
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective N1-ribosylation of hydantoin: synthesis and properties of the first contracted uridine analog†
Odai
Bsoul
,
Yakir
Lampel
 ,
Maayan
Rofe
,
Natalie
Pariente-Cohen
,
Chen
Timsit
and
Bilha
Fischer
*
,
Maayan
Rofe
,
Natalie
Pariente-Cohen
,
Chen
Timsit
and
Bilha
Fischer
*
Department of Chemistry, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: Bilha.fischer@biu.ac.il; Tel: +972-3-5318303
First published on 17th December 2024
Abstract
Modified nucleosides are vital in mRNA vaccines. We developed a contracted uridine analog, N1-hydantoinyl-ribose, HR, using steric shields to invert the regioselectivity of the classic Vorbrüggen reaction. We report synthetic routes and explore HR features such as acidity, stability, base pairing/stacking, and crystal/solution conformation compared to uridine.
Modified nucleos(t)ides have recently gained significant attention due to their crucial role in developing COVID-19 mRNA vaccines. The research by Kariko and Weissman on these modified nucleosides resulted in mRNA that is better suited for use in vaccines, ultimately contributing to the approval of the first mRNA vaccines.1,2 These vaccines utilize 1-methyl-pseudouridine, 1, containing mRNA, a seminal advancement recognized with the 2023 Nobel Prize in Physiology and Medicine.3 Over the past several years, multiple human clinical trials have explored therapeutic mRNA.4 To enhance the pharmacological properties, naturally modified nucleosides like pseudouridine, 2, and 1-methyl-pseudouridine are incorporated into therapeutic mRNA (Fig. 1A). The repertoire of nucleosides for fine-tuning mRNA properties primarily consists of naturally modified nucleosides.5 Currently, about 150 naturally modified nucleosides have been identified in RNA,6 including several metabolites of uridine, U, 3, e.g., 5-Me-U, 4, and 5-CH2OH-U, 5. Modified nucleosides play critical roles in cell regulation7 and increase mRNA translation levels8,9 by enhancing the metabolic stability of modified mRNA. Slow degradation of mRNA,10 is related to an additional H-bond donor at N1H of 2, paired with adenine (A),11 allowing water-coordinated H-bonding.12 In addition, in therapeutic mRNAs, U is often replaced by 1 or 2, to reduce immune response and cytotoxicity in cells.13 Importantly, modified U analogs have potential applications beyond improving therapeutic mRNAs. They could be useful in developing drugs based on ligands of uridine-nucleotide receptors, anticancer DNA polymerase inhibitors,1–11 and antiviral uridine nucleosides.14 Synthetic modifications of the uracil ring involve one of the following (Fig. 1B): (i) alkyl/aryl/functional groups are substituted at the uracil C5 or C6 positions (as shown in 3).15,16 (ii) Replacement of one of the atoms constituting the uracil ring by another atom, e.g., C6 carbon atom is replaced by a nitrogen atom,17 N1 nitrogen atom is replaced by a carbon atom,18 or O2/4 oxygen atoms are replaced by sulfur atoms.19 (iii) Variation of the size of the ring, e.g., by diazepine ring, a seven-membered ring analog of uracil.20 (iv) Transformation of the uracil ring to a chain – acyclic U mimetic – as in biuret riboside.21,22 These modifications alter the properties of uracil, including its volume, H-bonds, degree of aromaticity, acidity, and H-bonds. H-bonds are the dominant interaction of U – uracil N3H acts as H-bond donor and uracil O2 and O4 atoms as H-bond acceptors in 98%, 80% and 93% of the cases, respectively (Fig. S1A, ESI†).23 We concluded that O2, N3H, and O4 atoms are mandatory for recognition and should not be modified in synthetic U mimics. To identify analogs with greater potential for binding interactions with biomolecules than those of U, we recently