
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organic molecular design for high-power density sodium-ion batteries
Ying
Qi
,
Huaping
Zhao
and
Yong
Lei
 *
*
Fachgebiet Angewandte Nanophysik, Institut für Physik & IMN MacroNano, Technische Universität Ilmenau, Ilmenau 98693, Germany. E-mail: yong.lei@tu-ilmenau.de
First published on 13th January 2025
Abstract
Organic materials, with abundant resources, low cost, high flexibility, tunable structures, lightweight nature, and wide operating temperature range, are regarded as promising candidates for sodium-ion batteries (SIBs). Unfortunately, their poor electronic and ionic conductivity remain significant challenges, hindering the achievement of high power density for sodium storage. Power density, a critical factor in battery performance evaluation, is essential for assessing fast charging capabilities. Therefore, it is essential to summarize strategies for high-power density SIBs in further development. To address these limitations and guide future development, this highlight summarizes key advancements in SIB research over the past decade. We outline the effective molecular design strategies for improving high-power-density sodium storage, with a focus on structural optimizations ranging from the backbone to the side chains. Additionally, we propose future perspectives on electrodes, electrolytes, and potential applications to enhance the power density of organic sodium-ion batteries. This review is intended to give a comprehensive guideline on the future design of organic materials for fast-charge ability and overall performance.
 Ying Qi | Ying Qi received her MS degree in Chemical Engineering from Jiangsu University in 2020. She is currently a PhD candidate under the supervision of Prof. Yong Lei at the Technical University of Ilmenau in Germany. Her research focuses on the design and functionalization of organic materials for energy storage and conversion. |
 Huaping Zhao | Huaping Zhao obtained his PhD in materials science from the State Key Laboratory of Crystal Materials at the Shandong University in 2007. Following two years of postdoctoral research at the Institute of Chemistry (Chinese Academy of Sciences, 2007–2009), he was employed as a scientist by the University of Muenster from 2009 to 2011. Since 2012, he has been a senior scientist (permanent) in Professor Yong Lei's group at the Technical University of Ilmenau, Germany. His current research focus is the design and fabrication of functional nanostructures for energy storage and conversion. |
 Yong Lei | Yong Lei is Professor and Head of Department of Applied Nano-Physics at the Technical University of Ilmenau. He worked at Karlsruhe Institute of Technology during 2003–2006 as an Alexander von Humboldt Fellow. From 2006 he worked at University of Muenster as a group leader and Junior Professor. In 2011 he joined TU Ilmenau as Professor. His research focuses include template nanostructures for energy conversion and storage devices. He has received a few prestigious research funding including two European Research Council Grants. He is editorial board member Advanced Energy Materials, Energy & Environmental Materials, InfoMat, Science China Materials and Carbon Energy. |
1. Introduction
The growing global energy demand and the urgent need for carbon reduction necessitate a shift towards green energy solutions.1 Electrochemical energy storage (EES) devices, particularly rechargeable batteries, have gained significant traction and are rapidly developed and applied in both industry and households mainly attributed to the stability and continuity of power delivery.2 Recently, sodium-ion batteries (SIBs) have gained much attention due to the abundant sodium resources, low cost, high safety, and good low-temperature performance. So far, numerous active materials have been investigated for SIBs.3 Generally, inorganic compounds such as metal sulfides provide high energy density and long cycling life for sodium storage.4 As the demands for SIBs are projected to soar, the consumption of these metal compounds seems to increase significantly, potentially leading to supply problems. The limited availability of metal resources leads to high production costs, and the use of metal compounds in battery systems contributes to severe global warming. These factors make large-scale applications neither sustainable nor environmentally benign. Therefore, alternatives have been explored for metal compounds and proceed sustainable battery chemistry.Organic materials have emerged as potential active materials for SIBs, owing to their inherent physical and chemical characteristics and unique electrochemical properties.5 First, abundant raw material sources. Organic materials are mainly composed of non-metal elements such as carbon (C), hydrogen (H), oxygen (O), and nitrogen (N), which are extracted from sustainable and low-cost natural resources with minimal energy consumption.5a Second, flexibility and processability. Organic materials possess a soft molecular skeleton freely rotated and vibrating carbon chains as well as weak intermolecular force (e.g., van der Waals forces, hydrogen bonds), endowing them with excellent flexibility and processability. They can be fabricated into films or other shapes, suitable for flexible electronic devices.6 Third, structural diversity and tunability. The highly adjustable organic molecular structures allow for the optimization of electrochemical performance through molecular design. For example, halogen functional groups can be introduced to adjust the voltage plateau or stabilize specific capacity.7 Fourth, lightweight. Organic materials usually possess low density, which can reduce the overall weight of batteries and increase the energy density of full cells.8 Furthermore, green synthesis. The synthesis process of organic materials is environmentally friendly, and they can easily degrade after disposal, imposing a smaller burden on the environment.5d Last but not least, wide working temperature range. Organic electrode materials exhibit good electrochemical performance across a broad temperature range (−70 to 150 °C), enabling SIBs to operate under extreme environmental conditions.9 So far, organic materials with redox-active parts, such as carbonyl compounds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), imines (CN), azo derivatives (NN), nitriles (NC), thiocarbonyl compounds (CS), disulfide compounds (S–S) and nitroxide radical compounds (N–O) have been investigated over the last decade.10
O), imines (CN), azo derivatives (NN), nitriles (NC), thiocarbonyl compounds (CS), disulfide compounds (S–S) and nitroxide radical compounds (N–O) have been investigated over the last decade.10
As we know, energy density, power density, lifetime, and safety are four crucial factors in the evaluation of energy storage systems.5e Among these, energy density and power density are fundamental as they directly influence the electrochemical performance of the battery, battery management system, and practical application conditions.11 However, most works focus on increasing energy density, and fewer of them pay attention to power density. For product users, high power density enables rapid charging and discharging, shortening charging times and improving user experience. It also meets the demands of applications requiring powerful instantaneous energy output, such as drones and power tools. For enterprise manufacturers, high-power density batteries facilitate thermal management due to the relatively less heat generation during high-rate charge and discharge processes. Besides, it also can simplify system design by reducing the number of parallel batteries required. These can help improve the safety and lifespan of the battery, decrease system complexity and cost, and improve the overall energy utilization efficiency of the system. Therefore, high power-density batteries are beneficial for the customers and industry.
However, there are two stumbling blocks to achieving high power density in organic sodium-ion batteries (OSIBs): slow electron and ion diffusion. Although some conductive polymers (polyaniline (PANI), polyacetylene (PA), and polypyrrole (PPy)) possess a high conductivity range from 1 to 10−4 S cm−1.12 It might be difficult for them to reach half of the theoretical capacity due to the irreversible reactions.13 Even though a high proportion of conductive additives (such as carbon black, conductive polymers, etc.) can be added to enhance the electrical conductivity of organic electrode materials, this method will reduce the proportion of active materials in the electrode, and thereby lowering the overall energy density and power density.14 Additionally, poor porous structures and lower specific surface areas of organic materials also lead to impeding ion transfer. Organic materials collapse even falling down caused by solubility further hinders electron and ion transport pathways during cycling, thus limiting to improvement of power density.15
It is found that molecular design is one of the most straightforward and efficient ways to construct molecular structure precisely and economically and achieve high-power density for OSIBs.16 Efficient strategies on molecular design for high-power density OSIBs have been proposed. On the one hand, improving poor electrical conductivity is the primary target to accelerate electron transfer. The functional groups' introduction and conjugation construction can change the electrical conductivity.17 It is proved that the introduced functional groups and conjugation can increase the internal electron polarization, and narrow the band gap for the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), thus improving the electrical conductivity.18 On the other hand, enhanced ionic conductivity is another way to improve power density for OSIBs. The construction of porous organic materials with high specific surface area benefits ionic transport. Intramolecular and intermolecular interactions help reorder organic molecules, shortening the diffusion pathways and facilitating ionic transfer on the interfacial surface and within active materials, thus promoting rate capability.19
Although significant progress on organic molecules has been proposed for SIBs according to molecule types, morphology engineering, and reaction mechanisms.20 Further research on the high-power density of organic molecules for sodium storage is required, as high-power density enables the fast charge and discharge process in a large current, allowing high efficiency for the sodium storage system. In this highlight, we give an overview of high-power density OSIBs with various strategies from the aspects of molecular design in recent decades. A comprehensive understanding of these strategies provides a foundation for the future application of high-power density OSIBs, paving the way for more efficient, sustainable, and environmentally friendly energy storage solutions.
