
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sensitization experiments of ultrasmall gold nanoclusters: determination of triplet quantum yields and molar absorption coefficients†
Hayato
Sakai
 a,
Sunao
Hiramatsu
a,
Aoi
Akiyama
b,
Yuichi
Negishi
*bc and
Taku
Hasobe
*a
a,
Sunao
Hiramatsu
a,
Aoi
Akiyama
b,
Yuichi
Negishi
*bc and
Taku
Hasobe
*a
aDepartment of Chemistry, Faculty of Science and Technology, Keio University, Yokohama 223-8522, Japan. E-mail: hasobe@chem.keio.ac.jp
bDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
cInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: yuichi.negishi.a8@tohoku.ac.jp
First published on 6th December 2024
Abstract
We demonstrated for the first time the determination of triplet quantum yields and molar absorption coefficients of ultrasmall gold nanoclusters specifically [Au25(PET)18]− with phenylethanethiolate (PET) ligands using two sensitization experiments.
Recently, metal nanoclusters have attracted considerable interest in various basic and applied research fields owing to their size-dependent structures and properties derived from their discrete electronic structures.1–12 In particular, ligand-protected ultrasmall metal nanoclusters exhibit characteristic molecular behaviours associated with the energy gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO).13 For example, regarding [Au25(PET)18]− (denoted as Au25(PET)18) with phenylethanethiolate (PET) ligands, single-crystal X-ray diffraction demonstrated that Au25(PET)18 is composed of an icosahedral Au13 kernel and six Au2(PET)3 staple motifs on the surface.14,15 The molecular behaviour of these metal nanoclusters is attributed to singlet or triplet excited states,16 but the detailed excited-state dynamics associated with the molar absorption coefficients (εT) and quantum yields (ΦT) in the triplet states are yet to be determined. The singlet–triplet (S1–T1) energy gaps (ES–T) of ultrasmall gold nanoclusters such as Au25 clusters (e.g., E(S1) = ∼1.4 eV and E(T1) = ∼1.2 eV) have been reported to be much smaller than those of organic materials according to theoretical calculations and spectroscopic measurements.17,18 Thus, the values of E(S1) and E(T1) can be controlled simply and mainly by the number of metal atoms and types of ligands, making it much easier to establish a strategy for material design as compared to organic materials. Therefore, gold nanoclusters are useful for photo-functionalities based on absorption, emission,19–24 and electron/energy transfer in the visible (red) and NIR regions, and are expected to be explored in various fields such as energy conversion, catalysis, electronics, and biomedical applications.25–28
The photophysical behaviours associated with triplet excited states (T1) of ultrasmall gold nanoclusters (e.g., Aun: n = 25, 38) include phosphorescence emission through intersystem crossing (ISC),19,20 reverse intersystem crossing (RISC)29 and singlet oxygen (1O2) generation in oxygen-containing solutions.30 Additionally, the bimolecular rate constant (∼106 M−1 s−1) is extremely small, although the intermolecular triplet-triplet energy transfer (T-TEnT) from Aun to organic chromophores was observed.31 Time-resolved spectroscopy, such as transient absorption spectra (TAS) and emission lifetimes, suggested that TAS of Au25 from the longest lifetime component and photoluminescence decay at ∼1100 nm involve the excited state derived from the same T1.16,32–34 These lifetimes of Au25 (ca. 50–200 ns) are much shorter than those of the typical T1 lifetimes of organic chromophores (∼microsecond region). However, no attention has been paid to the determination of the εT and ΦT of such gold nanoclusters. Although sensitization experiments using T-TEnT with an energy donor (a sensitizer) are useful for assigning the triplet TAS and corresponding εT values, the T1 lifetimes of Aun are extremely short. Accordingly, conventional methods are difficult to sufficiently evaluate.