characterized cyanuryl–ribose, barbituryl–ribose, and 6-azauridine (Fig. S1B, ESI†).24 Our findings reveal that while these analogs retain the base-pairing selectivity of U, their properties, such as ionization, solution conformation, and base-stacking, differ from those of U. The quest for new modified uridine analogs for multiple uses prompted us to develop and characterize novel U mimetics with unique features. Specifically, a contracted version of U, N1-hydantoinyl ribose, HR, has never been reported before, neither as a natural modification of U nor as a synthetic nucleoside. Yet, hydantoin derivatives have been reported as RNA oxidation products,25,26 or as enzyme inhibitors or receptor ligands.27,28
 | ||
| Fig. 1 (A) Naturally modified uridine analogs. (B) Types of synthetically modified uridine analogs containing: (i) C5/C6- Substituted uracil. (ii) Various other atoms replacing the original uracil ring atoms. (iii) Expanded uracil ring. (iv) Acyclic uridine. | ||
Motivated by the above potential applications of HR, we describe here several synthetic routes to HR, culminating in a regioselective and facile synthesis. We successfully reversed the natural N3 vs. N1 regioselectivity of hydantoin ribosylation by employing steric shields. In addition, we report features of HR which may impact the structure of HR-modified RNA, or the therapeutic activity of HR,27,28 such as acidity, hydrolytic stability, base pairing, base stacking, and solution conformation vs. those of U.
Synthesis of HR: despite its apparent simple structure, the synthesis of HR was found challenging. N1-regioselective ribosylation of hydantoin was difficult due to two nucleophilic nitrogen atoms in the hydantoin ring, N1 and N3 (Scheme 1). Additionally, the basic conditions required for the deprotection of the ribose at the end of the synthesis tend to degrade the hydantoinyl moiety in HR. Initially, we attempted a conventional coupling of hydantoin, 6, with protected ribose, 7, under Vorbrüggen ribosylation conditions.29 Specifically, hydantoin was persilylated with HMDS and TMSCl in acetonitrile, followed by adding 7 and TMSOTf at RT. Unfortunately, the only product obtained was the undesired protected N3-hydantoinyl ribose, 8, rather than protected N1-hydantoinyl ribose, 9 (Scheme 1, 1st approach). Although the N1 nitrogen atom was expected to be more reactive than the sterically hindered N3, the latter proved significantly more reactive. The absence of an interaction between H-5 and C-1′ in the HMBC spectrum (Fig. S2 and S3, ESI†) of the product indicated the formation of 8 rather than 9. The reason for this regiospecificity is related to the greater nucleophilicity of N3 (pKa ∼ 9.1) vs. N1.30 To boost up N1 nucleophilicity vs. N3, we investigated additional synthetic approaches: a 2nd approach, using parabanoyl ribose as a starting material; a 3rd approach, protecting N3; and last, a modification of the Vorbrüggen reaction (modification of 1st approach) by increasing steric hindrance around N3. The 2nd synthetic approach involved the conjugation of parabanoyl ribose, 10,22 with chloroacetyl chloride in a two-phase reaction.31 However, like the Vorbrüggen reaction, this high-yield reaction also formed the protected N3-ribosylated hydantoin, 8, indicating that the carbonyl groups were first cleaved from the parabanic ring (yielding 11) before alkylation with chloro-acetyl chloride occurred. If the reactions had occurred in the opposite sequence, the desired product, 9, would have been obtained. The formation of product 832,33 indicates that the acylation occurred at nitrogen N3 (N in red, 2nd approach), suggesting that this N–H is more acidic, hence the nucleophilic reaction preferentially takes place through the N3 atom.