2. High-power density
For a battery, power density is an assessment of the ability to deliver a large amount of power in a short period, thus enabling high-demand applications.21 The power density of battery systems can be evaluated through rate performance to demonstrate the ability of fast charging and discharging behaviors. The United States Advanced Battery Consortium (USABC) proposed that fast-charge batteries are capable to recharging to 80% of the energy capacity within 15 min.22 In practical application, vehicles like Tesla take half an hour to recharge up to 80% under a supercharging system.23 There is still a long way to go. Organic materials possessing flexible structures from intramolecular and intermolecular interactions allow fast charge and discharge at high C rates, However, achieving high power density often comes at the cost of energy efficiency, due to internal resistances within the battery.According to followed equations of Power output (P) (eqn (1)) and voltage (V) (eqn (2)),21,24
| P = I·V | (1) |
| V = (μC − μA)/nF − IR | (2) |
Among them, internal battery resistance is one of the main phenomena affecting power density, caused by unsatisfactory electron and ion transport through the organic electrolyte, the electrolyte and electrode interface, and the electrode material. As the charge or discharge rate increases, transport resistance grows bringing about the potential drops. Consequently, electrochemical polarization, which occurs due to the difference between cut-off voltage and the charge/discharge state of active material, results in irreversible capacity loss.
2.1 Two factors for high power density
The low intrinsic electrical conductivity and sluggish ionic diffusion commonly hinder the achievement of the high-power density of OSIBs. Therefore, accelerated electron and ion transfer are necessary to improve the fast charge/discharge capability.Some conducting polymers such as polyaniline show as good conductivity as metal.25 However, most pure organic molecules exhibit lower electron conductivities (<10−10 S cm−1).26 The poor electrical conductivities of organic materials decelerate the electron transfer and increase the internal electronic resistance, thus reducing capacity loss and limiting rate capability. The tunability of organic materials allows the introduction of functional groups and conductive polymer segments to increase electrical conductivity, and consequently, improve power density.27 Additionally, π-conjugated and π–d conjugated systems in organic materials can enhance intramolecular and intermolecular interactions, thus facilitating charge transfer.28
On the other hand, ionic conductivity plays a critical role in the electrochemical performance of OSIBs. According to Fick's law, the ion diffusion coefficient (D) follows the equation:29
| D = L2/τ | (3) |
Therefore, it is found that functionally altering the structure of organic materials through molecular design can significantly enhance electron and ion diffusion in OSIBs. However, most reports focus on the active storage sites and morphology of organic materials.31 There are no reviews that systematically summarize the strategies for high-power density sodium storage via molecular design. It is necessary to illustrate the methods of molecular design for high-power density sodium storage. In this review, we have summarized three strategies according to the influence of atom substitution on the backbone, backbone framework modulation, and side chain optimization, to enhance rate performance for sodium storage, as shown in Scheme 1. First, backbone atom substitution. The replacement of atoms such as nitrogen, fluorine, and sulfur can accommodate the electron density of redox-active centers, further affecting the electrical conductivity. Second, backbone framework modulation. The ordered organic frames can be constructed through π-conjugation, coordination, and covalent bonds with stabilized charge states and enriched space channels, which can facilitate ion/electron diffusion. Third, side chain optimization. The decoration of electron-withdrawing groups and electron-donating groups can influence the changeability of optimized organic materials, thus changing the ability of electron/ion affinity.
 | ||
| Scheme 1 The strategies are based on high-power density organic sodium-ion batteries. (The inserted crystal structures of Na2C6O6 are visualized by VESTA32). | ||
3. Molecular design for high-power density
The evaluation of high-power density performance depends on the rate capability. However, organic materials generally have intrinsically poor electrical conductivity, and amorphous molecular structure, which limit the availability of fast rate capability. The development of high-rate capability SIBs has always been accompanied by modulating molecular structure, improving electrical conductivity, and fastening redox reaction kinetics.33 Over the past decades, organic materials have been developed and applied to pursue high-power density SIBs.34 From subtle variations in side chains to radical differences in molecular frames, they will significantly change the rate capability of organic materials.35 Therefore, understanding the relationship between molecular structure and rate performance remains a challenge. In this section, we will summarize the molecular design strategies for high-power density sodium storage, starting with backbone atom substitution, followed by the construction of the organic material's main framework, and concluding with the optimization of side chains.3.1 Backbone atom substitution
Atom substituents have been reported to adjust the intrinsic electronic and ionic characteristics, because of the redistribution ability of charge, especially for improving those distortions on the surface, and redox reaction kinetics for sodium storage.36 From a solid-state perspective, atom substitution is well understood: n-type dopants contribute extra electrons to donor states near the conduction band, generating free electrons and altering the Fermi level closer to the conduction band by increasing electron density. Conversely, p-type dopant atoms create empty acceptor states, introducing free holes that modify the band structure.37 In organic materials, atom replacement in the backbone can modulate molecular structure, and change the HOMO, LUMO, and bandgap. In particular, the repetitive appearance of doped organic chains in polymers can form electron concentrations, further enhancing the electrical conductivity significantly. Those functional groups formed via n-type and p-type atom substitution can be divided into electron-withdrawing groups (EWGs) and electron-donating groups (EDGs) in organic materials. They can affect the valence electron state of Na, influencing the values of Na binding energy, and the rate of sodiation/desodiation reaction.In organic backbone construction, the introduced functional groups formed by atomic substitution are generally those that can store ions during the cycle, beneficial to improve the storage performance and enhance the conductivity of the organic framework at the same time. EDGs, such as carborane, applied to organic materials can enhance voltage and capacity with the increasing concentration of substituted atoms, which can create a higher potential for the cathode and an active potential for the anode.38 On the other hand, EWGs such as imide, are functional groups that tend to be the backbone of organic molecules for sodium storage.39 The electron-withdrawing redox-active-center groups absorb/desorb ions and transfer electrons during sodium storage, which can have a great influence on electron transfer ability. For example, sulfur-substituted functional groups in Fig. 1(a) were found to display a high conductivity of above 4 μS cm−1, after stepwise replacement of oxygen atoms with sulfur atoms in the carboxylate groups.40 This is mainly because the completely sulfur-substituted molecules contribute higher electron density, thus promoting electron delocalization and facilitating electron transfer. As a result, a fully sulfur-substituted molecule serves as a sodium reservoir and involves an eight-electron redox reaction, four electrons more than that of terephthalate sodium salt (PTA-Na). The sulfur-substituted cathode exhibited a high capacity of 567 mA h g−1 at 50 mA g−1 and maintained high rate performance with a large current rate. Besides, pyridine as an EDG was introduced into the Na2C14H8N2O2 anode.41 It is found that the nitrogen atoms can promote electrical conductivity and even increase electrochemical activation for SIBs. On the contrary, inactive electrochemically Na2C14H8O4 without N atoms forms an unstable sodiated product, showing poor electrochemical performance. It is likely that the backbone atom substitutions can not only improve electrical conductivity but also alter inactive molecules for reversible sodium storage.
 | ||
| Fig. 1 Atom substitution in the backbone of organic materials for sodium storage. (a) Stepwise sulfur doping sodium salt anodes for sodium storage. Adapted with permission from ref. 40. Copyright 2017, Wiley-VHC. (b) Electron-rich and electron-deficient centers in IISERP-COF16, IISERP-COF17, and IISERP-COF18; (c) rate performance of IISERP-COF16, IISERP-COF17, and IISERP-COF18 at 0.05–1.0 A g−1, and (d) IISERP-COF18 anode even under large current density of 15 A g−1. Adapted with permission from ref. 42. Copyright 2020, Royal Society of Chemistry. | ||
Moreover, it is found that the different substitution positions of atoms influence the electrical conductivity and particle size.43 Jia et al. found that nitrogen atoms at the meta-position on the phenylpyridine moiety showed the strong reaction kinetics of Na-CPP, exhibiting a high rate capacity of 159.8 mA h g−1 at 10 C (1870 mA g−1).44 Recently, it has been revealed that the combination of EWGs and EDGs in main chains can provide a driving force to push and attract electrons and ions during sodium storage, creating ion reservoirs for anodes. As shown in Fig. 1(b), the inclusion of nitrogen atoms in phenyl, tetrazine, and bispyridine-tetrazine segments of three organic frameworks gains electronic improvement.42 Especially in IISERP-COF18, the adjacent relatively electron-rich (pyridine ring) and electron-deficient (s-tetrazine ring) centers worked as a push–pull model, thus lowering the LUMO energy levels, and assisting smooth electron transfer. Such e−-accumulated LUMO levels drove Na+ into organic frameworks from the electrolyte. Therefore, the IISERP-COF18 anode delivered an excellent rate capability and maintained a capacity of 127 mA h g−1 at an extremely high current density of 15 A g−1 (Fig. 1(c) and (d)). These demonstrate that the redox-active-center atom substitution is one of the molecular design methods to promote electron transfer and enhance active redox sites simultaneously.
In conclusion, the rational molecular design of backbone atom substitution effectively regulates the electronic distribution, enhancing the electrical conductivity and fastening rate performance. Besides, the anchored position and dopant types in the molecular structure can further influence the electrochemical activity, reaction kinetics, and potential plateau of organic active materials. Therefore, atom substitution is an effective strategy to enhance electron transfer and redox activity in OSIBs.