Here we demonstrate two different sensitization experiments to evaluate the triplet character of [Au25(PET)18]− (denoted as  ) for the determination of the εT and ΦT. First, the diffusion-controlled intermolecular T-TEnT from a sensitizer (C60) to Au25(PET)18 (∼1010 M−1 s−1) was observed in toluene. Then, the diffusion-controlled quenching trend of
) for the determination of the εT and ΦT. First, the diffusion-controlled intermolecular T-TEnT from a sensitizer (C60) to Au25(PET)18 (∼1010 M−1 s−1) was observed in toluene. Then, the diffusion-controlled quenching trend of  in O2-saturated toluene (∼1010 M−1 s−1) demonstrated the intermolecular T-TEnT from Au25(PET)18 (energy donor) to 3O2 (energy acceptor), yielding 1O2 generation with the triplet yield (ΦT: 24%). Based on these results, the εT of
in O2-saturated toluene (∼1010 M−1 s−1) demonstrated the intermolecular T-TEnT from Au25(PET)18 (energy donor) to 3O2 (energy acceptor), yielding 1O2 generation with the triplet yield (ΦT: 24%). Based on these results, the εT of 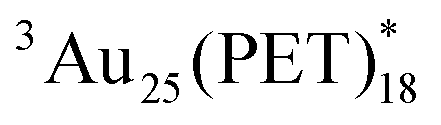 were successfully assigned. Additionally, the triplet behaviour of Au38(PET)24 was compared with Au25(PET)18.
were successfully assigned. Additionally, the triplet behaviour of Au38(PET)24 was compared with Au25(PET)18.
The syntheses and characterization of Au25(PET)18 and Au38(PET)24 were performed according to the reported method (Schemes S1 and S2 and Fig. S1 and S2 and Table S1 in ESI†).35,36 To determine the singlet and triplet energies (E(S1) and E(T1)), we also measured luminescence spectra of Au25(PET)18 at low temperatures following the reported methods20 (Fig. S2 and S3 in ESI†). Consequently, E(S1) and E(T1) of Au25(PET)18 were determined to be 1.5 eV and 1.2 eV, respectively. These values are comparable to the above-mentioned theoretical and experimental values of the related Au25.18,20 Additionally, the E(S1) and E(T1) of Au38(PET)24 should be slightly smaller than those of Au25(PET)18 according to previous report.18
According to previous reports,16,37 the ISC pathways of Au25(PET)18 and Au38(PET)24 are expected to be extremely fast. To examine the singlet and triplet characters, femtosecond transient absorption spectra (fs-TAS) were systematically measured in toluene (Fig. 1 and Fig. S4–S10 in ESI†). Although the origin of the absorption properties is generally dependent on the excitation wavelengths, we mainly chose 650 nm because there was no significant difference in the triplet behaviours upon either 350 or 650 nm excitation (Fig. S4–S6, ESI†). In the case of Au25(PET)18, the ultrafast relaxation occurred within ca. 4.0 ps after laser-pulse excitation (Fig. 1A and B), and long-lived excited species, extending from the visible to near-infrared (NIR) region (ca. 400–900 nm), remained within the measurement time-range of fs-TAS up to ∼6.0 ns. Assuming that this process is associated with ISC, the corresponding species-associated spectra (SAS) of Au25(PET)18 were analysed by target analysis (see: Fig. S11 and S12 in ESI†),38 yielding the SAS of S1 and T1 (Fig. 1C). The time-dependent concentration profiles (inset figure in Fig. 1C) demonstrate the ultrafast deactivation process. Note that the behaviour of the initial species is beyond the time-resolution of fs-TAS. The first species (trace a) and second species (trace b) should be attributed to S1 and T1, respectively. The longer and second species continue to be undecayed in the above-mentioned time-range (∼6 ns) (Fig. S4 in ESI†). Regarding Au38(PET)24, a similar relaxation process occurs within ca. 1 ps, whereas second and longer species decayed with the lifetime of ∼6 ns. This is in sharp contrast with Au25(PET)18. Noted that the singlet character derived from RISC process from T1 to S1 could not be obtained in Au25(PET)18 and Au38(PET)24 (see: the temperature-dependent fs-TAS in Fig. S13 in ESI†).
 | ||
| Fig. 1 (A) fs-TAS of Au25(PET)18 in toluene (λex: 650 nm) and (B) the time profile at 500 nm (298 K). (C) Species-associated spectra (SAS) of (a) first (black) and (b) second (red) species. Inset: The time-dependent concentration profiles of first and second species. | ||
To further observe the longer timescale in Au25(PET)18, we also measured picosecond transient absorption spectra (ps-TAS) of Au25(PET)18 (Fig. S14 and S15 in ESI†). The resulting excited species of Au25(PET)18 remained and the lifetime was calculated to be 160 ns at 298 K. No excitation wavelength-dependent spectral changes of S1 were observed for both Au25(PET)18 and Au38(PET)24 (Fig. S14–S17 in ESI†). From these results, triplet lifetimes of 160 ns for Au25(PET)18 and 5.0 ns for Au38(PET)24 were predicted, which are much shorter than those of typical organic molecules.