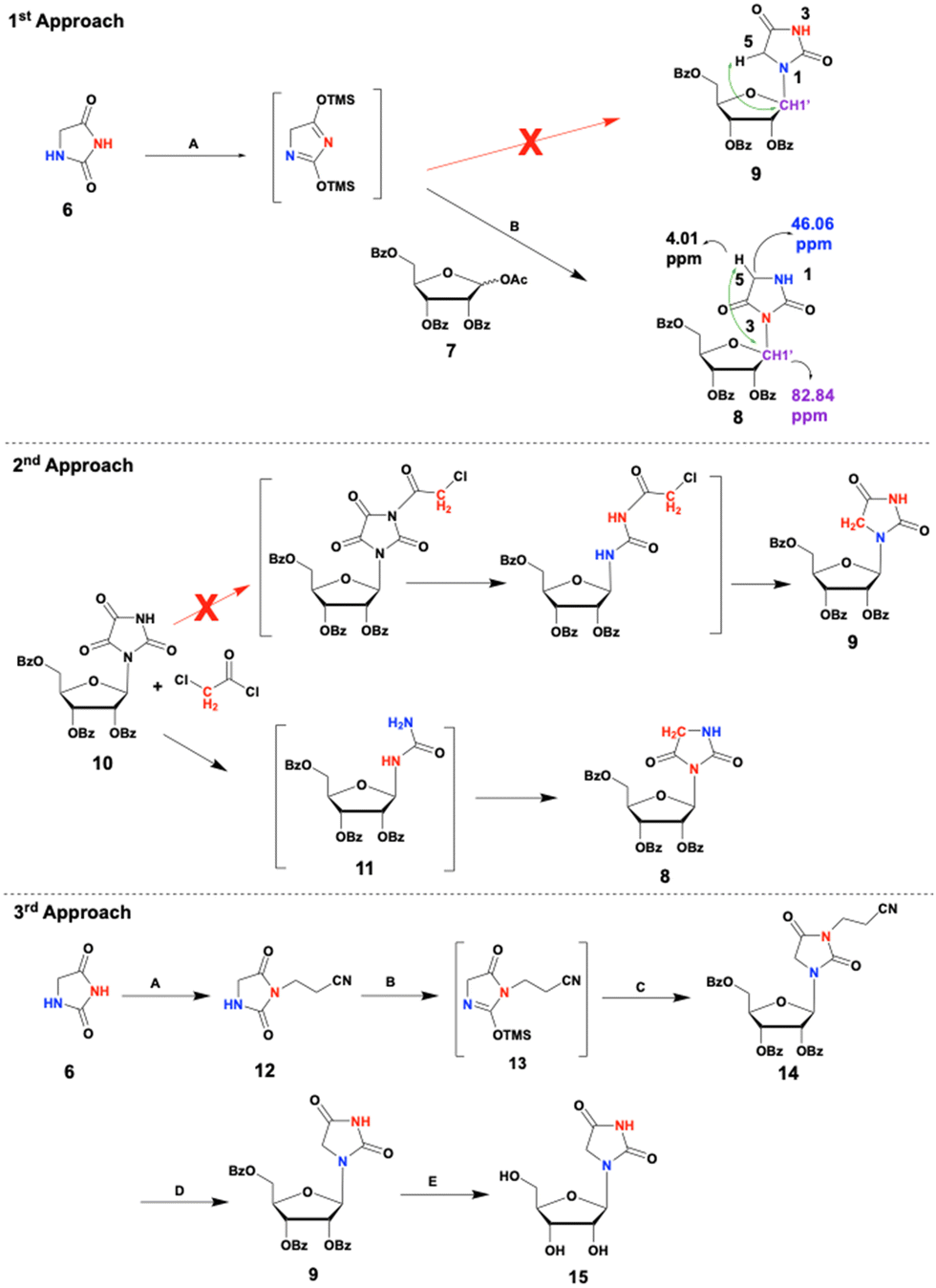 | ||
| Scheme 1 Reagents and conditions: 1st approach: (A) HMDS, TMSCl, MeCN, reflux, 45 min, Ar. (B) 7, TMSOTf, MeCN, overnight, RT, Ar. 2nd approach: 0.5 M NaOH, DCM, 30 min, RT. 3rd approach: (A) DIPEA, acrylonitrile in H2O, reflux, N2, overnight. (B) HMDS, TMSCl, MeCN, reflux, N2, 1 h. (C) 7, TMSOTf, MeCN, 0–25 °C, N2, overnight. (D) 20% tBuOK in THF, t-BuOH, 40 °C, 30 min, N2. (E) 7 N NH3/MeOH, 72 h, RT. | ||
The 3rd synthetic approach involved protecting hydantoin's more reactive N3 nitrogen atom before ribosylation under Vorbrüggen reaction conditions. Michael addition of hydantoin to acrylonitrile resulted in a regiospecific N3 protection, giving compound 12 in a good yield (Scheme 1, 3rd approach). Although a small amount of undesired N1, N3-di-substituted hydantoin byproduct was formed, using an excess of hydantoin effectively mitigated this issue. Under the Vorbrüggen ribosylation conditions, the N3-protected hydantoin 12 and protected ribose 7 successfully produced the desired protected N1-riboside 14 in a satisfactory 70% yield. However, removing the benzoyl and the propionitrile-protecting groups proved challenging. Hydantoins are susceptible to hydrolytic degradation under strong basic conditions.34 Seeking milder conditions, we treated compound 14 with methanolic ammonia. While this approach successfully removed the benzoyl groups, the propionitrile group remained intact. Further treatment with stronger bases was avoided due to the risk of hydantoin ring opening. Hence, we first removed the propionitrile group with a non-nucleophilic strong base that left the benzoyl groups unaffected. This was followed by deprotection of the benzoyl groups using methanolic