3.2 Backbone framework modulation
The backbone framework controls the morphology of organic materials. Therefore, to realize the fast charge process, the optimization of diffusion channels, and conductive networks are the main targets in structure design, since electron and ion diffusion are the key points for high-power density sodium storage. For ion diffusion, the construction of large layer distance, large pore size, and small-sized structure morphology allow rich porosity and short ion pathways, thus promoting fast ion diffusion during cycling.45 For electron diffusion, conjugated molecular structure helps delocalize the charges to allow fast electron transport.46 It is found that the conjugation system, coordination interaction, and covalent interaction can help create porous structures to provide size-controlled pores, enriched channels, and dimensional structure, which can fasten the charge and ion transport and present high rate performance.For one thing, the larger conjugated system can delocalize the surrounding charges, thus stabilizing +1/−1 charge/discharge states. At the same time, delocalization can strengthen the intermolecular and intramolecular interactions to further facilitate charge transport.
For the other, the enhanced intermolecular interactions are expected to form the layer-by-layer structure, thus establishing diffusion pathways for rapid ion transport. Therefore, the extended conjugated system enhances the electrical conductivity of active materials and facilitates reaction kinetics for sodium storage.
Wang et al. first proposed the π-conjugated system in sodium storage.47 It is found that sodium 4,4′-stilbene-dicarboxylate (SSDC) shows extended π-conjugations, enhancing the intermolecular interaction, thus stacking as layer-by-layer mode in Fig. 2(a). This unique packing mode with a large spacing distance allows fast diffusion pathways for Na insertion/extraction behaviors. Fig. 2(b) and (c) exhibited a remarkable rate capacity of 100 mA h g−1 at 2 A g−1, and even maintained a high capacity at 10 A g−1 with an obvious plateau around 0.2 V. The extended π-conjugated system paves a way to achieve high-rate capability through molecular design. Especially, disodium rhodizonate (Na2C6O6) is the smallest conjugation structure with a high theoretical specific capacity of 957 mA h g−1 for six-electron storage.49 The hexagonal C6O62− is alternatively packed with Na+ to form a layered structure. This layered structure and conjugated effect are ideal for Na+ insertion/extraction.50 Besides, 2D (two dimensional) planar-structured 1,4,5,8,9,11-Hexaazatrip henylenehexacarbonitrile (HAT-CN) small molecular with cyano (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) groups exhibited a remarkable electrochemical performance, as shown in Fig. 2(d).51 The introduction of six redox-active electron-withdrawing CN groups and extended conjugation framework results in high theoretical capacity (837 mA h g−1) and better electron affinity. Additionally. As a result, the layered HAT-CN displayed a high rate capacity of 277.7 mA h g−1 at 10 A g−1 (Fig. 2(e)).
N) groups exhibited a remarkable electrochemical performance, as shown in Fig. 2(d).51 The introduction of six redox-active electron-withdrawing CN groups and extended conjugation framework results in high theoretical capacity (837 mA h g−1) and better electron affinity. Additionally. As a result, the layered HAT-CN displayed a high rate capacity of 277.7 mA h g−1 at 10 A g−1 (Fig. 2(e)).
 | ||
| Fig. 2 Backbone framework modulation via conjugated systems in organic molecules. (a) Schematic molecular packing of SSDC for SIBs; (b) rate performance of SSDC, SBDC, and super-P electrodes at different current densities. (c) Charge and discharge curves of rate capability of SSDC from 0.05, 0.1, 0.2, 1, 2, 5, and 10 A g−1. Adapted with permission from ref. 47. Copyright 2015, American Chemical Society. (d) Crystal structure of HAT-CN. (e) The rate capability of HAT-CN as electrodes in SIBs. Adapted with permission from ref. 51. Copyright 2024, Wiley-VCH. | ||
Later, Wang et al. found that conjugated effect also worked in polymers. The synthesized poly(pentacenetetrone sulfide) (PPTS) possessed a large and rigid π-conjugated system.52 It showed a long-range layer-by-layer π–π arrangement with a d spacing distance of ∼3.4 Å in Fig. 3(a) and (b). Such large layer-by-layer arrangement makes it easy to insert/extract Na ions, thus facilitating the ionic migration (10−9 cm2 s−1 for ionic conductivity). The PPTS electrodes showed a superior fast-charge–discharge capacity of 160 and 100 mA h g−1 at a big current density of 10 and 50 A g−1. The polymer chains enhance the π–π interactions, leading to high performance in SIBs.53
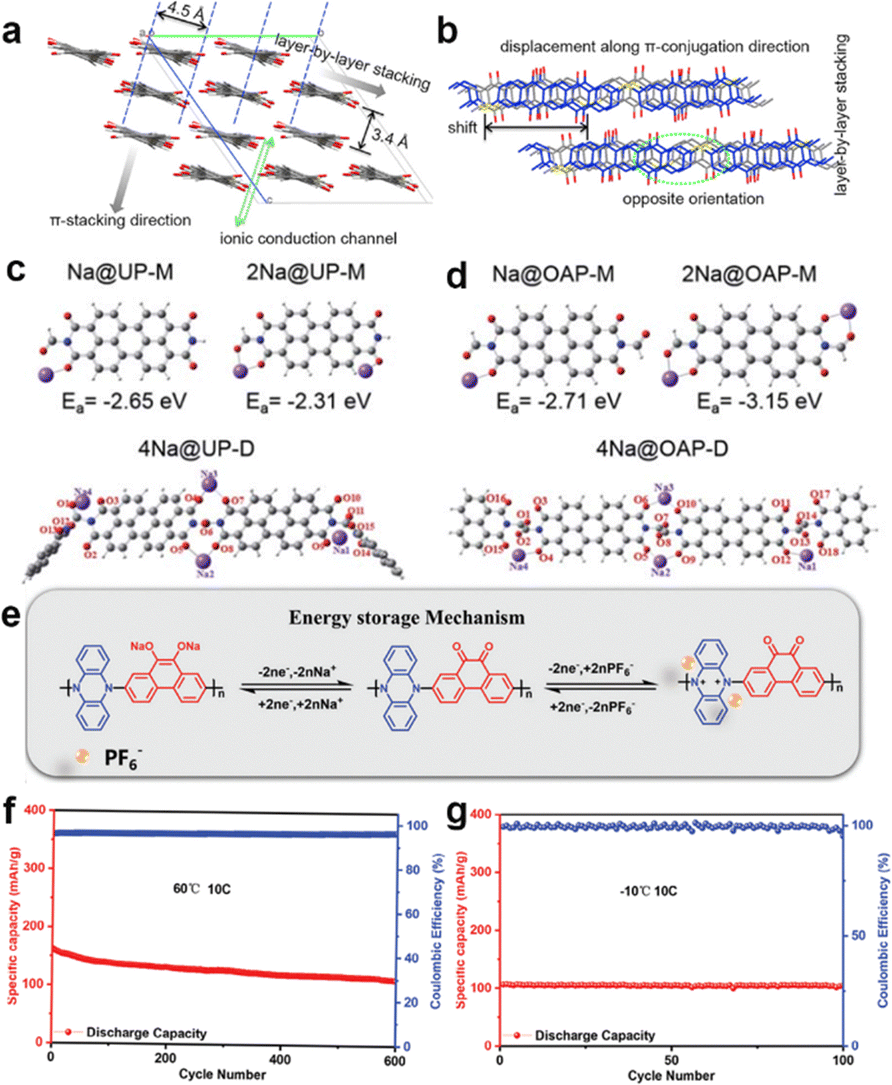 | ||
| Fig. 3 Backbone framework modulation via conjugated systems in organic polymers. Simulated molecular packing of PPTS along (a) the polymer chain and (b) π-conjugation direction. Adapted with permission from ref. 52. Copyright 2018, Elsevier. The possible structures of the simulated molecular structures of (c) UP and (d) OAP after attracting one or two Na ions (H, C, N, O, Na atoms are marked in white, grey, blue, red, and purple). Adapted with permission from ref. 54. Copyright 2020, Royal Society of Chemistry. (e) The reaction mechanism of PQPZ materials. The cycling performance of PQPZ materials at (f) 60 °C and (g) −10 °C at 10C (1C = 277 mA g−1) Adapted with permission from ref. 55. Copyright 2024, Wiley-VHC. | ||
Besides, the introduction of functional groups is capable of influencing the redox kinetics for sodium storage. It is demonstrated in Fig. 3(c) and (d) that electrochemical inactive carbonyl groups are decorated on perylenetetracarboxylic acid dianhydride (PTCDA) with oxamide (OAP) and perylenetetracarboxylic acid dianhydride (PTCDA) with urea (UP).54 The non-conjugated diketone in the linkages displayed stronger adsorption energy than that of UP, which can enhance reaction kinetics during cycling. While the nonplanar functional groups are anchored in the middle of the active center, they will break the electron transfer, thus causing the loss of the conjugated system and poor rate capability. Furthermore, the fabrication of donor–acceptor (D–A) conjugated organics is beneficial to tuning bandgap for charge transfer.56 The bipolar electrode design allows excellent cycling stability in an organic symmetric cell.57 Yao et al. synthesized 2,3,7,8-tetraamino-5,10-dihydrophenazine-1,4,6,9-tetraone (TDT), consisting of π electrons in quinone and p-electrons in piperazine, allowed electron delocalization throughout the entire molecular to highly extend conjugation system.29a Besides, the D–A bipolar organic electrode PQPZ can store Na+ and anions during the charge and discharge processes, respectively (Fig. 3(e)). These extended conjugation systems allow a narrower bandgap between HOMO and LUMO, delivering a remarkable rate of performance. Consequently, the PQPZ exhibited a satisfied electrochemical performance, and even stable cycling performances were displayed when tested at a wide range of working temperatures (Fig. 3(f) and (g)).55
In all, the extended conjugated system is beneficial for the +1/−1 charge state stabilization, and intermolecular and intramolecular force, thus strengthening layer-by-layer structure and promoting electron/ion transport. The conjugation effect can be strengthened through the following aspects: 1. coplanarity. It is revealed that the coplanarity of organic molecular structure can strengthen the conjugation effect for smooth electron transport. Besides, the polymerization of coplanar molecules can further enhance the coplanarity. 2. Functional group combination. The introduction of terminal functional groups, especially donor–acceptor groups can enhance electron delocalization, thus promoting the conjugation system. With enhanced conjugation, the molecular structures can be arranged in layers, thus promoting electron and ion transport.