Thus, the above-mentioned fs-TAS allowed us to predict the triplet TAS of Aun (n = 25, 38), although it is necessary to confirm whether these predictions are essentially correct or not. Therefore, we first observed triplet TAS of Au25(PET)18 using T-TEnT with a sensitizer (C60). Fig. 2A presents ps-TAS in a mixed toluene solution of C60 and Au25(PET)18. The excitation wavelength chosen was 355 nm to mainly excite C60. The molar ratio between C60 and Au25(PET)18 was also optimized after preliminary experiments in which the ps-TAS were measured by varying the ratios. After laser-pulse excitation, we initially observed the triplet spectrum of C60 at ca. 750 nm. Note that 355 nm excitation includes a small amount of absorption in Au25(PET)18, resulting in the appearance of  due to the direct excitation of Au25(PET)18 in addition to
due to the direct excitation of Au25(PET)18 in addition to  produced by T-TEnT from C60 (Fig. 2A and Fig. S2 in ESI†). Another serious problem arises from the fact that the lifetime of
produced by T-TEnT from C60 (Fig. 2A and Fig. S2 in ESI†). Another serious problem arises from the fact that the lifetime of 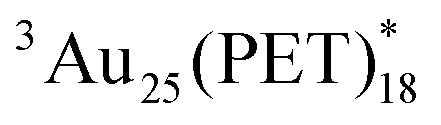 is much shorter than that of
is much shorter than that of  . Therefore, it is difficult to distinguish the triplet absorption spectrum of Au25(PET)18 by the direct excitation from that by intermolecular T-TEnT from C60. Although the εT cannot be determined by this method, the strong quenching process of
. Therefore, it is difficult to distinguish the triplet absorption spectrum of Au25(PET)18 by the direct excitation from that by intermolecular T-TEnT from C60. Although the εT cannot be determined by this method, the strong quenching process of  was clearly observed at 750 nm (Fig. 2B). The time profile at 750 nm showed the significant quenching of
was clearly observed at 750 nm (Fig. 2B). The time profile at 750 nm showed the significant quenching of  with increasing the concentrations of Au25(PET)18. Fig. 2C also shows a linear plot of the observed decay rate constants (kobs) at 750 nm based on the concentrations of Au25(PET)18. From the slope of the linear plot, the second-order rate constant of intermolecular T-TEnT from C60 to Au25(PET)18 was calculated to be 1.0 × 1010 M−1 s−1. This is quite similar to the diffusion-limited value in toluene (kdiff: 1.1 × 1010 M−1 s−1).39 The similar photophysical trend of Au38(PET)24 was successfully observed (Fig. S18 in ESI†). These results clearly demonstrate the triplet characters of Au25(PET)18 and Au38(PET)24.
with increasing the concentrations of Au25(PET)18. Fig. 2C also shows a linear plot of the observed decay rate constants (kobs) at 750 nm based on the concentrations of Au25(PET)18. From the slope of the linear plot, the second-order rate constant of intermolecular T-TEnT from C60 to Au25(PET)18 was calculated to be 1.0 × 1010 M−1 s−1. This is quite similar to the diffusion-limited value in toluene (kdiff: 1.1 × 1010 M−1 s−1).39 The similar photophysical trend of Au38(PET)24 was successfully observed (Fig. S18 in ESI†). These results clearly demonstrate the triplet characters of Au25(PET)18 and Au38(PET)24.