ammonia. While attempts to remove the N3-propionitrile group with DBU or DBN in MeCN were unsuccessful, potassium-t-butoxide in an extra dry solution of t-butanol and THF yielded product 9, which, upon treatment with methanolic ammonia gave the desired nucleoside, HR, 15. Yet, this 5-steps synthesis of HR results in only 16% overall yield of 15. To further improve the yield of HR we implemented a new protection-free strategy to reverse the natural regioselectivity of the Vorbrüggen reaction. By bis-silylation of hydantoin with bulky silyl groups, we effectively shielded the N3 position, allowing for selective N1 nucleophilic attack (Scheme 2). We used bulky silyl reagents – TBDMSCl, TBDPSCl, and TIPSCl instead of HMDS/TMSCl, to form bis-silylated-hydantoin products, 16/17/18. Importantly, the latter exhibit significantly enhanced stability to moisture compared to the TMS counterpart, which facilitates their isolation. Reacting 2,4-bis-TBDMS-hydantoin, 16, with 7, resulted in the desired N1-product 9 with a 35% yield, vs. 51% yield of the N3-product, 8 (Fig. 2). To reverse N1 vs. N3 regioselectivity, we treated hydantoin with TBDPSCl or TIPSCl to obtain a more substantial steric shield around N3, resulting in compounds 17 and 18, respectively. Using 2,4-bis(TBDPS)-hydantoin, the desired N1-product 9 was obtained with a 51% yield vs. 44% yield for the N3 product. With 2,4-bis(TIPS)-hydantoin, a more significant reversal of regioselectivity was obtained (N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N3 ratio was 3.2:1; Fig. 2). The remaining silyl group was removed during the work-up of the reaction.
:N3 ratio was 3.2:1; Fig. 2). The remaining silyl group was removed during the work-up of the reaction.