In the last decades, a large number of MOFs have been reported for OSIBs.63 The stable crystal structure is one of the main essential factors for a faster charge and discharge process. Organic ligands are the pillars to support frames therein. Early in 2014, MnBpy MOF as an anode was first applied in SIBs.64 However, the bipyridine ligand was completely decomposed and the whole crystal structure collapsed after cycling. Studies find that multidentate bridging ligands can tailor polymer architectures.65 5-Aminoisophthalic acid, was used as the organic linker to form Co(L) MOF and Cd(L) MOF.66 After ball milling, these micro-sized MOFs possessed short ion diffusion pathways, and enriched interfacial contact areas, which promote rapid charge and discharge process. Recently, π-conjugated organic ligands such as tetraminobenzoquinone (TABQ) (Ni-TABQ),67 octahydroxyltetrabenzoanthracene (TBA) (Cu-TBA),68 and 2,3,6,7,10,11-hexaminotriphenylene (HITP) (Co-HITP-P)69 can create large conjugation to reduce LUMO energy level, narrow the energy gap, and strengthen in-plane electron delocalization, which could enhance the charge transfer during cycling. Combined with redox-active ligands and redox-active, the formation of porous MOFs can effectively improve ion passability.70 Additionally, coordinated metal ions are the mortise and tenon to strengthen the organic frames. Different coordinated metal ions will bring about diverse physical and chemical properties for electrochemical performance. Chen et al. found that the activity of coordinated metal centres affected the reaction kinetics.71 The small-sized Cu-HHTP with active Cu2+ involved only cation storage, showing higher electrical conductivity, and faster ion diffusion, compared with inactive Zn2+ in Zn-HHTP involving cation/anion storage (Fig. 4(a) and (b)). Thus, Cu-HHTP exhibited negligible activation process and better rate capability than that of Zn-HHTP. Both ligands and metal ions in Cu-HHTP underwent redox reactions accompanied only by Na+–cations insertion, leading to poor cyclability. Besides, when it comes to inactive coordinated metal centres, the radii of inactive coordinated metal ions matter. Dong et al. reveal that the smaller radii of coordinated metal ions show smaller steric hindrance effects towards sodium ions, which exhibited superior rate performance and fast ion diffusion kinetics.66
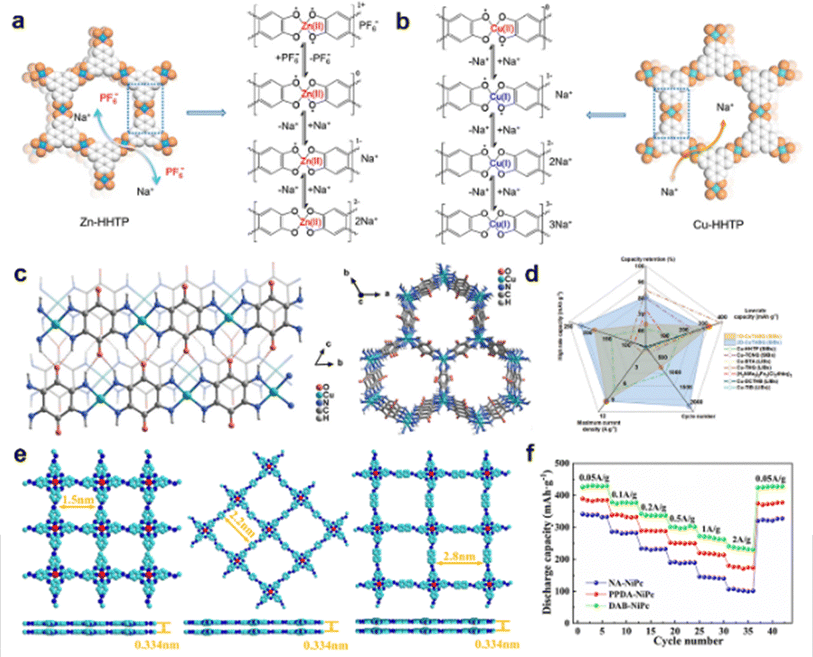 | ||
| Fig. 4 Backbone framework modulation via coordination interaction. Coordination interaction for illustration of the Na+ insertion/extraction in (a) ZnHHTP and (b) Cu-HHTP materials. Adapted with permission from ref. 71. Copyright 2021, Wiley-VHC. (c) The slipped π-stacking chain structure of 1D-CuTABQ and a two-dimensional layer of 2D-CuTABQ. (d) Comparisons of 1D-CuTABQ and 2D-CuTABQ with other works on electrochemical performance. Adapted with permission from ref. 72. Copyright 2023, Wiley-VHC. (e) Geometry optimization and (f) rate performance of NA-NiPc, PPDA-NiPc, and DAB-NiPc at 0.05–2 A g−1. Adapted with permission from ref. 73. Copyright 2021, Elsevier. | ||
The framework dimension of MOFs is one of the external factors to influence the porosity and diffusion paths. As shown in Fig. 4(c), chain-structure-like 1D-CuTABQ and layered 2D-CuTABQ are connected following variable valence states of metal ions (Cu2+ for 1D-CuTABQ and Cu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu+ of 1:1 for 2D-CuTABQ).72 Layered 2D-CuTABQ with honeycomb-like channels possessed rich porous structure, and larger specific surface area, facilitating the ionic diffusion during the charge/discharge process. 2D-CuTABQ displayed superior electrochemical performance than almost all the reported MOF cathodes at that time (Fig. 4(d)). Besides, the pore size of MOFs plays a vital role in the diffusion channels, considering the radium of Na ions during insertion/extraction. From the geometry structures in Fig. 4(e), NA-NiPc, PPDA-NiPc, and DAB-NiPc frameworks exhibited AA stacked multilayer network structures, with inner dimeters of pore sizes around 1.5, 2.2, and 2.8 nm, respectively.73 DAB-NiPc maintained better rate capability of 235 mA h g−1 at 2 A g−1 than NA-NiPc (176 mA h g−1) and PPDA-NiPc (103 mA h g−1) in Fig. 4(f). It mainly results from the large pore size that increases the number of ion transport and rate capability of sodium storage.
:Cu+ of 1:1 for 2D-CuTABQ).72 Layered 2D-CuTABQ with honeycomb-like channels possessed rich porous structure, and larger specific surface area, facilitating the ionic diffusion during the charge/discharge process. 2D-CuTABQ displayed superior electrochemical performance than almost all the reported MOF cathodes at that time (Fig. 4(d)). Besides, the pore size of MOFs plays a vital role in the diffusion channels, considering the radium of Na ions during insertion/extraction. From the geometry structures in Fig. 4(e), NA-NiPc, PPDA-NiPc, and DAB-NiPc frameworks exhibited AA stacked multilayer network structures, with inner dimeters of pore sizes around 1.5, 2.2, and 2.8 nm, respectively.73 DAB-NiPc maintained better rate capability of 235 mA h g−1 at 2 A g−1 than NA-NiPc (176 mA h g−1) and PPDA-NiPc (103 mA h g−1) in Fig. 4(f). It mainly results from the large pore size that increases the number of ion transport and rate capability of sodium storage.