 | ||
| Fig. 2 (A) ps-TAS of C60 (32 μM) in the presence of Au25(PET)18 (20 μM) in toluene (298 K). λex: 355 nm. (B) The corresponding time-profiles at 750 nm in the presence of different concentrations of Au25(PET)18. Inset: Pseudo-first order plot of kobs monitored at 750 nm versus concentrations of Au25(PET)18. | ||
Then, another way to reveal the triplet character of Aun involves a sensitization experiment in oxygen-saturated toluene because of the occurrence of intermolecular T-TEnT from Aun to the molecular oxygen (3O2). In addition to the 3O2- and Ar-saturated toluene solutions, air-saturated (21% 3O2)40 toluene solution was prepared according to the established method.41Fig. 3A shows ps-TAS of Au25(PET)18 in 3O2-saturated toluene. Comparing these triplet absorption spectra with ps-TAS of Au25(PET)18 in Ar-saturated toluene (Fig. S14 and S15 in ESI†), no change in shape was observed. Fig. 3B shows strong decays in 3O2-saturated and air-saturated toluenes compared to that in Ar-saturated toluene. These resulted in the occurrence of intermolecular T-TEnT because the triplet lifetimes in 3O2-saturated (τT: 47 ns) and air-saturated (τT: 120 ns) toluenes were much shorter than that in Ar-saturated toluene (τT: 160 ns). Since there is a linear relationship between kobs and 3O2 concentrations, the second-order rate constant can be calculated to be 1.0 × 1010 M−1 s−1. This is very close to the above-mentioned kdiff in toluene, which is in sharp contrast with the lack of quenching of 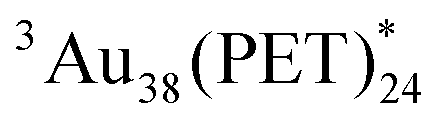 with 3O2 (Fig. S19 in ESI†). At the saturated oxygen concentration in toluene (∼10 mM), the decay rate constant (kd) of
with 3O2 (Fig. S19 in ESI†). At the saturated oxygen concentration in toluene (∼10 mM), the decay rate constant (kd) of  with 3O2 was determined to be 1.0 × 108 s−1. This is nearly two orders of magnitude greater than the rate constant of the triplet deactivation process of Au25(PET)18 (1/(160 ns) = 6.3 × 106 s−1), whereas the kd of
with 3O2 was determined to be 1.0 × 108 s−1. This is nearly two orders of magnitude greater than the rate constant of the triplet deactivation process of Au25(PET)18 (1/(160 ns) = 6.3 × 106 s−1), whereas the kd of  with 3O2 should be smaller than that of Au25(PET)18 (1/(5.0 ns) = 2.0 × 108 s−1). As a result, we can conclude that quantitative T-TEnT occurs between
with 3O2 should be smaller than that of Au25(PET)18 (1/(5.0 ns) = 2.0 × 108 s−1). As a result, we can conclude that quantitative T-TEnT occurs between  and 3O2.
and 3O2.
 | ||
| Fig. 3 (A) ps-TAS of Au25(PET)18 in O2-saturated toluene (λex: 355 nm) (298 K). (B) The time profiles at 600 nm in (a) Ar-saturated, (b) air-saturated (21% 3O2), (c) 3O2-saturated toluene. Inset: Pseudo-first order plot of kobs monitored at 600 nm vs.3O2 concentrations. | ||
The above-mentioned quantitative intermolecular T-TEnT from Au25(PET)18 to 3O2 resulted in 1O2 generation for evaluations of the ΦT. To examine the ΦT of Au25(PET)18via ISC, we employed 1O2 luminescence measurements using intermolecular T-TEnT from Au25(PET)18 to 3O2 in 3O2-saturated toluene at excitation wavelengths of 350 nm (Fig. 4A) and 650 nm (Fig. S20 in ESI†). By using intermolecular T-TEnT, we detected 1O2 luminescence at ca. 1270 nm, assuming negligible quenching processes from S1. The ΦT of Au25(PET)18 was calculated based on the reference compound: C60 (ΦT: 100%).42 Consequently, the ΦT of Au25(PET)18 (λex: 350 nm) was determined to be 24 ± 1.8%. Additionally, the ΦT of Au25(PET)18 (λex: 650 nm) is 24 ± 2.2%, and these values does not depend on the excitation wavelengths (Tables S2–S4 in ESI†). In contrast, in the case of Au38(PET)24, no T-TEnT occurred, as discussed above. Thus, no 1O2 peak at 1270 nm was observed in toluene (Fig. S21 in ESI†). The relatively small ΦT of Au25(PET)18 is likely attributable to the nonradiative process from S1 based on vibrational motion (vide infra). Based on the above ΦT and ps-TAS of Au25(PET)18, the εT were obtained (Fig. 4B, calculation process in ESI†) together with the kinetic parameters (Table 1).
