thus, we set out to examine the photochemical properties of the mono- and bimetallic compounds to determine the factors that underpin the enhanced reactivity on association with Cu-Xantphos.
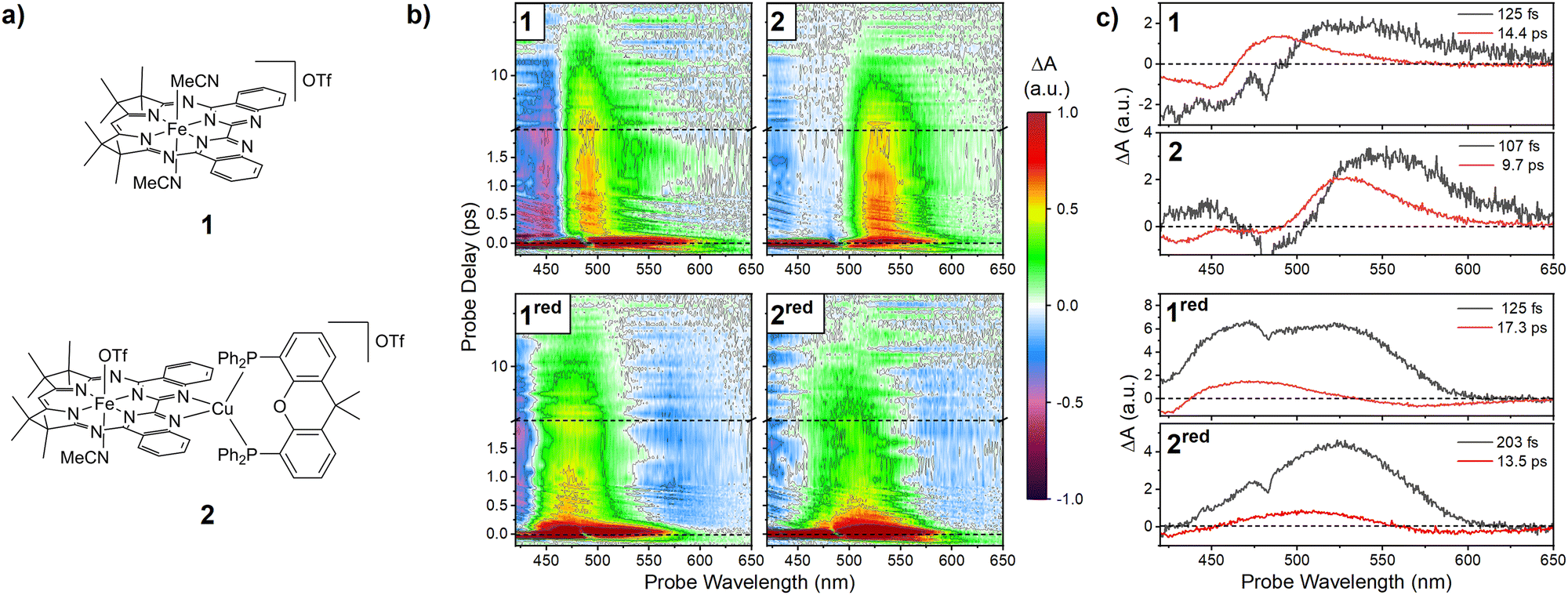 | ||
| Fig. 1 (a) Structure of 1 and 2. (b) Ultrafast relaxation dynamics of 1 and 2 and their reduced species after photoexcitation with < 50 fs pulses centred at 430 nm. Probe delay vs. probe wavelength transient spectra of all complexes in THF solution. Spectra shown on normalized scale to facilitate direct comparison. Note the change to logarithmic scale at a probe-delay of 2 ps. (c) species spectra (EAS, see text for details) recovered from a global kinetic fit of the TA data in panel b. The time-constants extracted from the fit are displayed in the panels. | ||
For the current studies, we examined the photochemistry and photodynamics of 1 and 2, as well as of their one-electron reduced forms, 1red ([Fe(Mabiq)]) and 2red ([Cu(Xantphos)Fe(Mabiq)(OTf)]). The latter are generated en route to the catalytically active forms, and result from photoreduction of the respective FeII precursors in the presence of a sacrificial electron donor (vide infra).8,9 We previously reported the synthesis and structural characterization of 1, 2 and 1red.8,10 Here we also report the synthesis of 2redvia the reaction of 1red with [Cu(Xantphos)OTf]. Although attempts to obtain a molecular structure of this complex were unsuccessful, we were able to isolate crystals of an analogue of 2red (2red,OMe, see Fig. S1, S2 and Tables S1–S3, ESI†) containing a methoxylated Xantphos ligand. The structure of this one-electron reduced form is similar to that of 2, but in the crystal the Fe centre is five-coordinate.
The effect of coordinating the Cu-Xantphos moiety on the redox properties of 2 is notable, as the first two reductions (i.e., Mabiq/Mabiq˙− and Mabiq˙−/Mabiq2−) are both shifted ∼+ 150 mV relative to the monometallic 1.8 The more positive redox potentials of 2 imply that its formal Fe0 form is less electron rich than that of 1. This is one likely reason why IPhOH is generated in the CO2 reduction pathway of 1, but a corresponding intermediate is absent in the mechanism of 2.8
Despite the significant differences in redox properties, the absorption spectra of 1 and 2 are virtually identical (Fig. S3, ESI†). The spectra of 1red and 2red are also highly similar (Fig. S3, ESI†), suggesting a similar electronic structure at the Fe-Mabiq centres also after reduction (For 1red, an intermediate spin ferrous centre is ligated to the dianionic Mabiq radical10).
The photophysics of molecular iron complexes are generally ultrafast and notoriously complicated, involving relaxation through a manifold of states of widely differing character.11–13
As a first step in understanding the photocatalytic activity of 1 and 2 and their reduced forms, we address the internal relaxation processes taking place after light absorption. For this we rely on ultrafast transient absorption (TA) spectroscopy, which allows us to follow photoinduced dynamics by the time-dependence of the UV/Vis absorption spectra.
Our TA instrument is described in detail elsewhere14 (see ESI†, Section S6 for additional details). Briefly, we impulsively excite the complexes using ≤ 50 fs pulses centred at 430 nm. We subsequently monitor the time-evolution of the optical transmission using an uncompressed white-light supercontinuum pulse (pump and probe spectra in Fig. S4, ESI†). The time-resolution of the experiment, set largely by the non-resonant response of the solvent, is approximately 60 fs.
The TA spectra of the complexes are depicted in Fig. 1b (spectra extracted at selected delay times in Fig. S5, ESI†). In accordance with other molecular iron complexes,11 as well as that of other Mabiq complexes of open-shell transition metals,91 and 2 show an ultrafast initial relaxation phase and a short overall excited-state lifetime.
For both complexes, photoexcitation leads to the immediate formation of a negative-amplitude ground-state-bleach (GSB) band below 450 nm and a broad excited-state absorption (ESA) band covering the 500–650 nm range. Over a few hundred femtoseconds, the ESA narrows and blue-shifts towards the 500 nm region. Subsequent to these early-time dynamics, the overall signal decreases without further spectral changes, as the excited complexes return to their ground state. After a few tens of picoseconds, the ground-state recovery is complete.
This behaviour is analogous to our observations for a Ni-Mabiq complex.9 Based on this work, we assign the initial excitation to a ligand-localized (π–π*) transition. This excitation rapidly transfers to the metal centre, whereafter the ESA around 500 nm essentially reports on (π–π*) re-excitations of the ground-state Mabiq ligand near a photoexcited metal centre.