In conclusion, MOFs possessing inherent advantages including porous structure, ordered ion pathways, and high specific surface area, have been extensively studied for high power density sodium storage. Their tuneable structures are capable of offering efficient diffusion channels for ion transport. It is found that the active redox-active metals and π-conjugated ligands of MOFs can improve structural integrity and enhance charge transfer. Besides, the dimensional structure and pore size of MOFs can change the diffusion channels, thus affecting the ion passability. However, poor conductivity, thermodynamic instability in electrolytes remain significant challenges for sodium storage. Additionally, the yield, complex synthesis process, and long synthesis times continue to limit their potential for large-scale applications.74 Developing strategies to overcome these limitations will be crucial for their broader use in energy storage systems.
O (carbonyl),75 CN (imine, triazine, phenazine),76 C–N (imide, amide),77 and NN (azo)78 are used to form covalent bonds in the synthesis of COFs. π conjugations with hydrogen bonding interactions dominated the noncovalent interactions to stack the framework structure of COFs.79 These COFs with periodic skeletons and ordered nanopores show high specific surface area, open porous structure, and highly conjugated systems. These ensure sufficient charge transfer and fast ion transport.
So far, two-dimensional (2D) and three-dimensional (3D) COFs have been studied.80 The porous architecture of 3D-COFs provides multiple diffusion channels and more active sites for ions.81 However, there are only a limited number of 3D COFs, due to the complex structure construction and restricted range of available elements.
Recently, 2D COFs have been studied for sodium storage since the first application of a 2D triazine-based COF cathode for SIBs.82 The 2D COFs possess a unique sheet-like structure and one-dimensional (1D) pore channels, which can shorten the ion diffusion pathway and fasten ion transport. For the repeated units in COFs, the functional groups of organic ligands will change the pore size and electronic conductivity, thus affecting the electrochemical performance. The nitrogen-rich COFs can reduce the energy gap between LUMO and HOMO, thus enhancing electronic and ionic conductivities.83 As shown in Fig. 5(a), 2D TQBQ-COF consists of carbonyls and pyrazine redox sites, delivering a high theoretical capacity of 515 mA h g−1.84 The TQBQ-COF layer (micropore of 11.4 Å) following a staggered stacking model, showed 5.6 Å pore size in TQBQ-COF (Fig. 5(b)). Due to the rich nitrogen atoms and conjugated structure, TQBQ-COF showed high electronic conductivity (∼10−9 S cm−1) and ionic conductivity (∼10−4 S cm−1). Consequently, The TQBQ-COF electrode kept a remarkable rate capacity of 134.3 mA h g−1 at a high current density of 10 A g−1 (Fig. 5(c)). To further adjust the bandgap between the HOMO and LUMO, fluorine was introduced to the covalent triazine framework (CTF). The calculation shown in Fig. 5(d), the fluorinated covalent triazine framework (FCTF) exhibited a smaller bandgap (1.45 eV) than that of CTF (2.35 eV).85 The smaller bandgap is beneficial to the fast electron/ion diffusion, bringing about improved rate capability. For COF structures, it is found that the extensive π-conjugation skeleton in COFs allows highly delocalized electrons, improving the conductivity and delivering a good rate performance.86 Besides, the stacking models in COFs turn the pore size. As shown in Fig. 5(e) and (f), fewer 2DP monolayers are superimposed over each other following the eclipsed AA and staggered AB arrangements.87 It is found that staggered AB few layer 2DP possess aligned open channels with an inner pore size of 0.6 nm, which allow rapid and smooth Na ion diffusion. The exfoliated 2DP sheets exhibited remarkably increased rate capacities of 262, 223, 196, 172, and 119 mA h g−1 at 0.1–5.0 A g−1 (Fig. 5(g) and (h)).
 | ||
| Fig. 5 Backbone framework modulation via covalent interaction. (a) The chemical structure and possible mechanism storage of TQBQ-COF. (b) Top and side views of AB stacking model of TQBQ-COF layers. (c) Rate performance of TQBQ-COF at 0.1, 0.3, 0.5, 1.0, 3.0, 5.0, 8.0, and 10.0 A g−1. Adapted with permission from ref. 84. Copyright 2020, Springer Nature. (d) The HOMO and LUMO energy levels and band gaps of CTF and FCTF COFs. Adapted with permission from ref. 85. Copyright 2019, American Chemical Society. Illustration for the (e) eclipsed AA and (f) staggered AB of 2DP. (g) Galvanostatic charge/discharge curves and (h) charge capacities of exfoliated 2DP at 0.1, 0.5, 1.0, 2.0, and 5.0 A g−1. Adapted with permission from ref. 87 Copyright 2018, Wiley-VHC. | ||
COFs for energy storage applications have been developed rapidly after COFs. Conjugated COFs show more active-redox centres, rich porosity, and multiple diffusion channels, resulting in fast electron and ion transport for SIBs. It is found that the heteroatoms doping and functional groups introduction can reduce the energy gap between LUMO and HOMO, improving electronic and ionic conductivities, accelerating electron and ion diffusion, and improving rate performance. Besides, the dimensional structure as well as pore size plays a vital role in ion transport. Therefore, the construction of COFs via covalent bonds is an efficient strategy of molecular design for high-power density.
3.3 Side chain optimization
Optimization with EWGs and EDGs groups on side chains can significantly alter the molecular coplanarity, electron distribution, as well as HOMO and LUMO energy levels, thus affecting the electrical conductivity and reaction kinetics of organic electrodes.88 Especially for polymerization optimization, it is capable of enhancing the concentration of functional groups, showing enhanced electrical conductivity. Therefore, it is an effective method to promote electron transfer during redox reactions.89EWGs are able to pull electron density away from the active-center sites. It is found that EWGs can lower the LUMO level and provide higher electron affinity. Early in 2012, Park et al. have reported that the addition of functional groups in benzene-carboxylate scaffolding can control the thermodynamic and kinetic properties of organic electrodes.90 The –SO3Na group possessing strong electron-withdrawing ability, has been optimized on organic electrodes.22 It is noticeable that the introduced –SO3Na group can act as a Na reservoir to provide rich Na+ during desodiation, thus promoting insertion–extraction reactions of Na ions.91 Similarly, fluorine (F) can effectively modify electronic properties and even adjust the band gap of the polymer.92 Sun et al. first introduced F into conductive PANI to form a fluoridized-polyaniline-H+/carbon nanotubes (FPHC) cathode via the covalent bond between –F and –NH.93 As shown in Fig. 6(a), the reduction of the HOMO–LUMO gap from 3.99 to 0.82 eV after PANI cooperated with PF6−, promoted the fast electron transfer between PANI and PF6−. Consequently, the FPHC possessed a high electronic conductivity of 0.162 S cm−1 (Fig. 6(b)). Therefore, the FPHC cathode exhibited an outstanding rate capability even under 3 A g−1, and delivered a high-power density of 7720 W kg−1 for sodium dual-ion batteries. However, Luo et al. found that the energy gap is not the only determining factor for electrochemical performance with the existence of –F groups.7 Although the energy gaps between HOMO and LUMO became smaller with the introduction of –F, –Cl, –Br halogen groups, respectively (Fig. 6(c)). The stable NaF-rich solid electrolyte interface (SEI) of the DFTP-Na anode could maintain a high rate capacity when the current density increased from 0.02 to 5 A g−1. Besides, fluorine assisted the interaction with Na+ when forming a Na–O bond with the carbonyl group in DFTP-Na. These results indicated that the –F introduction is better than that of –Cl and –Br.
 | ||
| Fig. 6 Optimization of functional groups on side chains for fast sodium storage. (a) The HOMO and LUMO energy gaps, and (b) electrical conductivities of PHC and FPHC. Adapted with permission from ref. 93. Copyright 2020, Wiley-VHC. (c) Energy levels of HOMO and LUMO for DTP-Na, DFTP-Na, DCTP-Na, and DBTP-Na, insides C, H, O, Na, Cl, Br, and F atoms are marked in grey, white, red, silver, pink, brown, and green, respectively. Adapted with permission from ref. 7. Copyright 2022, American Chemical Society. (d) The corresponding charge-density difference iso-surfaces of TP-T and TTHP-T binding with three sodium atoms, insides Na, C, N, O, and H atoms are purple, brown, blue, red, and white. (e) The rate performance of TP-T and TTHP-T at 0.05–8 A g−1. Adapted with permission from ref. 94. Copyright 2022, Elsevier. | ||
On the other hand, the EDGs such as alkyl groups (–R), amino (–NH2), hydroxyl (–OH), and alkoxy (–OCH3) are favorable to donate electrons to the reactive centre. They can provide electrochemical affinities for trapping Na ions, and facilitate insertion/deinsertion reactions for Na ions.95 As shown in Fig. 6(d), the existence of trihydroxyl groups in triazine-based anode (TTHP-T) is favorable to facilitating the trapping of Na ions between N and O atoms.94 The difference in charge densities of TP-T and TTHP-T anodes clearly showed that the presence of trihydroxyl-functional substitutions promotes the coupling between the benzene rings and the triazine centre, thus attracting electrons from Na. Besides, the trihydroxyl-functional substitutions can narrow the band gap, significantly improving the electrical conductivity. Benefiting from the faster Na+ and electron transfer, the TTHP-T anode maintained a high rate capacity of 406 mA h g−1 at 8 A g−1 (Fig. 6(e)). Further investigation explored that the –OH groups can allow the chelation with Na+ after deprotonation to boost sodium storage.96
All in all, the optimization of side chains including EWGs and EDGs, can regulate charge, which can adjust the HOMO and LUMO energy levels, and affect the energy bandgaps, thus affecting electron transfer and rapid reaction kinetics for sodium storage. However, it is found that the decoration of EWGs in the backbone can enhance the potential plateau of organic electrodes. Therefore, it is essential for anodes and cathodes to achieve a balance between voltage platform and rate performance. Additionally, the synergistic effect of both EWGs and EDGs is unclear and has rarely been explored recently.