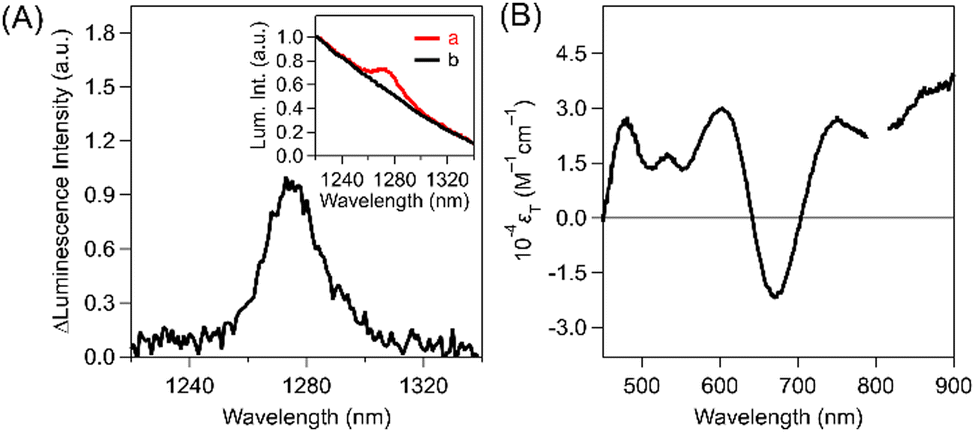 | ||
| Fig. 4 (A) A luminescence differential spectrum of Au25(PET)18 in 3O2-saturated toluene (298 K). The inset shows the parent luminescence spectra of Au25(PET)18 in (a) O2- and (b) Ar-saturated toluenes. λex: 350 nm. (B) The estimated εT values of Au25(PET)18 in toluene. | ||
To further examine the detailed dynamics of  , we also measured the temperature-dependent ps-TAS (Fig. S22 in ESI†). Although the T1 lifetimes increased with decreasing temperatures, the RISC process was negligible considering the single-exponential decay curves of
, we also measured the temperature-dependent ps-TAS (Fig. S22 in ESI†). Although the T1 lifetimes increased with decreasing temperatures, the RISC process was negligible considering the single-exponential decay curves of 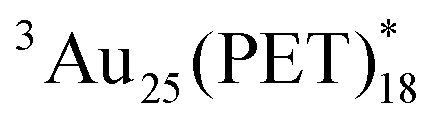 together with the above-mentioned S1–T1 gap of Au25(PET)18 (∼0.3 eV). This energy gap is much larger than those of the reported Aun with observations of RISC (∼0.03 eV).29 Hence, considering the above results of the temperature-dependent fs-TAS (Fig. S13 in ESI†) and ps-TAS, the origin of the changes in the excited lifetimes of the singlet and triplet states in temperature-dependent experiments is mainly due to the vibrational motions originating from Au13 kernel and/or six Au2(PET)3 staple motifs. This should be associated with the small ΦT.43 The proposed excited-state dynamics of Au25(PET)18 is also shown in Fig. S23 in in ESI.†
together with the above-mentioned S1–T1 gap of Au25(PET)18 (∼0.3 eV). This energy gap is much larger than those of the reported Aun with observations of RISC (∼0.03 eV).29 Hence, considering the above results of the temperature-dependent fs-TAS (Fig. S13 in ESI†) and ps-TAS, the origin of the changes in the excited lifetimes of the singlet and triplet states in temperature-dependent experiments is mainly due to the vibrational motions originating from Au13 kernel and/or six Au2(PET)3 staple motifs. This should be associated with the small ΦT.43 The proposed excited-state dynamics of Au25(PET)18 is also shown in Fig. S23 in in ESI.†
In conclusion, we demonstrated the triplet behavior of ultrasmall gold nanoclusters together with the determination of the ΦT and εT using sensitization experiments. The simple and easy control of excited states in metal nanoclusters will stimulate basic and applied researches on excited triplet states in the future.
This work was partially supported by JSPS KAKENHI Grant-in-Aid for Transformative Research Areas, “Materials Science of Meso-Hierarchy”(JP23H04876 to T. H.).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- R. W. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- M. A. Abbas, P. V. Kamat and J. H. Bang, ACS Energy Lett., 2018, 3, 840–854 CrossRef CAS.