As illustrated in the spectra in Fig. 1b, the photophysics of 1red and 2red are analogous to those of their non-reduced counterparts. Again, we observe a broad blue/green ESA which rapidly decays to reveal a narrower ESA feature below 500 nm. Quantitative analysis of the decay kinetics (using a global sum-of-exponentials model15) agrees with these observations. For all four species, two exponentials are sufficient to fit the dynamics after the initial ∼100 fs. Imposing a compartmental model with strictly sequential population transfer allows the extraction of the so-called “evolution associated spectra” (EAS). These are spectra of the compartments (or “states”) associated with the time-constants in the observed decay (Fig. 1c). In all cases the quantitative analysis agrees with our qualitative observations: the initial state displays broad ESA in the visible range. On a timescale of ∼100 fs, the population leaves this state, entering a less strongly absorbing state with a lifetime of 10–20 ps. The short lifetimes observed here are typical of most iron-based coordination complexes,11–13 with only a few notable exceptions.12,16,17
The similarity of the catalysts’ spectral dynamics is striking, and carries implications for their electronic structure: While coordination of a Cu centre induces minor shifts in the optical spectra, this is clearly not sufficient to alter the state ordering or character. Similarly, as reduction of the complexes is ligand-localized, this also does not qualitatively alter the dynamics: although the early-time spectra are notably different due to the different ligand redox state, energy-transfer to the metal centre remains ultrafast. The remarkably similar photodynamics of the complexes imply that the origin of the marked differences in photochemical behaviour of 1 and 2 is elsewhere. These functional differences are evinced not only by their photocatalytic CO2 reduction activity, but by their photoredox behaviour, as described herein.
To further explore the photoredox properties, we conducted a series of quantitative photoreduction experiments. 50 μM solutions of 1 and 2 in MeCN were irradiated at 455 nm in the presence of the reductant, 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH), at concentrations ranging from 25 mM to 100 mM. The experiments were performed under rigorous oxygen exclusion and in the absence of PS.
Before irradiation, we observed that BIH induced the appearance of a weak shoulder at the red edge of the absorption spectra of 1 and 2. While this implies ground-state interaction between part of the catalyst population and BIH (see Fig. S6 and S7, ESI†), this did not affect the photoreduction products. Further, the products obtained in the one-electron reductions of 1 or 2 are identical to those obtained using Et3N as a sacrificial donor, where no ground-state association is apparent (Fig. S6, S8–S11, ESI†).
As expected for a bimolecular process, while the photoreduction rates correlate with the BIH concentration, the reduction products formed do not depend on [BIH] (Fig. S12–S16, ESI†). Thus, we limit the following discussion of reaction times and quantum yields (QYs) to experiments at 100 mM BIH, corresponding to our photocatalytic CO2 reduction experiments.8
The spectral evolution of 1 (Fig. 2a, black) upon irradiation in the presence of 100 mM BIH shows clean conversion to 1red (Fig. 2a, red) within 25 min, as evinced by the loss of the 464 nm band of 1 concomitant with the growth of the characteristic 623 nm 1red absorption. After irradiation for 130 min, we observe the conversion to a spectrum similar to that of IPhOH (Fig. 2b, blue. See Fig. S17, ESI† for reference). This indicates not only that BIH is a strong enough reductant to enable formation of the formal ‘Fe0’ form of the complex (denoted as 12×red), but that BIH˙+ is also a sufficiently strong acid to protonate 12×red. (We denote the two-electron reduced, protonated product as 12×redH+, since here, protonation occurs in the absence of PhOH.) Interestingly, further irradiation of 12×redH+ yields yet another species at 220 min, which can be identified as the unprotonated 12×red (Fig. 2b, green), based on comparison with the spectrum of electrochemically generated 12×red (Eapp = −2.10 VFc, see Fig. S18–S20, ESI†). This loss of the proton after prolonged irradiation may coincide with H2 evolution. In the presence of CO2, this side reaction is not observed and was not further investigated. Irradiation of the highly reduced 12×red beyond 220 min led to an overall signal loss, which we attribute to decomposition (Fig. S21, ESI†).