4. Conclusion and perspective
4.1 Conclusion
OSIBs have gained significant attention due to the high scalability and sustainability of organic materials, which show abundant reserve, low price, eco-friendly synthesis and biodegradability, tuneable structures, better flexibility and processability, lightweight design, and extreme operating temperatures for electrochemical energy storage. However, the poor ionic and electronic conductivities of organic materials suppress rapid charging and discharging, thus limiting the achievement of high-power density for the fast-charging era. Molecular design is an efficient method to improve electron and ionic transfer for anodes and cathodes via backbone and side chains optimization.In this review, three strategies have been proposed to enhance power density for SIBs through molecular design as follows:
1. Backbone atom substitution. Substituting active-center atoms (e.g., with nitrogen or sulfur) help narrow the bandgaps of HOMO and LUMO levels, thus enhancing electrical conductivity and exhibiting excellent rate capabilities for SIBs. Besides, the combination of both n-type and p-type atoms in organic materials creates a push–pull effect to improve electron transfer and excellent rate performance.
2. Backbone framework modulation. The structural design for high-power density sodium storage can facilitate electron and ion movement. Through conjugated effect, coordinated bonds, and covalent bonds, there are three common organic materials have been explored. Conjugation systems, MOFs, and COFs with large ordered pathways and large coplanarity networks allow efficient sodium ion and electron diffusion, thus achieving impressive rate capacities. Besides, the introduction of terminal function groups decreases energy gaps, thus improving electrical conductivity and reaction kinetics. These frameworks leverage structural and electronic properties to optimize ion transport, offering promising avenues for high-performance sodium-ion batteries.
3. Side chain optimization. Adjusting side chains with electron-withdrawing groups like –SO3, and halogen functional groups can increase electron affinity, thus improving conductivity. The decoration of electron-donating groups such as hydroxyl and amino groups can enhance sodium ion trapping, thus enabling high-rate performance.
Molecular design can be applied widely for high-power density sodium storage, and organic materials can be divided into n-type, p-type, and bipolar-type (both n-type and p-type) materials according to the charge state of active organic groups. In practical application, establishing extended conjugated systems allows all organic electrodes to display stability and fast charger transfer for SIBs. Besides, p-type organics as cathodes are expected to have higher potentials through backbone p-typed atom substitution and the introduction of EWGs side chains. On the other hand, n-type organics as anodes can be designed as ion reservoirs through the combination of n-type and p-type dopants, and porous structure construction to achieve fast adsorption and desorption.
4.2 Perspectives
Most research is currently focusing on inorganic materials for SIBs, but there is also increasing attention on organic materials recently. A superior rate capability enables fast charging and discharging process for OSIBs and eliminates the risk of thermal runaway to enhance operational safety. According to the urgent requirement for high-power OSIBs, suggestions and perspectives to further promote fast ion and charge transport are presented as following aspects:1. Electrodes synthesis: the realization of fast charge and discharge performance requires rapid electron diffusion in conductive networks and smooth ion transport in porous channels. In the design of organic active materials, it is suggested to optimize the inherent characteristics of organic materials, such as 2D organic materials, P–N constructed organic molecules. The combination with conductive subtracts such as reduced graphene oxides (rGOs), carbon nanotubes (CNTs), MXenes, and some conductive polymers, could establish a conductive network for fast electron transfer.97 However, the combination of more carbonate materials could lower the capacity of electrodes. Additionally, conductive additives and binders are needed in preparing organic electrodes, which can increase electrode mass without enhancing electrode capacity. Therefore, the content of active organic materials has to increase, even when synthesizing binder-free electrodes in the future. Besides, advanced in situ or operando techniques cooperating with theoretical calculations should be applied to clearly explore reaction mechanisms and guide the design of electrode materials.
2. Electrolytes. Investigating new electrolytes with high ionic conductivity can further enhance fast-charging performance. At the same time, electrolytes that possess flame retardancy, thermal and chemical stability, electrochemical robustness, and dendrite suppression can provide high safety in battery systems.
3. Practical application. The most promising field of organic materials is flexible devices, like wearable devices, and skin sense. These devices have low requirements for voltage and capacity, which make organic materials acceptable. However, further development and optimization for organic full cells are needed to allow them a promising candidate for sustainable storage devices.
Although many challenges lie on the way, it is reasonable to devote more efforts to this field to pursue greener and cheaper energy devices. This review aims to give some suggestions on the future molecular design of organic materials for high-power density sodium storage.
Data availability
Data availability is not applicable to this article, as no new data were created or analysed during this study.Conflicts of interest
There are no conflicts to declare regarding to the content of this article.Acknowledgements
The authors gratefully acknowledge support from the German Research Foundation (DFG, Project number 501766751) and the Sino-German Center for Research Promotion (GZ1579).References
- C. Xu, J. Qiu, Y. Dong, Y. Li, Y. Shen, H. Zhao, U. Kaiser, G. Shao and Y. Lei, Energy Environ. Mater., 2023, 7, e12626 CrossRef.
- (a) Y. Wang, R. Ou, J. Yang, Y. Xin, P. Singh, F. Wu, Y. Qian and H. Gao, J. Energy Chem., 2024, 95, 407–427 CrossRef; (b) Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef; (c) M. Sha, L. Liu, H. Zhao and Y. Lei, Carbon Energy, 2020, 2, 350–369 CrossRef.
- (a) J. C. Hyun, H. M. Jin, J. H. Kwak, S. Ha, D. H. Kang, H. S. Kim, S. Kim, M. Park, C. Y. Kim, J. Yoon, J. S. Park, J.-Y. Kim, H.-D. Lim, S. Y. Cho, H.-J. Jin and Y. S. Yun, Energy Environ. Sci., 2024, 17, 2856–2863 RSC; (b) Y. Wan, B. Huang, W. Liu, D. Chao, Y. Wang and W. Li, Adv. Mater., 2024, e2404574, DOI:10.1002/adma.202404574.
- Y. Dong, C. Xu, Y. Li, C. Zhang, H. Zhao, U. Kaiser and Y. Lei, Adv. Energy Mater., 2023, 13, 2204324 CrossRef.
- (a) Y. Xu, M. Zhou and Y. Lei, Mater. Today, 2018, 21, 60–78 CrossRef; (b) B. Sun, Z. Sun, Y. Yang, X. L. Huang, S. C. Jun, C. Zhao, J. Xue, S. Liu, H. K. Liu and S. X. Dou, ACS Nano, 2024, 18, 28–66 CrossRef; (c) J. Hu, Y. Hong, M. Guo, Y. Hu, W. Tang, S. Xu, S. Jia, B. Wei, S. Liu, C. Fan and Q. Zhang, Energy Storage Mater., 2023, 56, 267–299 CrossRef; (d) J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2022, 8, 54–70 CrossRef; (e) C. Wang, C. Yang and Z. Zheng, Adv. Sci., 2022, 9, e2105213 CrossRef.
- X.-X. He, X.-H. Liu, Z. Yang, H. Zhang, L. Li, G. Xu, Y. Qiao, S.-L. Chou and M. Wu, Electrochem. Commun., 2021, 128, 107067 CrossRef.
- J. Huang, K. I. E. Callender, K. Qin, M. Girgis, M. Paige, Z. Yang, A. Z. Clayborne and C. Luo, ACS Appl. Mater. Interfaces, 2022, 14, 40784–40792 CrossRef PubMed.
- C. Yuan, Q. Wu, Q. Li, Q. Duan, Y. Li and H.-G. Wang, ACS Sustainable Chem. Eng., 2018, 6, 8392–8399 CrossRef.
- Y. Hong, Z. Ma, K. Li, J. Li, S. Tang, Z. Xu, D. Yu, D. Chen, L. Qin, J. Xie and Q. He, J. Mater. Chem. A, 2023, 11, 7898–7923 RSC.