 | ||
| Scheme 2 Synthesis and mechanism of HR formation using steric shields. Reagents and conditions: (A) tBDMSCl (for 16); or tBDPSCl (for 17); or TIPSCl (for 18); triethylamine, RT, Ar, 48 h. (B) 7, TMSOTf, MeCN, 0–25 °C, Ar, overnight. (C) 7N NH3/MeOH, 72 h, RT. | ||
 | ||
| Fig. 2 Shifting regioselectivity of ribosylation of hydantoin from N3 to N1 by silyl steric shields. | ||
Conformation of HR in crystal and solution states: hydantoinyl ribose, HR, was crystallized from water, forming colorless needles. X-ray crystal structure analysis indicated a ribose S-conformation facilitated by the small size of the hydantoin ring (Fig. 3A and Tables S1–S7, ESI†). The torsional angle around C4′, C5′-bond is 159°, placing the 5′-OH group above the ribose ring as a gg rotamer. Additionally, the hydantoinyl ring adopts the anti-position with a χ angle of 255°. In addition to its crystal structure, analysis of the solution conformation of HR is essential for estimating its binding potential to biomolecules and for designing new therapeutic agents based on the HR scaffold. Hence, we determined the solution conformation of HR (Fig. 3B), using 1H-, and 13C-, –1D and 2D NMR spectra (Fig. S4, ESI†).35,36

Ribose pucker: in solution, the ribose ring can adopt Northern, N, and Southern, S, conformations which exist in rapid equilibrium (Fig. S5A. ESI†). By analyzing the coupling constants (JHH, JCH) of HR (Table S8 and eqn (S1) and (S2), ESI†),36,37 we found that unlike U, which adopts S conformation in a 44% occurrence, HR exhibits a 67% preference for the S conformer (Table 1).24
| Compound | Sugar puckering %S | Rotamer population around C4′–C5′ bond | ||
|---|---|---|---|---|
| %gg | %tg | %gt | ||
| HR | 67 | 55 | 30 | 15 |
| U | 44 | 58 | 20 | 22 |
Conformation around the ribose –CH
2
OH exocyclic group: the position of the ribose exocyclic –CH2OH group is a mixture of staggered rotamers around the C4′–C5′ bond (Fig. S5B, ESI†). The percentage of gg, gt, and tg rotamer populations in HR were determined using the  and
and  coupling constants (calculated based on eqn (S2), ESI†).37 We found that the gg rotamer is the predominant one for both HR and U, with populations of 58% and 55%, respectively (Table 1).
coupling constants (calculated based on eqn (S2), ESI†).37 We found that the gg rotamer is the predominant one for both HR and U, with populations of 58% and 55%, respectively (Table 1).
Glycosidic angle: the glycosidic angle of HR was calculated using the 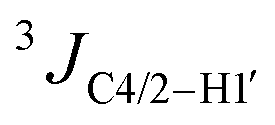 values and eqn (S3) (ESI†).38 Two solutions were obtained: 218° (anti) and 262° (high-anti). The latter value is consistent with the crystal structure of HR (Fig. 3B(ii)).
values and eqn (S3) (ESI†).38 Two solutions were obtained: 218° (anti) and 262° (high-anti). The latter value is consistent with the crystal structure of HR (Fig. 3B(ii)).
pH-dependent stability of HR and its acid–base equilibrium: pH-dependent stability of HR under pH 1.4 and 13 was monitored by 13C NMR at RT. After 1 week at pH 1.4, no decomposition was observed (Fig. S6A.1, ESI†). However, under pH 13, HR began degrading immediately (Fig. S6A.2, ESI†). Acid–base equilibrium is a key characteristic of nucleobases as it affects their charge, tautomeric structure, and mode of participation in H-bonds (Fig. S6.B, ESI†).39 This, in turn, determines how a nucleobase interacts with and within nucleic acids, thus dictating the structure and function of the nucleic acid. We determined the pKa value of HR through pH titration monitored by 13C NMR (Fig. S6C, ESI†). In the pH range of 9–12, a downfield shift of 2 ppm was observed in carbonyl C2. We determined a pKa value of 9.3, attributed to the hydantoin N3–H, identical to that of U.
Base pairing and base stacking of HR: H-bonding between nucleobases is essential for base-pairing, which in turn dictates the structure and functionality of nucleic acids. In addition to Watson–Crick base pairing, RNA structures accommodate various alternative base pairings, such as the GU wobble pair.39 We investigated the mode of base pairing between the contracted uridine, HR, and adenosine, A, or guanosine, G, using 1H NMR-monitored titration.
Specifically, 0.1 M HR in DMSO-d6 was added to 0.1 M A in DMSO-d6, and the N3H signal of HR, which may participate in H-bond with A, was inspected at 10.5 ppm (Fig. S7.A and S7.B, ESI†). HR: A ratio was gradually changed from 1:0.25 up to 1:2. During the titration we observed a significant widening of the N3H proton signal of HR, until the signal almost disappeared, implying the formation of an H-bond between HR and the exocyclic amine of A. The exocyclic amine of A alone was observed as a very broad signal (Fig. S7.B, ESI†). Unexpectedly, de-shielding the protons of the A's exocyclic amine was insignificant. This is presumably due to the averaging of both signals of the N6-amine protons. Furthermore, we tested the HR ability to selectively recognize A vs. G. For this purpose, we gradually added 0.1 M HR in DMSO-d6 to 0.1M G in DMSO-d6, observing no change in width of the N3H proton signal. The peaks obtained for HR:G = 4:1 and HR:G = 3:2 slightly shifted downfield. Those signals were the weighted average of both HR and G NH signals. The more G there is, the more downfield is the weighted average of the signal. π–π Interactions between pairs of nucleobases provide an additional driving force for shaping the structure of nucleic acids.39 Hence, we investigated the possibility of self-base stacking interactions of HR by NMR, comparing the chemical shifts at high concentrations (0.4 M, δ∞) with those in a diluted monomeric solution (0.003 M, δ0) in D2O. We monitored the chemical shifts of H5 and H1′ of the ribose proton in HR, as these shifts may indicate stacking interactions between the base moieties (Table S9, ESI†). While small concentration-dependent Δδ values due to inter-molecular interactions were observed for U,24 no changes were detected for HR, indicating the absence of base-stacking interactions in water.
Motivated by the need for novel modified U analogs to optimize mRNA vaccine properties and develop new drugs, we designed the synthesis of HR, 15, a contracted U analog, the first of its kind. Initially, ribosylation of hydantoin under the Vorbrüggen reaction conditions,29 exclusively yielded the undesired N3-hydantoinyl ribose. By introducing a steric shielding strategy around the N3 position, rather than relying on N3 protection, we successfully reversed the natural regioselectivity to obtain the desired N1-hydantoinyl ribose, thus, marking a significant milestone in the classic Vorbrüggen synthesis. This new approach can be broadly applied to the ribosylation of heterocycles having multiple nucleophilic nitrogen atoms, allowing the synthesis of various new nucleosides, as well as numerous heterocyclic compounds.
In addition, we characterized the unique properties of HR, which are a prerequisite for understanding its potential as an enzyme inhibitor,27 receptor–ligand,28 or its potential role in RNA structure and function. We found that HR mimics U in some respects yet, it possesses distinct structural and chemical features due to the compact nature of the hydantoin ring, making HR a promising steric probe for biochemical studies and investigating RNA properties. Further studies are necessary to understand how HR behaves when incorporated into RNA oligonucleotides.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- K. Karikó and D. Weissman, Curr. Opin. Drug Discovery Dev., 2007, 10, 523–532 Search PubMed.
- Y. N. Lamb, Drugs, 2021, 81, 495–501 CrossRef.
- The Nobel Prize in Physiology or Medicine 2023, https://www.nobelprize.org/prizes/medicine/2023/prize-announcement/ (Accessed August 20, 2024).
- N. Pardi, et al. , Nat. Rev. Drug Discovery, 2018, 17(4), 261–279 CrossRef PubMed.
- U. Sahin, et al. , Nat. Rev. Drug Discovery, 2014, 13(10), 759–780 CrossRef PubMed.
- M. Schaefer, et al. , Open Biol., 2017, 7(5), 2046–2441 Search PubMed.
- H. Shi, et al. , Molecular, 2020, 19(1), 1–17 CrossRef.
- B. Li, et al. , Bioconjug. Chem., 2016, 27, 849–853 CrossRef.
- Y. T. Yu and U. T. Meier, RNA Biol., 2014, 11(12), 1483–1494 CrossRef.
- D. R. Davis, Nucleic Acids Res., 1995, 23(24), 5020–5026 CrossRef.