 | ||
| Fig. 2 Representative UV/Vis absorption spectra recorded during the photoreduction of 50 μM 1 (panels a and b) and 2 (panels c and d) in the presence of 100 mM BIH as a sacrificial reductant‡. | ||
Overall, despite the short excited-state lifetimes, the photoreduction of both 1 and 1red with BIH is possible; the latter process coinciding with ligand protonation (Scheme 1). The QY for photoconversion of 1 to 1red is 1.1 × 10−4 while the subsequent electron and proton transfer steps to yield 12×redH+ are significantly slower with a QY of 4.0 × 10−5.‡
 | ||
| Scheme 1 Center: Reduction pathways in the photoreduction of 1 and 2 with BIH. Fe oxidation states denote formal charges. Electrons are provided by the sacrificial reductant (BIH or BI˙ radical). The transformation from 12×redH+ to 12×red may proceed via H2 evolution. E0(12×redH+/1red) taken from ref. 7. Side: CO2 reduction pathways determined in previous work.7,8 | ||
Irradiation of 2 (Fig. 2c, black) in the presence of BIH rapidly produces the spectrum of 2red (Fig. 2c, red, Fig. S22, ESI†). The formation of 2red is remarkably fast compared to that of 1red (2.0 min vs. 25 min), with a corresponding ∼9-fold increase in QY (9.7 × 10−4vs. 1.1 × 10−4).
The subsequent transformation of 2red to 22×red (Fig. 2d, blue) is also substantially faster than the photoconversion of 1red (4.5 min vs. 105 min, corresponding to a total reaction time of 6.5 and 130 min), and proceeds with a much higher QY (9.0 × 10−5vs. 4.0 × 10−5). The spectrum of the electrochemically generated 22×red (Eapp = −1.76 VFc, see Fig. S22–S24, ESI†) confirms our assignment. BIH can thus transfer two electrons to 2 but does not protonate this compound. In accord, we also could not protonate the electrochemically generated 22×red; and no reaction or oxidation could be observed in the presence of a range of proton sources (Fig. S25, ESI†).
After continued irradiation the bimetallic 22×red further converts, within the following ∼4 min, as indicated by the growth of new spectral features at 590 and 1000 nm (Fig. 2d, green). Based on good qualitative agreement between this spectrum and spectro-electrochemical data, we attribute this third photoreduction product to the three-electron reduced species 23×red (electrochemically generated at Eapp = −2.24 VFc, Fig. S22, S23 & S26, ESI†). The photoreduction of 2 by BIH thus uniquely proceeds with the transfer of three electrons (Scheme 1), even in the absence of PS. We note that after prolonged irradiation of this highly reduced species, further well-defined spectral changes are observed, as as-of-yet unidentified species are formed (Fig. S27 and S28, ESI†). In the presence of CO2 as a substrate, however, the formation of these additional species is suppressed, as 23×red is continually removed from solution via the CO2 reduction reaction.
With respect to the catalytic capacity of the Fe-Mabiq complexes, the ∼15 ps excited-state lifetimes present a bottleneck to the per-photon QY for formation of photo-reduced intermediates. The reductions of both 1 and 2 by BIH also are mechanistically non-trivial. However: the chemical yields of the reduced intermediates are generally high; the clear isosbestic points indicate negligible formation of side-products, thus implying a high degree of catalyst stability.
Our results concerning photoreduction of 2 explain the ability of the bimetallic complex to act as a self-sensitized photoredox catalyst for CO2 reduction. Rapid formation of the reduced catalytic intermediates favours rapid turnover and mitigates catalyst decomposition and unproductive pathways. In the mechanisms of CO2 reduction by 1 and 2, a third reduction step occurs prior to, or coincident with, CO2 binding. Therefore, the singular ability to photochemically generate the formal Cu+IFe−1 species 23×red in the absence of an added PS is highly significant. The data imply that 23×red can either directly be photogenerated and then bind CO2, or that a CO2-bound intermediate could be photoreduced. For the monometallic 1, these final light-induced electron transfer steps, and efficient transfer between the reduced forms, are only feasible with an added PS.6,8
The reason for faster photoreduction of 2 is not clear, and will require further study. The ground state redox potentials of 1 and 2 differ substantially (vide supra), however, and the effect of the Cu centre on the excited state redox potentials and thus the electron transfer driving force may be a principal factor. Structural characteristics may also contribute: steric constraints imposed by the Cu-Xantphos unit could result in lower reorganization energies.18
Current work includes extending the range of photo-accessible redox-states of M-Mabiq complexes, and accordingly, their catalytic capacity. In fact, 1 can be photo-generated from FeIII(Mabiq)Cl2via homolytic cleavage of the Fe–Cl bond (Fig. S29, ESI†), paralleling the behaviour of CoIII(Mabiq)Cl2.19 The ability to photochemically access such a broad span of redox states is unprecedented among photoredox catalysts; and the formally low valent forms are potent reductants. We find the bimetallic Mabiq complexes especially promising given their enhanced photochemical properties.
We acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG): TRR 325 and project 547756633 (ET, JH and AK), 426785626 (CRH and MH), 507868493 (CRH).
Data availability
Supporting data for this article have been included in the ESI.† Crystallographic data for 2red,OMe has been deposited at the CCDC under 2391634.Conflicts of interest
There are no conflicts to declare.Notes and references
- N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CAS.
- A. Rosas-Hernández, C. Steinlechner, H. Junge and M. Beller, in Chemical Transformations of Carbon Dioxide, ed.X.-F. Wu and M. Beller, Springer International Publishing, Cham, 2018, DOI:10.1007/978-3-319-77757-3_7, pp. 229–253.
- P. Gotico, W. Leibl, Z. Halime and A. Aukauloo, ChemElectroChem, 2021, 8, 3472–3481 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- Y.-K. Zhang, L. Zhao, W.-J. Xie, H.-R. Li and L.-N. He, ChemSusChem, 2024, 17, e202400090 CrossRef CAS PubMed.
- Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS.
- K. Rickmeyer, L. Niederegger, M. Keilwerth and C. R. Hess, ACS Catal., 2022, 12, 3046–3057 CrossRef CAS.
- K. Rickmeyer, M. Huber and C. R. Hess, Chem. Commun., 2024, 60, 819–822 CAS.
- R. Lauenstein, S. L. Mader, H. Derondeau, O. Z. Esezobor, M. Block, A. J. Römer, C. Jandl, E. Riedle, V. R. I. Kaila, J. Hauer, E. Thyrhaug and C. R. Hess, Chem. Sci., 2021, 12, 7521–7532 CAS.
- P. Banerjee, A. Company, T. Weyhermüller, E. Bill and C. R. Hess, Inorg. Chem., 2009, 48, 2944–2955 CAS.
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 Search PubMed.
- K. S. Kjær, et al. , PCCP, 2018, 20, 4238–4249 Search PubMed.
- B. C. Paulus, K. C. Nielsen, C. R. Tichnell, M. C. Carey and J. K. McCusker, J. Am. Chem. Soc., 2021, 143, 8086–8098 CAS.
- A. Kumar, P. Malevich, L. Mewes, S. Wu, J. P. Barham and J. Hauer, J. Chem. Phys., 2023, 158, 144201 CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 1657, 2004, 82–104 Search PubMed.
- J. Wellauer, F. Ziereisen, N. Sinha, A. Prescimone, A. Velić, F. Meyer and O. S. Wenger, J. Am. Chem. Soc., 2024, 146, 11299–11318 CAS.
- Y. Ye, P. Garrido-Barros, J. Wellauer, C. M. Cruz, R. Lescouëzec, O. S. Wenger, J. M. Herrera and J.-R. Jiménez, J. Am. Chem. Soc., 2024, 146, 954–960 CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CAS.
- O. Z. Esezobor, W. Zeng, L. Niederegger, M. Grübel and C. R. Hess, J. Am. Chem. Soc., 2022, 144, 2994–3004 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2391634. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06104g |
| ‡ Representative photoreduction data (Fig. 2) and QY values are shown for 1. Slight variation in the quantitative, but not qualitative, rates is observed depending on batches of 1 (see Section S7, Fig. S15, ESI†). |
| This journal is © The Royal Society of Chemistry 2025 |