- (a) T. Banerjee and R. Kundu, Energy Fuels, 2024, 38, 12487–12509 CrossRef; (b) X. Gan and Z. Song, Sci. China: Chem., 2023, 66, 3070–3104 CrossRef.
- (a) C. Friebe and U. S. Schubert, Top. Curr. Chem., 2017, 375, 19 CrossRef; (b) T. He, X. Kang, F. Wang, J. Zhang, T. Zhang and F. Ran, Mater. Sci. Eng., R, 2023, 154, 100737 CrossRef.
- (a) R. Balint, N. J. Cassidy and S. H. Cartmell, Acta Biomater., 2014, 10, 2341–2353 CrossRef PubMed; (b) M. Yang, M. Guo, E. Xu, W. Ren, D. Wang, S. Li, S. Zhang, C. W. Nan and Y. Shen, Nat. Nanotechnol., 2024, 19, 588–603 CrossRef PubMed; (c) Y. Wang, Y. Ding, X. Guo and G. Yu, Nano Res., 2019, 12, 1978–1987 CrossRef.
- L. Zhu, Y. Shen, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2013, 49, 11370–11372 RSC.
- C. Wei, L. Tan, Y. Zhang, B. Xi, S. Xiong and J. Feng, ACS Appl. Mater. Interfaces, 2022, 14, 2979–2988 CrossRef.
- X. Yin, S. Sarkar, S. Shi, Q. A. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef.
- H. Guo, H. Dai and C. Wang, ChemPlusChem, 2023, 88, e202300026 CrossRef PubMed.
- S. Jindal, Z. Tian, A. Mallick, S. Kandambeth, C. Liu, P. M. Bhatt, X. Zhang, O. Shekhah, H. N. Alshareef and M. Eddaoudi, Small, 2024, 2407525, DOI:10.1002/smll.202407525,.
- (a) L. Chen, S. Liu, L. Zhao and Y. Zhao, Electrochim. Acta, 2017, 258, 677–683 CrossRef CAS; (b) H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- L. Kong, M. Liu, H. Huang, Y. Xu and X. H. Bu, Adv. Energy Mater., 2021, 12, 2100172 CrossRef.
- (a) R. Rajagopalan, Y. Tang, C. Jia, X. Ji and H. Wang, Energy Environ. Sci., 2020, 13, 1568–1592 RSC; (b) Z. Wu, Q. Liu, P. Yang, H. Chen, Q. Zhang, S. Li, Y. Tang and S. Zhang, Electrochem. Energy Rev., 2022, 5, 26 CrossRef CAS.
- P. V. Braun, J. Cho, J. H. Pikul, W. P. King and H. Zhang, Curr. Opin. Solid State Mater. Sci., 2012, 16, 186–198 CrossRef CAS.
- (a) R. Wang, L. Wang, R. Liu, X. Li, Y. Wu and F. Ran, ACS Nano, 2024, 18, 2611–2648 CrossRef CAS PubMed; (b) Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS PubMed.
- M. Amer, J. Masri, A. Dababat, U. Sajjad and K. Hamid, Energy Convers. Manage.: X, 2024, 24, 100751 Search PubMed.
- H. Li, M. Xu, Z. Zhang, Y. Lai and J. Ma, Adv. Funct. Mater., 2020, 30, 2000473 CrossRef CAS.
- X. Cao, J. Liu, L. Zhu and L. Xie, Energy Technol., 2019, 7, 1800759 CrossRef.
- Q. Wang, T. O’Carroll, F. Shi, Y. Huang, G. Chen, X. Yang, A. Nevar, N. Dudko, N. Tarasenko, J. Xie, L. Shi, G. Wu and D. Zhang, Electrochem. Energy Rev., 2024, 7, 15 CrossRef CAS.
- Y. Wu, R. Zeng, J. Nan, D. Shu, Y. Qiu and S.-L. Chou, Adv. Energy Mater., 2017, 7, 1700278 CrossRef.
- Y. Chen and C. Wang, Acc. Chem. Res., 2020, 53, 2636–2647 CrossRef CAS PubMed.
- (a) Y. Yao, M. Pei, C. Su, X. Jin, Y. Qu, Z. Song, W. Jiang, X. Jian and F. Hu, Small, 2024, 20, 2401481 CrossRef CAS; (b) C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2019, 5, 5–19 CrossRef.
- (a) S. Biswas, A. Pramanik, A. Dey, S. Chattopadhyay, T. S. Pieshkov, S. Bhattacharyya, P. M. Ajayan and T. K. Maji, Small, 2024, 20, 2406173 CrossRef CAS PubMed; (b) D.-G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- (a) X. Chen, W. Sun and Y. Wang, ChemElectroChem, 2020, 7, 3905–3926 CrossRef CAS; (b) Y. Xin, Y. Ge, Z. Li, Q. Zhang and H. Tian, Acta Phys.-Chim. Sin., 2023, 40, 2303060 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- Y. Li, M. Chen, B. Liu, Y. Zhang, X. Liang and X. Xia, Adv. Energy Mater., 2020, 10, 2000927 CrossRef.
- Z. Gan, J. Yin, X. Xu, Y. Cheng and T. Yu, ACS Nano, 2022, 16, 5131–5152 CrossRef PubMed.
- (a) S. Suriyakumar, A. Mazumder, P. S. Dilip, M. Hariharan and M. M. Shaijumon, Batteries Supercaps, 2023, 6, e202300111 CrossRef; (b) M. Zhou, L. Zhu, Y. Cao, R. Zhao, J. Qian, X. Ai and H. Yang, RSC Adv., 2012, 2, 5495–5498 RSC.
- D. Tomerini, C. Gatti and C. Frayret, Phys. Chem. Chem. Phys., 2016, 18, 2442–2448 RSC.
- L.-P. Ma, W. Ren and H.-M. Cheng, Acta Phys.-Chim. Sin., 2021, 38, 2012080 Search PubMed.
- J. Luder, M. H. Cheow and S. Manzhos, Phys. Chem. Chem. Phys., 2017, 19, 13195–13209 RSC.
- (a) A. Tufail, W. E. Price, M. Mohseni, B. K. Pramanik and F. I. Hai, J. Water Proc. Engineering, 2021, 40, 101778 CrossRef; (b) K. Sakaushi, E. Hosono, G. Nickerl, T. Gemming, H. Zhou, S. Kaskel and J. Eckert, Nat. Commun., 2013, 4, 1485 CrossRef PubMed.
- H. Zhao, J. Wang, Y. Zheng, J. Li, X. Han, G. He and Y. Du, Angew. Chem., Int. Ed., 2017, 56, 15334–15338 CrossRef PubMed.
- K. Holguin, K. Qin, E. P. Kamphaus, F. Chen, L. Cheng, G.-L. Xu, K. Amine and C. Luo, J. Power Sources, 2022, 533, 231383 CrossRef.
- S. Haldar, D. Kaleeswaran, D. Rase, K. Roy, S. Ogale and R. Vaidhyanathan, Nanoscale Horiz., 2020, 5, 1264–1273 RSC.
- J. Hong, M. Lee, B. Lee, D. H. Seo, C. B. Park and K. Kang, Nat. Commun., 2014, 5, 5335 CrossRef PubMed.
- K. Jia, L. Zhu and F. Wu, ChemSusChem, 2021, 14, 3124–3130 CrossRef PubMed.
- M.-M. Hu, H. Huang, Q. Gao, Y. Tang, Y. Luo, Y. Deng and L. Zhang, Energy Fuels, 2020, 35, 1851–1858 CrossRef.
- L. Liu, Y. Gong, Y. Tong, H. Tian, X. Wang, Y. Hu, S. Huang, W. Huang, S. Sharma, J. Cui, Y. Jin, W. Gong and W. Zhang, CCS Chem., 2024, 6, 1255–1263 CrossRef.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130 CrossRef PubMed.
- C. Wang, Energy Environ. Mater., 2020, 3, 441–452 CrossRef.
- (a) C. Wang, Y. Fang, Y. Xu, L. Liang, M. Zhou, H. Zhao and Y. Lei, Adv. Funct. Mater., 2016, 26, 1777–1786 CrossRef; (b) S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan, S. Yu, Z. Sun, Y. Liu and Z. Lu, Nat. Commun., 2016, 7, 13318 CrossRef PubMed.
- K. Chihara, N. Chujo, A. Kitajou and S. Okada, Electrochim. Acta, 2013, 110, 240–246 CrossRef.
- M. Zhang, Y. Zhao, F. Kang, W. Huang and Q. Zhang, Adv. Funct. Mater., 2024, 2415186, DOI:10.1002/adfm.202415186.
- M. Tang, S. Zhu, Z. Liu, C. Jiang, Y. Wu, H. Li, B. Wang, E. Wang, J. Ma and C. Wang, Chem, 2018, 4, 2600–2614 Search PubMed.