- S. Antonello, N. V. Perera, M. Ruzzi, J. A. Gascón and F. Maran, J. Am. Chem. Soc., 2013, 135, 15585–15594 CrossRef CAS.
- S. H. Yau, O. Varnavski and T. Goodson, III, Acc. Chem. Res., 2013, 46, 1506–1516 CrossRef CAS.
- T. Saegusa, H. Sakai, H. Nagashima, Y. Kobori, N. V. Tkachenko and T. Hasobe, J. Am. Chem. Soc., 2019, 141, 14720–14727 CrossRef CAS PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS.
- Q. Yao, T. Chen, X. Yuan and J. Xie, Acc. Chem. Res., 2018, 51, 1338–1348 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- Z. Liu, M. Zhou, L. Luo, Y. Wang, E. Kahng and R. Jin, J. Am. Chem. Soc., 2023, 145, 19969–19981 CrossRef CAS PubMed.
- X.-S. Han, X. Luan, H.-F. Su, J.-J. Li, S.-F. Yuan, Z. Lei, Y. Pei and Q.-M. Wang, Angew. Chem., Int. Ed., 2020, 59, 2309–2312 CrossRef CAS PubMed.
- P. N. Day, R. Pachter, K. A. Nguyen and R. Jin, J. Phys. Chem. A, 2019, 123, 6472–6481 CrossRef CAS PubMed.
- M. Sugiuchi, J. Maeba, N. Okubo, M. Iwamura, K. Nozaki and K. Konishi, J. Am. Chem. Soc., 2017, 139, 17731–17734 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, O. J. H. Chai, N. Ding, W. Xu, S. Zang and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 9934–9939 CrossRef CAS PubMed.
- M. Zhou and Y. Song, J. Phys. Chem. Lett., 2021, 12, 1514–1519 CrossRef CAS PubMed.
- S. Sharma, K. Kaushik, A. Salam, R. Garg, J. Mondal, R. Lamba, M. Kaur and C. K. Nandi, ACS Appl. Nano Mater., 2024, 7, 32–60 CrossRef CAS.
- W.-Q. Shi, L. Zeng, R.-L. He, X.-S. Han, Z.-J. Guan, M. Zhou and Q.-M. Wang, Science, 2024, 383, 326–330 CrossRef CAS PubMed.
- K. L. D. M. Weerawardene and C. M. Aikens, J. Am. Chem. Soc., 2016, 138, 11202–11210 CrossRef CAS.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed.
- X. Wen, P. Yu, Y.-R. Toh, A.-C. Hsu, Y.-C. Lee and J. Tang, J. Phys. Chem. C, 2012, 116, 19032–19038 CrossRef CAS.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS.
- M. Mitsui, Y. Wada, R. Kishii, D. Arima and Y. Niihori, Nanoscale, 2022, 14, 7974–7979 RSC.
- M. Zhou and R. Jin, Annu. Rev. Phys. Chem., 2021, 72, 121–142 CrossRef CAS PubMed.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS.
- M. Zhou, C. Yao, M. Y. Sfeir, T. Higaki, Z. Wu and R. Jin, J. Phys. Chem. C, 2018, 122, 13435–13442 CrossRef CAS.
- X. Kang, H. Chong and M. Zhu, Nanoscale, 2018, 10, 10758–10834 RSC.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- T. D. Green and K. L. Knappenberger, Nanoscale, 2012, 4, 4111–4118 RSC.
- J. J. Snellenburg, S. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Software, 2012, 49, 1–22 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, 2010 Search PubMed.
- M. Motalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Boca Raton, 3rd edn, 2006 Search PubMed.
- S. Fukuzumi, H. Imahori, H. Yamada, M. E. El-Khouly, M. Fujitsuka, O. Ito and D. M. Guldi, J. Am. Chem. Soc., 2001, 123, 2571–2575 CrossRef CAS PubMed.
- D. K. Palit, A. V. Sapre, J. P. Mittal and C. N. R. Rao, Chem. Phys. Lett., 1992, 195, 1–6 CrossRef CAS.
- K. Kwak, V. D. Thanthirige, K. Pyo, D. Lee and G. Ramakrishna, J. Phys. Chem. Lett., 2017, 8, 4898–4905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, synthesis, spectroscopic measurements. See DOI: https://doi.org/10.1039/d4cc06083k |
| This journal is © The Royal Society of Chemistry 2025 |