- H. Adachi, et al. , Biochim. Biophys. Acta, Gene Regul. Mech., 2019, 1862(3), 230–239 CrossRef.
- M. I. Newby and N. L. Greenbaum, PNAS, 2002, 99, 12697–12702 CrossRef.
- C. J. C. Parr, et al. , Nucleic Acids Res., 2020, 48(6), e35–e35 CrossRef.
- E. De Clercq, J. Med. Chem., 2016, 59(6), 2301–2311 CrossRef.
- R. Saladino, et al. , J. Med. Chem., 2001, 44(26), 4554–4562 CrossRef PubMed.
- K. A. Jacobson, et al. , Biochem. Pharmacol., 2006, 71(4), 540–549 CrossRef PubMed.
- G. P. Connolly and J. A. Duley, Trends Pharmacol. Sci., 1999, 20(5), 218–225 CrossRef PubMed.
- E. M. Mukhin, et al. , Russ. Chem. Bull., 2023, 72(2), 425–481 CrossRef PubMed.
- R. K. Kumar and D. R. Davis, Nucleic Acids Res., 1997, 25(6), 1272–1280 CrossRef PubMed.
- T. Kunieda and B. Witkop, J. Am. Chem. Soc., 2002, 91(27), 7751–7752 CrossRef PubMed.
- H. Okamura, et al. , Angew. Chem., 2019, 131(51), 18864–18869 CrossRef.
- M. Varaggoulis, et al. , Collect. Symp. Ser., 2005, 7, 253–257 Search PubMed.
- T. Ginsburg-Shmuel, et al. , J. Med. Chem., 2010, 53(4), 1673–1685 CrossRef PubMed.
- H. Salameh, et al. , ACS Omega, 2020, 5(48), 31314–31322 CrossRef PubMed.
- A. Tomaszewska-Antczak, et al. , Chem. – Eur. J., 2015, 21, 6381–6385 CrossRef PubMed.
- Y. Ye, et al. , J. Am. Chem. Soc., 2003, 125, 13926–13927 CrossRef PubMed.
- S. H. Cho, et al. , Eur. J. Med. Chem., 2019, 164, 517–545 CrossRef PubMed.
- L. Ma, et al. , Molecules, 2022, 27(18), 5867 CrossRef CAS PubMed.
- C. R.-P. Helmut Vorbrüggen, The Handbook of Nucleoside, John Wiley, American Chemical Society, NY, 2002, vol. 45 Search PubMed.
- G. P. Connolly, Uridine and Pyrimidine Nucleotides in Cell Function, Springer, Berlin, 2001, vol. 151, pp. 403–421 Search PubMed.
- S. Verma, et al. , Indo. Global J. Pharm. Sci., 2013, 3(1), 33–39 CrossRef CAS.
- A. J. Jones, et al. , Proc. Natl. Acad. Sci. U. S. A., 1970, 65(1), 27–30 CrossRef CAS.
- K. Hammerbacher, et al. , Chem. Biodiversity, 2018, 15(9), e1800173 CrossRef.
- M. Meusel and M. Gütschow, Org. Prep. Proced. Int., 2004, 36(5), 391–443 CrossRef CAS.
- N. Stern, et al. , Org. Biomol. Chem., 2010, 8(20), 4637–4652 RSC.
- M. Sundaralingam, et al. , J. Am. Chem. Soc., 1972, 94(23), 8205–8212 CrossRef PubMed.
- J. Donohue and K. N. Trueblood, J. Mol. Biol., 1960, 2(6), 363–371 CrossRef CAS PubMed.
- J. H. Ippel, et al. , NMR Chem., 1996, 34, 156–176 Search PubMed.
- Nucleic Acids in Chemistry and Biology, ed., G. M. Blackburn, M. Egli, M. J. Gait and J. K. Watts, The Royal Society of Chemistry, UK, 4th edn, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2383868. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06033d |
| This journal is © The Royal Society of Chemistry 2025 |