- D. Li, C. Wang, J. Hu, W. Tang, S. Jia, M. Guo and C. Fan, Chem. Eng. J., 2022, 449, 137745 CrossRef.
- X. Gao, Y. Chen, C. Gu, J. Wen, X. Peng, J. Liu, Z. Zhang, Q. Zhang, Z. Liu and C. Wang, J. Mater. Chem. A, 2020, 8, 19283–19289 RSC.
- J. Chen, H. Yin, Q. Xue, J. Zhang, X. Chen, X. Liu, R. He, L. Zhu and F. Wu, Adv. Funct. Mater., 2024, 2411362, DOI:10.1002/adfm.202411362.
- W. Li, H. Ma, W. Tang, K. Fan, S. Jia, J. Gao, M. Wang, Y. Wang, B. Cao and C. Fan, Nat. Commun., 2024, 15, 9533 CrossRef PubMed.
- (a) J. Zhang, J. Chen, F. Wu, G. Huang, X. Liu, R. He and L. Zhu, ACS Sustainable Chem. Eng., 2023, 11, 17849–17856 CrossRef; (b) M. Fu, Y. Chen, W. Jin, H. Dai, G. Zhang, K. Fan, Y. Gao, L. Guan, J. Chen, C. Zhang, J. Ma and C. Wang, Angew. Chem., Int. Ed., 2024, 63, e202317393 CrossRef PubMed.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef.
- K.-G. Liu, F. Bigdeli, A. Panjehpour, S. Hwa Jhung, H. A. J. Al Lawati and A. Morsali, Coord. Chem. Rev., 2023, 496, 215413 CrossRef.
- Y.-Z. Chen, R. Zhang, L. Jiao and H.-L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef.
- (a) D. Zhu, G. Xu, M. Barnes, Y. Li, C. P. Tseng, Z. Zhang, J. J. Zhang, Y. Zhu, S. Khalil, M. M. Rahman, R. Verduzco and P. M. Ajayan, Adv. Funct. Mater., 2021, 31, 2100505 CrossRef; (b) Y. An, S. Tan, Y. Liu, K. Zhu, L. Hu, Y. Rong and Q. An, Energy Storage Mater., 2021, 41, 354–379 CrossRef.
- J. Cao, X. J. Li and H. Q. Tian, Curr. Med. Chem., 2020, 27, 5949–5969 CrossRef PubMed.
- (a) J. Lin, R. Chenna Krishna Reddy, C. Zeng, X. Lin, A. Zeb and C.-Y. Su, Coord. Chem. Rev., 2021, 446, 214118 CrossRef; (b) Z. Ye, Y. Jiang, L. Li, F. Wu and R. Chen, Nano-Micro Lett., 2021, 13, 203 CrossRef PubMed.
- R. Fernández de Luis, A. Ponrouch, M. Rosa Palacín, M. Karmele Urtiaga and M. I. Arriortua, J. Solid State Chem., 2014, 212, 92–98 CrossRef.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef PubMed.
- C. Dong and L. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 7160–7168 CrossRef PubMed.
- L. Wang, Y. Ni, X. Hou, L. Chen, F. Li and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 22126–22131 CrossRef PubMed.
- M. Qi, L. Cheng, H. G. Wang, F. Cui, Q. Yang and L. Chen, Adv. Mater., 2024, 36, e2401878 CrossRef PubMed.
- W. Shuang, Y. Wang, Y. Wu, N. Ma, F. Chen, X. Wang, W. Yan, Z. Bai, L. Yang and J. Zhang, Adv. Funct. Mater., 2024, 2408962, DOI:10.1002/adfm.202408962.
- K. Wakamatsu, S. Furuno, Y. Yamaguchi, R. Matsushima, T. Shimizu, N. Tanifuji and H. Yoshikawa, ACS Appl. Energy Mater., 2023, 6, 9124–9135 CrossRef.
- Y. Chen, Q. Zhu, K. Fan, Y. Gu, M. Sun, Z. Li, C. Zhang, Y. Wu, Q. Wang, S. Xu, J. Ma, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 18769–18776 CrossRef PubMed.
- K. Fan, C. Fu, Y. Chen, C. Zhang, G. Zhang, L. Guan, M. Mao, J. Ma, W. Hu and C. Wang, Adv. Sci., 2023, 10, e2205760 CrossRef PubMed.
- J. Zhao, M. Zhou, J. Chen, L. Tao, Q. Zhang, Z. Li, S. Zhong, H. Fu, H. Wang and L. Wu, Chem. Eng. J., 2021, 425, 131630 CrossRef CAS.
- J. Lee, Y. Kim, S. Park, K. H. Shin, G. Jang, M. J. Hwang, D. Kim, K. A. Min, H. S. Park, B. Han, D. K. P. Ng and L. Y. S. Lee, Energy Environ. Mater., 2023, 6, e12468 CrossRef.
- W. Yuan, J. Weng, M. Ding, H.-M. Jiang, Z. Fan, Z. Zhao, P. Zhang, L.-P. Xu and P. Zhou, Energy Storage Mater., 2024, 65, 103142 CrossRef.
- Y. Zhang, Y. Wu, Y. Liu and J. Feng, Chem. Eng. J., 2022, 428, 131040 CrossRef.
- M. K. Shehab, K. S. Weeraratne, O. M. El-Kadri, V. K. Yadavalli and H. M. El-Kaderi, Macromol. Rapid Commun., 2023, 44, e2200782 CrossRef PubMed.
- V. Singh, J. Kim, B. Kang, J. Moon, S. Kim, W. Y. Kim and H. R. Byon, Adv. Energy Mater., 2021, 11, 2003735 CrossRef.
- A. Halder, M. Ghosh, M. A. Khayum, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef PubMed.
- Y. Xu, J. Gong, Q. Li, X. Guo, X. Wan, L. Xu and H. Pang, Nanoscale, 2024, 16, 11429–11456 RSC.
- A. Ghani, S. Ahmed, A. Murtaza, I. Muhammad, W. L. Zuo and S. Yang, J. Phys. Chem. C, 2023, 127, 16802–16810 CrossRef.
- L. Chen, Y. Li, Y. Zhang, S.-B. Ren, J. Bi, X. Xue, D.-M. Han, D. Wu, Y. Wang, X. Chen and Y. Wu, Chem. Eng. J., 2024, 497, 154743 CrossRef.
- B. C. Patra, S. K. Das, A. Ghosh, A. Raj K, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6, 16655–16663 RSC.
- R. Shi, L. Liu, Y. Lu, C. Wang, Y. Li, L. Li, Z. Yan and J. Che, Nat. Commun., 2020, 11, 178 CrossRef PubMed.
- H. Zhang, W. Sun, X. Chen and Y. Wang, ACS Nano, 2019, 13, 14252–14261 CrossRef CAS PubMed.
- M. Zhang, Y. Tong, Z. Sun, J. Wang, Y. Lin, F. Kang, Q. Zhang and W. Huang, Chem. Mater., 2023, 35, 4873–4881 CrossRef CAS.
- J. Liu, P. Lyu, Y. Zhang, P. Nachtigall and Y. Xu, Adv. Mater., 2018, 30, 1705401 CrossRef PubMed.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- C. Wang, H. Tang, H. Zhu, W. Tang, S. Jia, D. Li, Y. Liu, M. Guo and C. Fan, J. Power Sources, 2022, 546, 231962 CrossRef.
- Y. Park, D. S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef PubMed.
- M. Zhou, W. Li, T. Gu, K. Wang, S. Cheng and K. Jiang, Chem. Commun., 2015, 51, 14354–14356 RSC.
- M. Lee, S. Park, B. Bae, Y. K. Jeong, J.-M. Oh, J. K. Park and S.-M. Paek, Chem. Eng. J., 2023, 477, 147072 CrossRef.
- Z. Sun, K. Zhu, P. Liu, X. Chen, H. Li and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202211866 CrossRef PubMed.
- Y. Shan, Y. He, Y. Gu, Y. Sun, N. Yang, H. Jiang, F. Wang, C. Li, D.-E. Jiang, H. Liu, X. Zhu and S. Dai, Chem. Eng. J., 2022, 430, 133055 CrossRef.
- J. Y. Wang and X. W. Zhan, Acc. Chem. Res., 2021, 54, 132–143 CrossRef PubMed.
- C. Zhang, C. Fu, H. Guo, Y. Chen, K. Fan, Z. Li, J. Zou, H. Dai, G. Zhang, J. Ma and C. Wang, Energy Environ. Sci., 2024, 17, 6360–6367 RSC.
- (a) C. Yuan, Q. Wu, Q. Shao, Q. Li, B. Gao, Q. Duan and H. G. Wang, J. Colloid Interface Sci., 2018, 517, 72–79 CrossRef PubMed; (b) D. Zhou, T. Wu and Z. Xiao, J. Alloys Compd., 2022, 894, 162415 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |