
Macrocyclic catalysis mediated by water: opportunities and challenges
Dejun
Zhang
,
Lingyun
Wang
 ,
Wanqing
Wu
*,
Derong
Cao
* and
Hao
Tang
*
,
Wanqing
Wu
*,
Derong
Cao
* and
Hao
Tang
*
State Key Laboratory of Luminescent Materials and Devices, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, China. E-mail: cewuwq@scut.edu.cn; drcao@scut.edu.cn; haotang@scut.edu.cn
First published on 2nd December 2024
Abstract
Nanospaces within enzymes play a crucial role in chemical reactions in biological systems, garnering significant attention from supramolecular chemists. Inspired by the highly efficient catalysis of enzymes, artificial supramolecular hosts have been developed and widely employed in various reactions, paving the way for innovative and selective catalytic processes and offering new insights into enzymatic catalytic mechanisms. In supramolecular macrocycle systems, weak non-covalent interactions are exploited to enhance substrate solubility, increase local concentration, and stabilize the transition state, ultimately accelerating reaction rates and improving product selectivity. In this review, we will focus on the opportunities and challenges associated with the catalysis of chemical reactions by supramolecular macrocycles in the aqueous phase. Key issues to be discussed include limitations in molecular interaction efficiency in aqueous media, product inhibition, and the incompatibility of catalysts or conditions in “one-pot” reactions.
1. Introduction
In the complex chemical reactions of biological systems, enzymes play a crucial role due to their superior catalytic activity and precise substrate selectivity.1 Enzymes possess highly active sites and multiple compartmentalized cavities, enabling them to efficiently capture substrates or reaction intermediates from bulk solution, while providing unique internal nanoenvironments for these components. These discrete and confined spaces enhance the effective local concentration of specific reaction species, facilitate their preorganization, stabilize transition states, and promote efficient mass transfer of substrates, intermediates, and products, ensuring reactions proceed with high selectivity and continuity.2 To explore the highly efficient catalytic mechanisms of enzymes, an effective strategy involves initially constructing structurally simple systems with well-defined structure–activity relationships that mimic a single catalytic site of enzymes, and then gradually increase system complexity to understand the catalytic coupling mechanisms. For this purpose, various supramolecular macrocyclic host molecules have been developed as nanoreactors, including crown ethers,3 cyclodextrins,4 cucurbiturils,5 calixarenes6 and pillararenes.7 Subsequently, strategies for the precise control of non-covalent interactions between multiple components—such as hydrogen bonds, van der Waals forces, the hydrophobic effect, electrostatic interactions, π–π interactions, cation–π interactions, and anion–π interactions—are used to construct highly ordered, self-assembled aggregates with desired structures and functions, making the development of such approaches a significant research area in supramolecular chemistry.8,9 These aggregates are not only widely applied in fields such as chemical sensors, drug delivery, and separation but are also utilized in biomimetic studies of enzymatic catalytic coupling mechanisms.10 The core strategy of supramolecular catalysis lies in leveraging the interactions between supramolecular hosts (such as cucurbiturils, cages, and capsules) and guests (i.e., reactants or catalysts) to achieve precise control over chemical reactions. On the one hand, by designing and synthesizing supramolecular hosts with specific structures and functions, efficient recognition and capture of substrates, preorganization of reactants, and enhancement of local concentrations can be accomplished. On the other hand, supramolecular catalysis can accurately catalyze specific reaction pathways within complex reaction systems and stabilize particular transition states, thereby effectively lowering the activation energy and minimizing the generation of by-products, ultimately enhancing catalytic efficiency.11,12Currently, artificial supramolecular hosts have been widely employed in a variety of reactions, with numerous examples reported of using well-defined nanospaces to catalyze new reactions and generate molecules that are challenging to obtain through traditional catalytic methods. To date, several reviews have explored the diversity of supramolecular assembly,13,14 the size effects of nanospaces15,16 and variations in specific system regulation strategies from multiple perspectives.17,18 This review highlights representative studies from the past five years, elucidating how supramolecular chemists have addressed three key challenges in aqueous-phase macrocyclic catalysis: (1) limitations in molecular interaction efficiency in aqueous environments; (2) product inhibition; and (3) incompatibility of catalysts or conditions in “one-pot” reactions. For readers seeking a more comprehensive overview of the field and those interested in related topics, the aforementioned reviews are recommended.
2. Challenge 1: limitations in molecular interaction efficiency in aqueous medium
Aqueous catalytic reactions offer significant advantages, including environmental friendliness, mild reaction conditions, and ease of product separation, making them an important area of research.19,20 However, the limited interaction efficiency of molecules in aqueous environments has constrained the development of this field. This limitation arises from three primary factors, each of which is addressed by various supramolecular strategies. First, substrates dispersed in solution must diffuse towards each other or to the catalyst surface before interaction can occur. The low concentration of reaction species due to their low solubility can significantly limit reaction efficiency. In some cases, the diffusion rate can be much slower than the reaction rate, making the overall process limited by diffusion rather than the chemical reaction itself, resulting in a diffusion-controlled process with low reaction efficiency.21 Supramolecular hosts can act as phase transfer catalysts, effectively enhancing the local concentration of substrates and catalysts, facilitating a close association between these reaction species.22 Second, the formation of solvation shells—due to hydrogen bonding and other weak interactions between reaction species and polar water—hinders molecular proximity and effective collisions between these reaction species, and also destabilizes intermediates. These issues can be solved by encapsulating the reaction species within the hydrophobic cavity of supramolecular hosts. Third, the relatively high degree of freedom of molecules in solution reduces the probability of effective collisions at the reaction site.23–25 Encapsulating them within the confined cavity of supramolecular hosts restricts their free motion and facilitates their preorganization.2.1 Size matching of hosts and guests
Addressing the challenge of limited water solubility of reaction species, macrocyclic hosts in aqueous solution selectively recognize substrates and encapsulate them within their internal cavities, essentially functioning as phase transfer catalysts, effectively enhancing substrate solubility26 and increasing local concentrations.27 The core principle is precise size matching between hosts and guests,28 as quantified by the packing coefficient (PC), which is the ratio of guest(s) volume to macrocycle cavity volume. Rebek summarized that optimal binding between hosts and guests occurs when the PC is approximately 0.55.29 Guests that are too small must occupy the cavity along with several “high-energy” water molecules, which cannot form optimized hydrogen bonds as effectively as those in bulk solution. On the other hand, guests that are too large cause deformation in both the guest and host due to strain, with both scenarios leading to an unstable encapsulation process. Reinhoudt et al. observed that substrates around a PC of 0.55 achieve optimal template reactions, with a PC of 0.58 resulting in 50% yield, whereas the yield for substrates with a PC of 0.74 is less than 5%.30 Nau et al. conducted an in-depth investigation into the use of cucurbit[n]urils (CB[n]) with varying cavity sizes for catalyzing the Diels–Alder reaction (Fig. 1).31 Cucurbit[5]uril (CB[5]) resulted in 3.5% conversion due to its inability to effectively encapsulate substrates. Cucurbit[6]uril (CB[6]) could just accommodate a single cyclopentadiene (CPD), thereby hindering its dimerization. Cucurbit[7]uril (CB[7]) tightly bound two substrates, forming a ternary complex with a PC of 0.63. As the Diels–Alder reaction progressed, the PC of the transition state decreased to 0.6, effectively alleviating the tight packing state and internal pressure, which increased the reaction rate by approximately five orders of magnitude. However, cucurbit[8]uril (CB[8]) did not provide sufficient confinement for the substrate (PC ≈ 0.41), causing the reaction to proceed in a loosely packed state. Additionally, the packing of the product, endo-dicyclopentadiene (DCPD), was not efficient (PC ≈ 0.38), leading to a lack of catalytic effect. | ||
| Fig. 1 Cucurbit[n]urils with varying cavity sizes used for catalyzing the Diels–Alder reaction. Adapted from ref. 31 with permission from ACS Publishing Group. | ||
2.2 Activation of reaction components
In addition to the packing coefficient between the host and the guest, their geometric shapes and electronegativity, the structure and position of substituents, as well as the coexistence of ions and co-solvent all influence the host's ability to encapsulate and desolvate guests, which in turn enhances substrate reactivity, activates catalysts, and stabilizes transition states as well as final products.32–34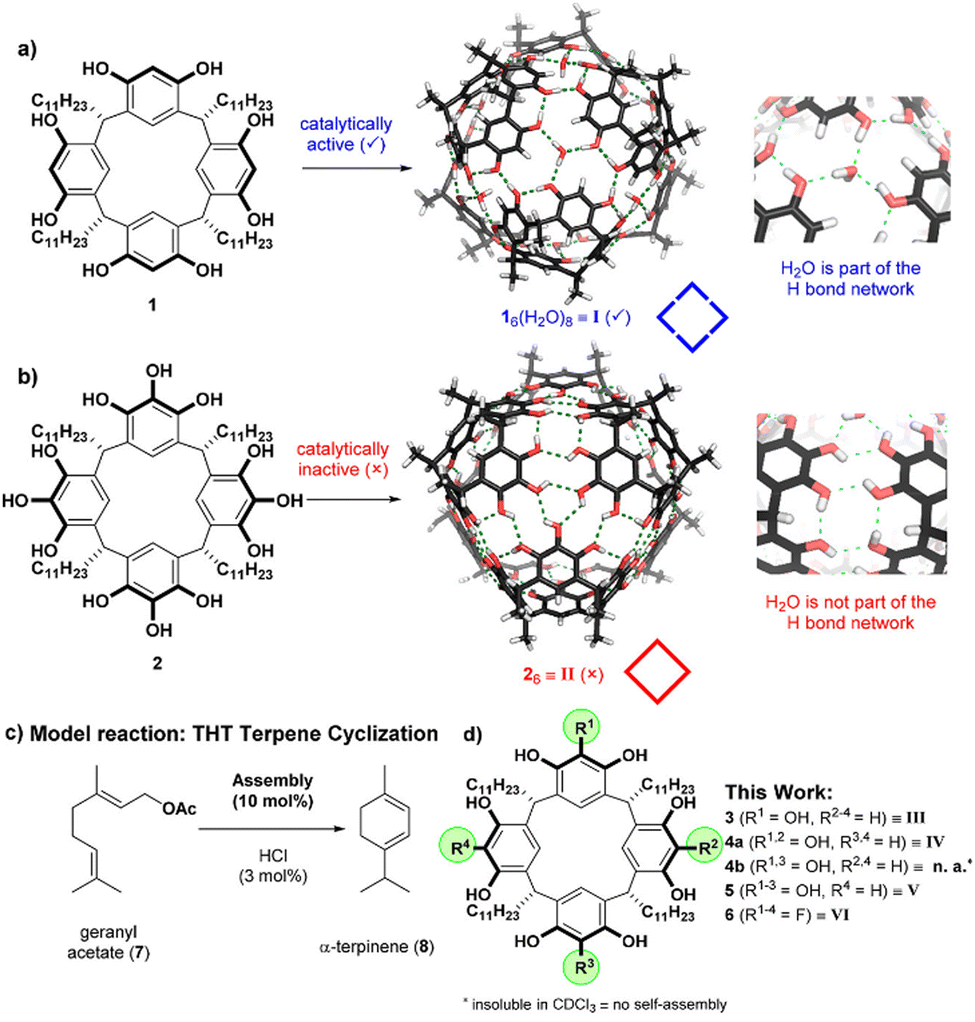 | ||
| Fig. 2 Two examples of capsules featuring hydrogen-bond networks with (a) and without (b) water embedded, as well as the catalysis of the tail-to-head terpene cyclization (c) by a series of capsules (d). Adapted from ref. 47 with permission from ACS Publishing Group. | ||
The binding mode49 between the macrocycle and substrates as well as the arrangement of binding sites50 can significantly influence substrate reactivity.51 Sashuk et al. reported that the aldehyde groups of azo pyridinium aldehyde were encapsulated within the cavity of CB[7], while the azo moiety remained exposed to the solution and was subsequently attacked by hydrazine, leading to the formation of hydrazone.52 Aliaga et al. found that the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complex between CB[7] and aromatic Schiff bases promoted hydrolysis, whereas the formation of a 2:1 host–guest complex resulted in an inhibitory effect.53
:1 host–guest complex between CB[7] and aromatic Schiff bases promoted hydrolysis, whereas the formation of a 2:1 host–guest complex resulted in an inhibitory effect.53
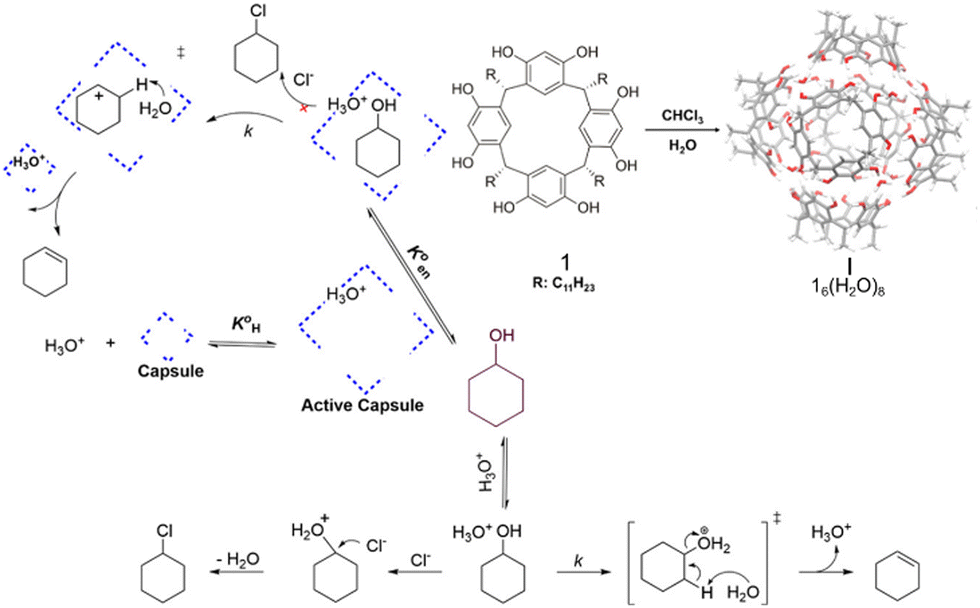 | ||
| Fig. 3 Catalysis of the dehydration of cyclohexanol to produce cyclohexene using the capsule I. Adapted from ref. 59 with permission, copyright 2020, American Chemical Society. | ||
The supramolecular host can fine-tune the protonation degree and electrophilicity of catalysts upon encapsulation into its hydrophobic cavity.68–71 Eelkema et al. discovered that the encapsulation of organic catalysts such as aniline, 1,4-diazabicyclo[2.2.2]octane, and prolinol within CB[7] significantly reduced their catalytic activity, while enhancing the catalytic activity of nornicotine.72 This difference was attributed to the fact that the former catalysts were completely isolated within the CB[7] cavity, whereas the aliphatic amine of nornicotine extended beyond the carbonyl portal of CB[7], altering its protonation equilibrium.
Additionally, various factors, including the host/catalyst ratio, the solvent polarity,73 and the presence of cofactors such as acid/base, metal ions, and guests,74 can collectively regulate the catalytic activity. These guests may function as synergists, such as radical stabilizing agents like C60,75 or as competitive agents. Eelkema et al. demonstrated that the dynamic assembly/disassembly process between hosts and catalysts could be modulated by the addition of hosts or a competitive guest, allowing for a switch from “off” to “on”.76 Reek et al. discovered that the addition of a small cofactor allowed the previously independent zinc porphyrin and pyridine ligand to form an effective catalyst, resulting in an eightfold enhancement of rhodium catalyst activity.77
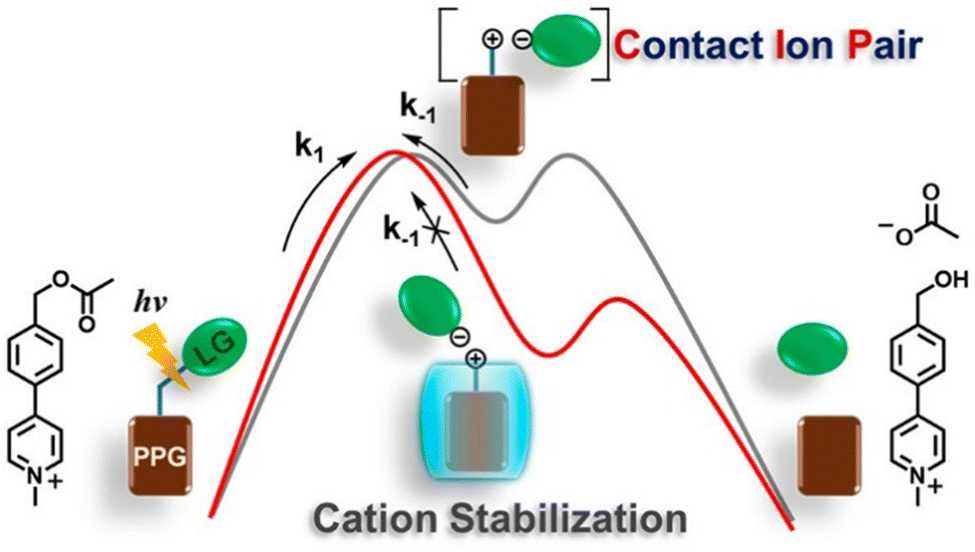 | ||
| Fig. 4 Catalysis of photolysis reactions of benzyl acetate using CB[7]. Reproduced with permission from ref. 79, copyright 2023, American Chemical Society. | ||
Supramolecular hosts effectively modulate the reactivity of various intermediates, including cationic,82–84 radical, and anionic85–87 intermediates, through their surface charge characteristics,88 host–guest interactions (such as electrostatic, cation–π, and anion–π interactions), solvent polarity,89 and external stimuli like exogenous salts. This mechanism alters the geometry and electronic properties of the reaction transition state, resulting in a significant reduction in the activation energy and influencing the mode of attack of reagents on the intermediates.90 Consequently, the reaction process becomes smoother, while significantly suppressing the premature quenching of intermediates,91,92 thereby ensuring that the reaction proceeds continuously and stably.93 Gibb et al. reported that the positively charged capsule stabilizes a negatively charged transition state through the coulombic force generated by its electrostatic potential (EP) field, thereby accelerating the rate of cyclization of α,ω-thioalkane halides.94 Moreover, exogenous salts, featuring counterions that complement the surface charge of the capsule, can bind to the outer wall of the capsule, weakening the EP field and influencing transition state reactivity.95
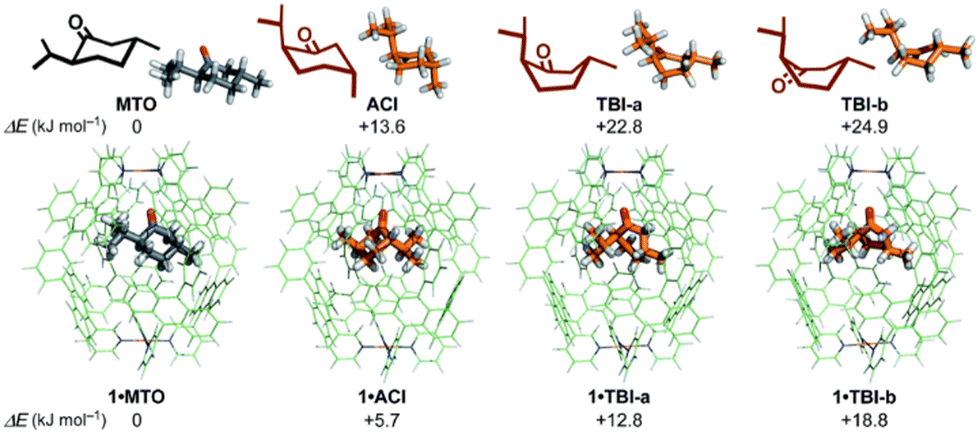 | ||
| Fig. 5 Stabilization of conformers of MTO using a polyaromatic capsule. Adapted from ref. 96 with permission from the Royal Society of Chemistry. | ||
2.3 Regulation of molecular freedom
Supramolecular hosts selectively recognize guests (i.e., reaction species), encapsulating them within a confined cavity. This process may involve conformational changes or structural adaptations in both the host and guest molecules to achieve optimal complementarity or an optimized arrangement of binding sites. The specific cavity shape, arrangement of binding sites, and electronegativity of the host help maintain the guests in a defined position and orientation, restricting their free motion and facilitating their preorganization. This preorganization optimizes the spatial relationship between substrates or between substrates and catalysts of the catalytic systems, lowers activation energy, promotes the desired reaction pathways, and allows for precise control over the reaction rate, selectivity, and efficiency under mild, tunable conditions.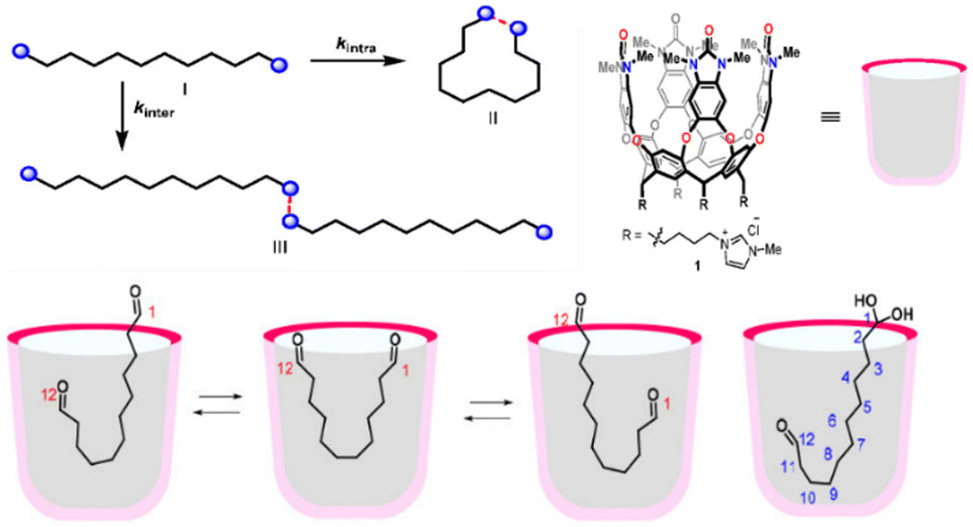 | ||
| Fig. 6 Enhancement of the conformational arrangement of the substrate by cavitand preorganization. Adapted from ref. 100 with permission, copyright 2021, American Chemical Society. | ||
Controlling the conformation of individual macrocycles may be achieved through structural design. Cacciapaglia et al. grafted the guanidine/guanidinium dyad onto a calixarene scaffold via carbonyl bridging, which moderately increased the conformational flexibility of the host, enabling it to transform into a highly catalytically active protonated form.107 Tiefenbacher et al. discovered that chirality transfer in the nerol cyclization was only observed when the involved capsule was attached at the edge to a linear alkyl chain containing an odd number of carbon atoms (enantiomeric excess >5%).108 This finding indicates an odd–even effect, wherein the structure and properties of the material exhibit alternating changes based on whether the number of structural units in the molecule is odd or even.109,110 Yang et al. successfully prevented the interconversion of conformers by grafting bulky β-cyclodextrin (β-CD) substituents at both ends of pillar[5]arene (P5), thereby obtaining a pair of enantiomers.111 The absolute configuration of the central P5 and the conjugating position on β-CD jointly determined regio- and stereoselectivity in the chiral catalytic reaction.
The conformation of macrocycles can also be regulated by forming aggregates. Salvio et al. systematically investigated multifunctional guanidinium-decorated calix[4]arenes derived from four guanidine or arginine units at the upper rim, which can self-assemble into aggregates in aqueous solution through hydrophobic effects.112 These aggregates can rearrange, mold, and flex to meet the geometric and electronic requirements of the substrates, significantly enhancing the efficiency of phosphodiester bond cleavage in the aqueous phase. We synthesized a series of monoester copillar[5]arenes, where the position of the ester group modulates their self-inclusion behavior, resulting in varying guest selectivity.113 Monoester copillar[5]arenes derived from acetate chains can form stable self-inclusion complexes in both low- and high-concentration solutions, exhibiting high guest selectivity. However, the butyrate chain-functionalized monoester copillar[5]arene could not form a self-inclusion complex and showed lower guest selectivity. Furthermore, the position of the ester group is critical for the generation of stable self-inclusion complexes.
During host/guest binding, in addition to inducing conformational changes in the guest, the host also undergoes conformational changes. Guan et al. observed that a metallocage (K12[Ga4L6], L = N,N′-bis(2,3-dihydroxybenzoyl)-1,5-diaminonaphthalene, KGaL) exhibits a unique “breathing” mechanism for substrate encapsulation.114 The naphthalene rings of KGaL rotated toward the closing direction of the guest-entry face to adapt to the shape of the substrate, resulting in a reduction of the cavity volume. As the guest gradually entered, a shape change was induced in one side of the “concave cup” structure for KGaL, leading to an increase in the cavity volume.
The solvent environment and coexisting species can also influence the conformation of macrocycles. Inoue discovered that by finely tuning the hydrophobicity and ionic strength of an aqueous solution, the penetration depth and orientation of the hydrophobic sensitizing moiety grafted onto the CD portal could be precisely controlled.115 This flexibility allows for the adjustment of the size and shape of the nanospace provided by the CD cavity to accommodate the requirements of different guests. Reek et al. found that introducing an additional cofactor such as water at the edge of the hydrogen bond network leads to the breaking of hydrogen bonds between adjacent capsule faces, consequently altering the connectivity of the entire supramolecular system and ultimately affecting the acidity and structure of the host.116 Lusby et al. employed a simple Pd2L4 capsule that utilizes endotopic and exotopic binding sites to separately bind substrates and effectors.117 When an effector binds to the outer surface of the capsule, it partially neutralizes charges on that surface, leading to subtle electronic effects. The effector can modulate the binding affinity of the capsule for substrates and their transition states through an allosteric regulation mechanism, wherein enzymes adjust the properties of their active sites by binding a control molecule on the protein exterior.118 We synthesized two novel copillar[5]arenes functionalized with ω-hydroxyalkoxy groups.119 Among them, the copillar[5]arene functionalized with a 6-hydroxyhexyloxy group exhibits reversible self-assembly behavior, leading to the formation of self-inclusion monomers and hugging dimers. By adjusting factors such as solvent, temperature, guest, and hydrogen-bond interactions, the reversible self-assembly behavior can be controlled. In contrast, the copillar[5]arene functionalized with a shorter 4-hydroxybutyloxy group does not display any self-assembly behavior.
In response to external stimuli such as light,120 acid and base,121 and the binding of effectors such as solvents,122 substrates,123,124 sensitizers,125 and exotopic ligands, the host may undergo deformation and experience dynamic reversible assembly and disassembly processes.126,127 Beves et al. designed and synthesized a heteroleptic coordination cage  featuring a photoswitchable azobenzene-derived ligand, which effectively catalyzed the Michael addition reaction between methyl vinyl ketone and benzoyl nitromethane (Fig. 7).128 After irradiation with a 530 nm LED light source for 10 minutes, the cage successfully disassembled, resulting in a tenfold reduction in the product formation rate. Conversely, when exposed to a 405 nm LED light source for 5 minutes, the cage was able to reassemble and restore its original catalytic activity, thereby enabling the on-and-off control of the catalytic reaction in the cage. Notably, the corresponding homoleptic cages exhibited no catalytic activity. Yoshizawa et al. designed and synthesized a heterocyclic capsule constructed from bent amphiphilic compounds containing two phenothiazine redox switches, which demonstrated reversible behavior in response to redox stimuli, facilitating the assembly and disassembly of the capsule.129
featuring a photoswitchable azobenzene-derived ligand, which effectively catalyzed the Michael addition reaction between methyl vinyl ketone and benzoyl nitromethane (Fig. 7).128 After irradiation with a 530 nm LED light source for 10 minutes, the cage successfully disassembled, resulting in a tenfold reduction in the product formation rate. Conversely, when exposed to a 405 nm LED light source for 5 minutes, the cage was able to reassemble and restore its original catalytic activity, thereby enabling the on-and-off control of the catalytic reaction in the cage. Notably, the corresponding homoleptic cages exhibited no catalytic activity. Yoshizawa et al. designed and synthesized a heterocyclic capsule constructed from bent amphiphilic compounds containing two phenothiazine redox switches, which demonstrated reversible behavior in response to redox stimuli, facilitating the assembly and disassembly of the capsule.129
 | ||
| Fig. 7 Reversible dynamic processes of cage assembly and disassembly enabled by light stimuli. Reproduced with permission from ref. 128, copyright 2024, American Chemical Society. | ||
Supramolecular strategies were used to increase the frequency of effective collisions between the substrates and accelerate the reaction process.132,133 Li et al. designed and synthesized a calix[4]squaramide organocatalyst functionalized with bis-squaramide and cyclohexanediamine scaffolds.134 The cooperative effect between the cavity and the chiral squaramide catalytic center effectively reduced the distance between two substrates, facilitating the asymmetric Michael addition between 1,3-dicarbonyl and α,β-unsaturated carbonyl compounds (Fig. 8). Liu et al. discovered that cucurbit[10]uril could bind two substrates to form ternary complexes with a 1:2 stoichiometry.135 This complex altered the distance and orientation between two substrates, placing them in a “reaction-ready” state, which enhances the selectivity and yield of the photodimerization of substrates. In contrast, the anthracene moieties of substrates incorporated within CB[8] adopt a head-to-tail orientation, which was unfavorable for photodimerization and stabilized the carbocation intermediate, unexpectedly yielding the photosolvolysis product of 9-anthracenemethanol. Furthermore, the supramolecular regulation strategy can be used to suppress unwanted reactions. Rescifina et al. found that CB[7] can bind nitrone and styrene separately to form a singular complex.136 This binding model effectively prevents close contact between two substrates, thereby reducing their reaction yield.
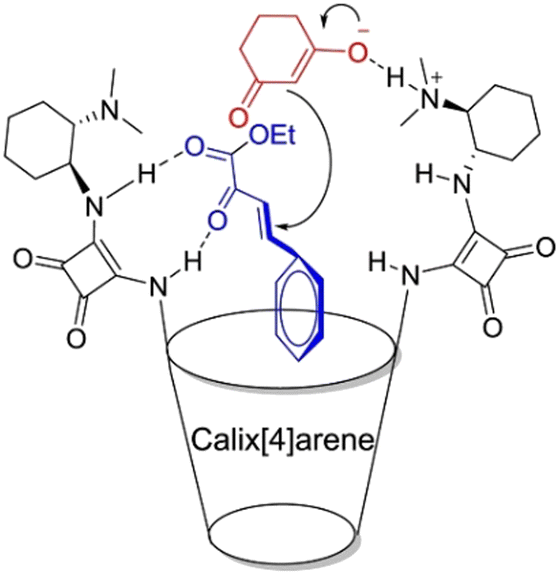 | ||
| Fig. 8 Control of the distance between two substrates by calix[4]squaramide. Reproduced with permission from ref. 134, copyright 2020 Elsevier Publishing Group. | ||
Anchoring of catalysts by the cavity effectively desolvates them, thereby lowering the reaction energy barrier,137 and regulates the spatial orientation between the catalysts and substrates, thus modulating chemoselectivity and regioselectivity.138,139 Anchoring can be achieved through weak interactions between the catalyst and the host. For example, by grafting coordinating groups onto a macrocyclic skeleton, metal catalysts can be bound either at the macrocycle portals140 or encapsulated within the cavities.141,142 Jiang et al. successfully constructed a highly active Fe(OTf)3/CD complex by binding Fe(OTf)3 to the primary hydroxyl groups located on the narrower rim of the cyclodextrin (CD) scaffold.143 The CD cavity effectively modulated the interaction between Fe(OTf)3 and encapsulated substrates, such as carbazoles, within the cavity. Podewitz et al. designed and synthesized a calix[8]arene modified with a phenanthroyl group capable of coordinating Cu(I) within its cavity.144 Substrates were enriched within the calixarene cavity, facilitating effective interactions with the Cu(I) center.145 Moreover, hosts can simultaneously encapsulate complementary substrates and catalysts, forming synergistic ternary complexes.146 Beves et al. utilized CB[10] to encapsulate [Ru(bpy)3]2+, the photocatalyst, along with substrates simultaneously, forming ternary complexes (Fig. 9).147 The arrangement facilitated intermolecular charge transfer to methyl viologen when the photocatalyst is in an excited state, thereby enhancing oxidative quenching efficiency.
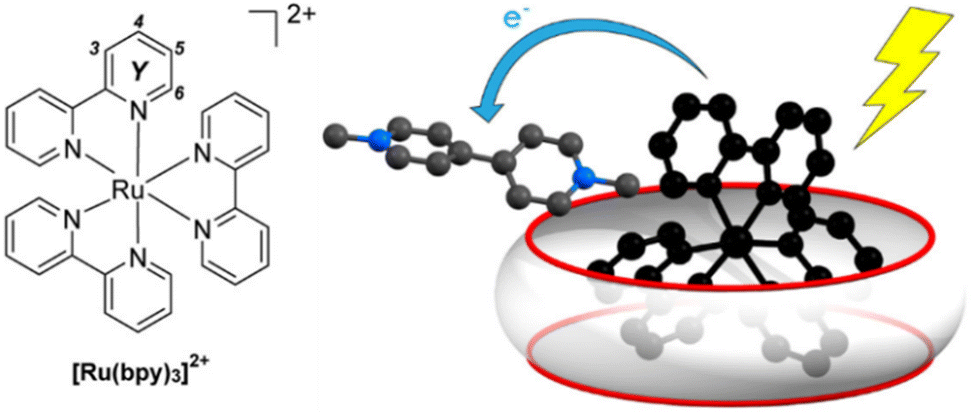 | ||
| Fig. 9 Simultaneous encapsulation of both the catalyst and the substrate by CB[10]. Adapted from ref. 147 with permission, copyright 2020, American Chemical Society. | ||
Anchoring of catalysts can be achieved by incorporating the catalytic active sites into the macrocyclic skeleton.148 Wang et al. adopted a strategy of incorporating catalytic active sites into a macrocyclic skeleton, successfully synthesizing a series of chiral bis-phosphate macrocycles.149 Through the synergistic effect of complementary ion-pair binding and cavity-directed non-covalent interactions, they adjusted the distance between the catalyst and the substrate, thereby enhancing the catalytic activity of the reaction. Duan et al. embedded NADH active sites within a metal–organic capsule, which was assembled using preorganization ligands and functionalized metallocorners (Fig. 10).150 In the external environment of the capsule, the presence of a reductant or photosensitizer facilitated a typical 1e− hydrogenation, enabling the highly selective reduction of nitro groups over carbonyl groups. Inside the capsule, the substrate was encapsulated within the cavity, allowing for preorganization that forced the active sites to closely contact the substrate. Consequently, the ADH active site achieved highly selective reduction of carbonyl groups through a typical 2e− hydride transfer hydrogenation, thereby enabling selective reduction between carbonyl and nitro groups in switchable bifunctional compounds.
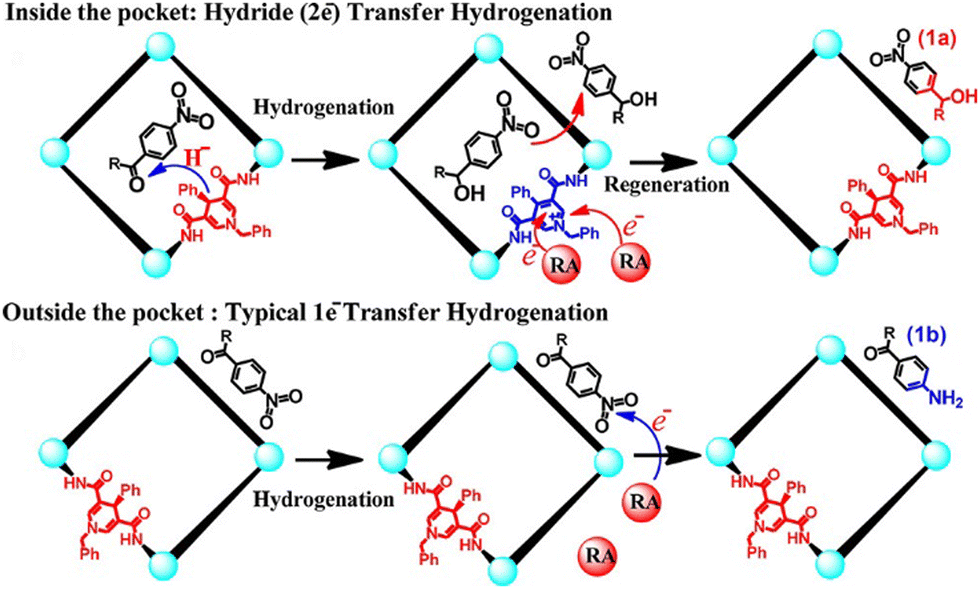 | ||
| Fig. 10 The regulation of selective reduction of carbonyl and nitro groups by the capsule. Reproduced with permission from ref. 150, copyright 2019, American Chemical Society. | ||
3. Challenge 2: product inhibition
Supramolecular catalysis enables efficient regulation of chemical reaction processes; however, it faces a significant challenge—product inhibition due to the difficulty of product release.151,152 Product inhibition typically arises from strong non-covalent interactions between the product and macrocycles, leading to the occupation of active sites. As a result, it prevents new substrates from entering, hindering subsequent reactions and potentially bringing the entire catalytic process to a halt. Several strategies have been employed to effectively address this issue and significantly enhance the turnover number (TON) of the catalyst: altering the binding affinity for substrates and products; selectively binding the catalyst rather than the substrate and the product; phase separation of the products or catalyst; and implementing self-catalysis.3.1 Altering the binding affinity for substrates and products
The design of ideal catalytic hosts should ensure that their binding affinity for substrates is significantly stronger than that for products, allowing substrates to readily displace products in the cavity, thereby facilitating subsequent reactions. The selective binding can inherently occur for certain reactions. For example, when hydrophilic reactants transform into hydrophobic products, the enhanced hydrophilicity drives the product to escape this unfavourable environment.59 Selective binding can be achieved by tuning host–guest interactions through the design of hosts tailored for specific reactions. For example, when positively charged substrates react to yield neutral products, negatively charged macrocycles are an effective choice due to their higher binding affinity for substrates compared to products.147 Similarly, when neutral substrates convert into negatively charged products, a hydrophobic cavity is ideal, as the negatively charged products are preferentially solvated by bulk water rather than remaining in the cavity.153 Moreover, appropriate size matching between the cavity and the substrates rather than the products may play a crucial role in preventing product inhibition.83,102Another strategy to tune the host–guest binding affinity is to adjust solvent polarity by adding a less polar solvent. With the decrease of solvent polarity, the binding affinity of the product to the host can be substantially reduced. Nau et al. demonstrated that introducing a less polar cosolvent, such as 10% methanol, effectively decreased the binding constant of highly hydrophobic products with the host, enabling the catalytic reaction to proceed with turnover numbers (TON) of 10 or higher, thereby addressing the issue of product inhibition.31
3.2 Selectively binding the catalyst
To prevent product inhibition, macrocycles can be used to selectively bind and activate catalysts instead of directly interacting with substrates or products. Wang et al. designed bis-diarylthiourea macrocycles that contain two cooperative diarylthiourea binding sites and two BINOL moieties (Fig. 11).154 The macrocycles selectively bind disulfonate especially ethanedisulfonate anions by the diarylthiourea groups, establishing a well-confined chiral microenvironment for protonated electrophilic substrates. Utilizing only 1 mol% macrocycles and acid, cooperative interactions in the Friedel–Crafts reaction of indoles with imines can achieve yields of up to 99% and an enantiomeric excess of 99%. Yang et al. successfully synthesized β-cyclodextrin derivatives modified with triazole functional groups, which exhibited a strong coordination capability with metals. These derivatives effectively bound Cu(I) catalysts, facilitating the reaction of Cu(I)-catalyzed azide–alkyne cycloaddition to yield 1,4-disubstituted 1,2,3-triazoles in aqueous environments.155 The reaction exhibited a TON of up to 45000, and the catalyst retained high catalytic activity even after multiple cycles.
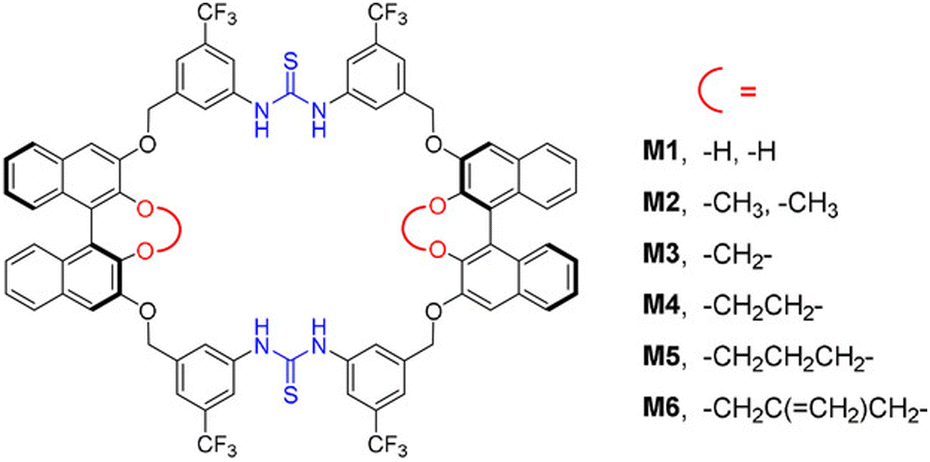 | ||
| Fig. 11 Structure of bis-diarylthiourea macrocycles. Reproduced with permission from ref. 154, copyright 2020, Wiley Publishing group. | ||
3.3 Phase separation of the products or catalyst
Phase separation methods such as distillation or extraction can also be applied in supramolecular catalytic systems to prevent product inhibition by separating products from the reaction system. This approach allows for the recovery and reuse of the host in a new solvent, maintaining the efficiency of the catalytic cycle.156 Dreimann et al. utilized methylated β-cyclodextrin in combination with a Rh-based catalyst for the continuous hydroformylation of 1-decene.157 The process employed vacuum distillation (at temperatures below 150 °C) to separate the products, enabling efficient turnover cycles with a high TON. This setup allowed the entire continuous process to operate stably for over 200 hours, achieving a chemical selectivity of more than 97% for the desired linear aldehyde product throughout this period. Bisht et al. developed spatially directional multivalent resorcin[4]arene cavitand glycoconjugates as phase transfer catalysts for organic reactions, including thiazole formation, thiocyanation, and Mannich reactions in aqueous media.158 After the reactions, the products were extracted with dichloromethane or ethyl acetate, allowing the aqueous solution containing the macrocyclic catalyst to be directly reused for up to five reaction cycles while maintaining high catalytic activity.Separation of the macrocyclic catalyst from the product can be also achieved through catalyst precipitation. By reducing the solubility of the catalyst to promote its precipitation, this method facilitated the separation of the product from the catalyst, thereby addressing the issue of product inhibition. Young et al. induced the precipitation of capsules in the reaction mixture by adding acetonitrile, followed by centrifugation, which effectively separated the majority of capsules with a recovery rate of up to 90%.159 The recovered capsule exhibited the same catalytic activity as the pristine sample.
3.4 Autocatalysis
Autocatalytic reactions use their products as catalysts for subsequent reactions, forming a self-sustaining catalytic system. This strategy not only avoids product inhibition but also significantly enhances reaction rates. Matile et al. reported the catalysis of epoxide-opening ether cyclizations within a series of capsules with π-basic but Brønsted acidic inner surfaces.160 Their study highlighted that autocatalysis was driven by the formation of hydrogen bonds between the transition state and the product on π-acidic surfaces, which activated both the nucleophile and the leaving group. This interaction enhances the reaction rate and promotes chemo- and diastereoselectivity, though enantioselectivity remains unattained. The unique structural properties of π-acidic surfaces of capsules facilitate these interactions, making them distinct from other catalytic environments. Matile et al. employed supramolecular capsules to catalyze House–Meinwald rearrangements to obtain new cyclic hemiacetals, discovering that the autocatalysis on anion–π catalysts was independent of substrate stereochemistry.1614. Challenge 3: incompatibility of catalysts or conditions in “one-pot” reactions
In the field of complex chemical synthesis, “one-pot” reactions in the aqueous phase have garnered significant attention due to their environmental friendliness, high efficiency, and ease of operation. However, a notable challenge arises from the incompatibility of catalysts or conditions in “one-pot” reactions. Specifically, the catalysts or reaction conditions required for two reactants or two consecutive steps often cannot coexist in the same aqueous environment. This issue severely restricts the development and application range of “one-pot” reactions in aqueous medium. Moreover, enzymes provide distinct catalytic microenvironments through cavity pockets to facilitate cascade reactions. Therefore, studying the catalysis of “one-pot” reactions also aids in understanding the relationship between the structure and function of enzymes. Supramolecular macrocycles inherently possess two distinct microenvironments—inside and outside the cavity—making it possible for two mutually exclusive catalytic conditions to coexist within a single system. Moreover, arranging different catalytic active sites on the macrocycle allows for the creation of a diverse and compatible catalytic environment where multiple catalytic conditions can coexist. Su et al. reported a [(Pd/Pt)6(RuL3)8]28+ nanocage for cascade reaction. They discovered that the highly positive charge (+28) on the surface of the nanocage induces protonation of the 24 imidazole groups on its framework (Fig. 12(a)), shifting ionization equilibrium and pKa, thus creating positively charged confined nanospaces in aqueous solution. This nanocage acted as a bifunctional acid–base catalyst, with extrinsic Brønsted acidity facilitating acid-catalyzed acetal hydrolysis in the bulk solution and intrinsic basicity promoting Knoevenagel condensation in cage-confined nanospaces.162 Li et al. synthesized chiral p-tert-butylcalix[4]crown-5 with a mononitro bridge substituent in a 1,3-alternate conformation, serving as a bifunctional organocatalyst for the asymmetric Henry reaction of aromatic aldehydes and nitromethane (Fig. 12(b)).163 The catalyst demonstrated good catalytic activity, achieving yields of up to 95% and enantioselectivity up to 22.3% ee. The secondary amino group functioned as a base to activate nitromethane, while the sulfonamido group acted as an acid to activate the aromatic aldehyde. | ||
| Fig. 12 Structure of (a) [(Pd/Pt)6(RuL3)8]28+ nanocage highlighting the protonation of the imidazole groups on the framework; (b) a bridging calix[4]crown-5 with two well-designed binding sites. Adapted from ref. 162 with permission, copyright 2021, National Science Review and ref. 163 with permission, copyright 2021, American Chemical Society. | ||
5. Conclusions and outlook
This minireview focuses on the challenges encountered for macrocyclic catalysis in the aqueous phase and their corresponding solutions. The specific issues addressed include (1) limited molecular interaction efficiency in aqueous environments, (2) product inhibition, and (3) incompatibility of catalysts or conditions in “one-pot” reactions. To tackle the first issue, the matching of hosts and substrates can be optimized, enabling the hosts to specifically recognize and effectively encapsulate substrates within their cavities. During the reaction process, the charge distribution and the characteristic of the binding site of macrocycles can influence the interaction efficiency of reaction species involved, including substrates, catalysts, transition states, and products. These effects on the reaction species may include, but are not limited to, facilitating desolvation, increasing local concentration, modifying photophysical properties, and adjusting their acidity and basicity. The macrocyclic cavity can preorganize two reactive groups or intramolecular fragments of the substrate, facilitating their adaptation to the internal environment of the host. Additionally, the host can undergo conformational changes or dynamic reversible assembly/disassembly processes to better accommodate the shape of the substrates. Furthermore, the host can facilitate interactions between two substrates or the catalytic active center and the substrate. For the second issue, one of the key factors is to ensure that the binding affinity of hosts for substrates is greater than for products, allowing substrates to displace products in the host cavity. Moreover, the host can be designed to preferentially bind to catalysts rather than to substrates or products, facilitating the smooth release of products from the macrocycle upon formation. Techniques such as distillation or extraction can be employed to separate products, or the addition of organic solvents and guest molecules can induce catalyst precipitation for effective separation. Additionally, employing an autocatalytic system may be a promising strategy to mitigate product inhibition. To address the third issue, developing macrocyclic catalysts with dual catalytic active sites or hosts featuring distinct internal and external environments can enable “one-pot” reactions that are otherwise hindered by incompatible catalysts or reaction conditions needed for two reactants or sequential steps.Despite significant progress in addressing the three issues of aqueous catalysis within macrocycle chemistry, further exploration of strategies to solve these issues remains a critical and important area of research. Additionally, a key challenge lies in monitoring and regulating the mass transfer of intermediates within catalytic systems, critical for preventing the intermediate deactivation and the inhibition of active sites. Supramolecular macrocycle catalysis has potential advantages in monitoring and regulating the mass transfer of intermediates: macrocycles have simple structures, well-defined cavity microenvironments, stable functionality resistant to deactivation, ease of design and synthesis, and clear structure–activity relationships. These features make them ideal simplified models for complex systems like enzymes, where substrate mass transfer channels are also crucial in catalysis research, thereby facilitating fundamental studies on chemical kinetics at the elementary reaction level. Moreover, kinetic studies of macrocycle systems have also advanced. For example, in-depth studies have been conducted on the regulation mechanisms of conformational changes in macrocyclic aromatics by small guest molecules or coexisting ions.164–166 Additionally, kinetic methodologies have been developed for elucidating the host–guest binding mechanisms in ternary and more complex systems.167–170 However, despite these research advancements, no related research has been reported in the field of macrocyclic catalysis regarding intermediate mass transfer. This may be due to two reasons: (1) in single-macrocycle catalysis systems, the specific stepwise reactions are carried out in aqueous media, making it difficult to precisely control the relative positions of the two catalytic sites; intermediates diffuse through the aqueous phase, which is relatively inefficient. (2) In complicated systems such as the supramolecular and biocatalytic coupled systems, the diversity and complexity of components, binding sites, interaction forces, binding extent and speed, as well as structural changes during the binding process are often too complex and varied, making it challenging to study the mass transfer of intermediates at the elementary reaction level. Future research should focus on developing advanced macrocyclic systems capable of controlling intermediate mass transfer more efficiently, emulating the complexity and specificity of enzyme systems. Furthermore, integrating supramolecular catalysts with dynamic, adaptable features could unlock new catalytic pathways, expanding the scope and applicability of macrocyclic catalysis in sustainable and green chemistry. We hope that this review will inspire new research directions to better facilitate macrocyclic catalysis in aqueous environments.
Author contributions
D. C. and H. T. designed the scope of the manuscript. D. Z. and H. T. wrote the original draft of the manuscript. L. W. and W. W. discussed and helped revise the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Key Research & Development Program of China (2016YFA0602900), the National Natural Science Foundation of China (22071066, 22071065), and the Natural Science Foundation of Guangdong Province (2024A1515011664).Notes and references
- R. Wolfenden, Chem. Rev., 2006, 106, 3379–3396 CrossRef CAS.
- A. Stank, D. B. Kokh, J. C. Fuller and R. C. Wade, Acc. Chem. Res., 2016, 49, 809–815 CrossRef CAS PubMed.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- K. A. Connors, Chem. Rev., 1997, 97, 1325–1358 CrossRef CAS PubMed.
- W. A. Freeman, W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367–7368 CrossRef CAS.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1991, 91, 1721–2085 CrossRef CAS.
- D. Cao, Y. Kou, J. Liang, Z. Chen, L. Wang and H. Meier, Angew. Chem., Int. Ed., 2009, 48, 9721–9723 CrossRef CAS PubMed.
- J.-M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS.
- Z. Dong, Q. Luo and J. Liu, Chem. Soc. Rev., 2012, 41, 7890–7908 RSC.
- B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC.
- M. Otte, ACS Catal., 2016, 6, 6491–6510 CrossRef CAS.
- K. Wang, J. H. Jordan, X. Y. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 132, 13816–13825 CrossRef.
- G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC.
- Z. Laughrey and B. C. Gibb, Chem. Soc. Rev., 2011, 40, 363–386 RSC.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2022, 123, 5262–5296 CrossRef PubMed.
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed.
- X. Yang, Q. Zhang, Y. Liu, M. Nian, M. Xie, S. Xie, Q. Yang, S. Wang, H. Wei, J. Duan, S. Dong and H. Xing, Angew. Chem., Int. Ed., 2023, 135, e202303280 CrossRef.
- J. R. Pliego, J. Phys. Chem. B, 2008, 113, 505–510 CrossRef.
- C.-P. Li and M. Du, Chem. Commun., 2011, 47, 5958–5972 RSC.
- K. Kanagaraj, M. Alagesan and C. Yang, Comprehensive Supramolecular Chemistry II, Elsevier, Oxford, 2017, pp. 11–60 Search PubMed.
- X. Lu, R. Kawazu, J. Song, Y. Yoshigoe, T. Torigoe and Y. Kuninobu, Org. Lett., 2021, 23, 4327–4331 CrossRef CAS.
- Z. Rapi, T. Nemcsok, P. Bagi, G. Keglevich and P. Bakó, Tetrahedron, 2019, 75, 3993–4004 CrossRef CAS.
- Q.-Q. Wang, Handbook of Macrocyclic Supramolecular Assembly, 2020, pp. 829–875 Search PubMed.
- S. Mecozzi and J. J. Rebek, Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- A. M. A. van Wageningen, P. Timmerman, J. P. M. van Duynhoven, W. Verboom, F. C. J. M. van Veggel and D. N. Reinhoudt, Chem. – Eur. J., 2006, 3, 639–654 CrossRef.
- F. N. Tehrani, K. I. Assaf, R. Hein, C. M. E. Jensen, T. C. Nugent and W. M. Nau, ACS Catal., 2022, 12, 2261–2269 CrossRef CAS.
- H. Lambert, Y.-W. Zhang and T.-C. Lee, J. Phys. Chem. C, 2020, 124, 11469–11479 CrossRef CAS.
- S. He, B. Huang, B. Xiao, S. Chang, M. Podalko and W. M. Nau, Angew. Chem., Int. Ed., 2023, 62, e202313864 CrossRef CAS PubMed.
- R. L. Spicer, A. D. Stergiou, T. A. Young, F. Duarte, M. D. Symes and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 2134–2139 CrossRef CAS PubMed.
- W. C. Geng, D. Zhang, C. Gong, Z. Li, K. M. Barraza, J. L. Beauchamp, D. S. Guo and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 12684–12688 CrossRef CAS PubMed.
- H. Xu and Q. Wang, Chin. Chem. Lett., 2019, 30, 337–339 CrossRef CAS.
- Y. Xia, Z. Song, Z. Tan, T. Xue, S. Wei, L. Zhu, Y. Yang, H. Fu, Y. Jiang, Y. Lin, Y. Lu, A. L. Ferguson and J. Cheng, Nat. Commun., 2021, 12, 732 CrossRef CAS PubMed.
- A. K. Das, N. Sepay, S. Nandy, A. Ghatak and S. Bhar, Tetrahedron Lett., 2020, 61, 152231 CrossRef CAS.
- N. i Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398–5401 CrossRef CAS.
- B. Mitra, G. Chandra Pariyar and P. Ghosh, RSC Adv., 2021, 11, 1271–1281 RSC.
- T. Uchikura, M. Oshima, M. Kawasaki, K. Takahashi and N. Iwasawa, Angew. Chem., Int. Ed., 2020, 59, 7403–7408 CrossRef CAS PubMed.
- Y. Yang, X. Jing, Y. Shi, Y. Wu and C. Duan, J. Am. Chem. Soc., 2023, 145, 10136–10148 CrossRef CAS.
- T.-R. Li, F. Huck, G. Piccini and K. Tiefenbacher, Nat. Chem., 2022, 14, 985–994 CrossRef.
- I. Némethová, D. Schmid and K. Tiefenbacher, Angew. Chem., Int. Ed., 2023, 62, e202218625 CrossRef.
- Z. Osman, L. Sperni, F. Fabris and A. Scarso, Tetrahedron Lett., 2024, 139, 154985 CrossRef CAS.
- K. Kanagaraj, J. Rebek and Y. Yu, Green Synth. Catal., 2023, 4, 7–9 CrossRef CAS.
- S. Merget, L. Catti, G. Piccini and K. Tiefenbacher, J. Am. Chem. Soc., 2020, 142, 4400–4410 CrossRef CAS.
- Q. Zhang and K. Tiefenbacher, Angew. Chem., Int. Ed., 2019, 58, 12688–12695 CrossRef CAS PubMed.
- E. V. Silveira, V. Nascimento, E. H. Wanderlind, R. F. Affeldt, G. A. Micke, L. Garcia-Rio and F. Nome, J. Org. Chem., 2019, 84, 9684–9692 CrossRef CAS PubMed.
- A. Fierro, L. García-Río, S. Arancibia-Opazo, J. J. Alcázar, J. G. Santos and M. E. Aliaga, J. Org. Chem., 2020, 86, 2023–2027 CrossRef.
- G. Olivo, G. Capocasa, O. Lanzalunga, S. Di Stefano and M. Costas, Chem. Commun., 2019, 55, 917–920 RSC.
- N. Rad, O. Danylyuk and V. Sashuk, Angew. Chem., Int. Ed., 2019, 58, 11340–11343 CrossRef CAS PubMed.
- J. J. Alcázar, N. Geue, V. Valladares, A. Cañete, E. G. Pérez, L. García-Río, J. G. Santos and M. E. Aliaga, ACS Omega, 2021, 6, 10333–10342 CrossRef.
- S. L. Silva, M. S. Valle and J. R. Pliego, J. Org. Chem., 2020, 85, 15457–15465 CrossRef CAS PubMed.
- L. J. Jongkind, M. Rahimi, D. Poole, S. J. Ton, D. E. Fogg and J. N. H. Reek, ChemCatChem, 2020, 12, 4019–4023 CrossRef CAS.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- C. Schöttle, E. Guan, A. Okrut, N. A. Grosso-Giordano, A. Palermo, A. Solovyov, B. C. Gates and A. Katz, J. Am. Chem. Soc., 2019, 141, 4010–4015 CrossRef.
- M. P. van der Helm, T. de Beun and R. Eelkema, Chem. Sci., 2021, 12, 4484–4493 RSC.
- W. Zhang, G. Cheng, G. L. Haller, Y. Liu and J. A. Lercher, ACS Catal., 2020, 10, 13371–13376 CrossRef CAS.
- D. M. Kaphan, M. D. Levin, R. G. Bergman, K. N. Raymond and F. D. Toste, Science, 2015, 350, 1235–1238 CrossRef CAS PubMed.
- T. Hong, Z. Zhang, Y. Sun, J.-J. Tao, J.-D. Tang, C. Xie, M. Wang, F. Chen, S.-S. Xie, S. Li and P. J. Stang, J. Am. Chem. Soc., 2020, 142, 10244–10249 CrossRef CAS.
- J. Tang, C. Chen, T. Hong, Z. Zhang, C. Xie and S. Li, Org. Lett., 2022, 24, 7955–7960 CrossRef CAS PubMed.
- M. Fairley, L. Davin, A. Hernán-Gómez, J. García-Álvarez, C. T. O'Hara and E. Hevia, Chem. Sci., 2019, 10, 5821–5831 RSC.
- H. M. Dodge, M. R. Kita, C.-H. Chen and A. J. M. Miller, ACS Catal., 2020, 10, 13019–13030 CrossRef CAS.
- J. M. Barlow, J. W. Ziller and J. Y. Yang, ACS Catal., 2021, 11, 8155–8164 CrossRef CAS.
- J.-J. Tao, J.-D. Tang, T. Hong, J.-W. Ye, J.-Y. Chen, C. Xie, Z. Zhang and S. Li, ACS Omega, 2021, 6, 35093–35103 CrossRef CAS PubMed.
- A. M. Camp, M. R. Kita, P. T. Blackburn, H. M. Dodge, C.-H. Chen and A. J. M. Miller, J. Am. Chem. Soc., 2021, 143, 2792–2800 CrossRef CAS.
- A. Najafian and T. R. Cundari, Inorg. Chem., 2019, 58, 12254–12263 CrossRef CAS.
- Y. Luo, G. Ouyang, Y. Tang, Y.-M. He and Q.-H. Fan, J. Org. Chem., 2020, 85, 8176–8184 CrossRef CAS PubMed.
- B. M. Bizzarri, A. Fanelli, L. Botta, C. Sadun, L. Gontrani, F. Ferella, M. Crucianelli and R. Saladino, RSC Adv., 2020, 10, 17185–17194 RSC.
- F. Wang, Y. Guo, Y. Zhang and P. Tang, ACS Catal., 2021, 11, 3218–3223 CrossRef CAS.
- G. Li, F. Trausel, M. P. van der Helm, B. Klemm, T. G. Brevé, S. A. P. van Rossum, M. Hartono, H. H. P. J. Gerlings, M. Lovrak, J. H. van Esch and R. Eelkema, Angew. Chem., Int. Ed., 2021, 60, 14022–14029 CrossRef CAS PubMed.
- Y. Wang, H. Zheng, G. Zhang, H. Wang, Y. Xiong and W. Liao, Eur. J. Inorg. Chem., 2022, e20220677 Search PubMed.
- T. W. Traut, Allosteric regulatory enzymes, Springer Science & Business Media, 2007 Search PubMed.
- Z. Lu, R. Lavendomme, O. Burghaus and J. R. Nitschke, Angew. Chem., Int. Ed., 2019, 58, 9073–9077 CrossRef CAS PubMed.
- T. G. Brevé, M. Filius, C. Araman, M. P. Helm, P. L. Hagedoorn, C. Joo, S. I. Kasteren and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 9340–9344 CrossRef.
- L. J. Jongkind, J. A. A. W. Elemans and J. N. H. Reek, Angew. Chem., Int. Ed., 2019, 58, 2696–2699 CrossRef CAS PubMed.
- A. L. J. Beckwith, Chem. Soc. Rev., 1993, 22, 143–151 RSC.
- S. Mei, Q. Ou, X. Tang, J.-F. Xu and X. Zhang, Org. Lett., 2023, 25, 5291–5296 CrossRef CAS PubMed.
- X. Tang, S. Mei, J.-F. Xu and X. Zhang, Chem. Commun., 2024, 60, 5286–5289 RSC.
- N. Luo, Y. F. Ao, D. X. Wang and Q. Q. Wang, Chem. – Eur. J., 2021, 16, 3599–3603 CAS.
- S. Gambaro, C. Talotta, P. Della Sala, A. Soriente, M. De Rosa, C. Gaeta and P. Neri, J. Am. Chem. Soc., 2020, 142, 14914–14923 CrossRef CAS PubMed.
- D. Liu, Y. Fan, M. Liu, Q. Ge, R. Gao and H. Cong, Org. Lett., 2024, 26, 3896–3900 CrossRef CAS.
- F. Bordignon, R. Calmanti, A. Perosa, F. Fabris and A. Scarso, ChemCatChem, 2024, 16, e202400278 CrossRef CAS.
- Y. Zhu, J. Rebek Jr and Y. Yu, Chem. Commun., 2019, 55, 3573–3577 RSC.
- J. Wang, T. A. Young, F. Duarte and P. J. Lusby, J. Am. Chem. Soc., 2020, 142, 17743–17750 CrossRef CAS PubMed.
- L.-D. Syntrivanis, I. Némethová, D. Schmid, S. Levi, A. Prescimone, F. Bissegger, D. T. Major and K. Tiefenbacher, J. Am. Chem. Soc., 2020, 142, 5894–5900 CrossRef CAS.
- Q. Liu, X. Tian, Y. Shen, X. Huang, K. Wang and X.-Y. Hu, RSC Adv., 2021, 11, 38115–38119 RSC.
- V. V. Welborn, W.-L. Li and T. Head-Gordon, Nat. Commun., 2020, 11, 415 CrossRef CAS.
- L. Xu, G. Fang, Y. Yu, Y. Ma, Z. Ye and Z. Li, Mol. Catal., 2019, 467, 1–8 CrossRef CAS.
- A. M. Raj, G. Sharma, R. Prabhakar and V. Ramamurthy, Org. Lett., 2019, 21, 5243–5247 CrossRef CAS.
- Y. Jin, M. Li, M. Liu, Q. Ge, H. Cong and Z. Tao, Eur. J. Org. Chem., 2022, e202101446 CrossRef CAS.
- Y. Kojima, M. Miura and K. Hirano, ACS Catal., 2021, 11, 11663–11670 CrossRef CAS.
- K. Wang, X. Cai, W. Yao, D. Tang, R. Kataria, H. S. Ashbaugh, L. D. Byers and B. C. Gibb, J. Am. Chem. Soc., 2019, 141, 6740–6747 CrossRef CAS.
- W. Yao, K. Wang, Y. A. Ismaiel, R. Wang, X. Cai, M. Teeler and B. C. Gibb, J. Phys. Chem. B, 2021, 125, 9333–9340 CrossRef CAS PubMed.
- R. Sumida, Y. Tanaka, K. Niki, Y. Sei, S. Toyota and M. Yoshizawa, Chem. Sci., 2021, 12, 9946–9951 RSC.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- K. Wang, J. H. Jordan and B. C. Gibb, Chem. Commun., 2019, 55, 11695–11698 RSC.
- X. Zhang, X. Guo and D. Liu, J. Mol. Liq., 2023, 384, 122266 CrossRef CAS.
- J.-M. Yang, Y. Yu and J. Rebek, J. Am. Chem. Soc., 2021, 143, 2190–2193 CrossRef CAS.
- M. Petroselli, V. Angamuthu, F.-U. Rahman, X. Zhao, Y. Yu and J. Rebek, J. Am. Chem. Soc., 2020, 142, 2396–2403 CrossRef CAS PubMed.
- K. de la Vega-Hernández, M. G. Suero and P. Ballester, Chem. Sci., 2024, 15, 8841–8849 RSC.
- N. Kishida, K. Matsumoto, Y. Tanaka, M. Akita, H. Sakurai and M. Yoshizawa, J. Am. Chem. Soc., 2020, 142, 9599–9603 CrossRef CAS PubMed.
- T. A. Young, V. Martí-Centelles, J. Wang, P. J. Lusby and F. Duarte, J. Am. Chem. Soc., 2019, 142, 1300–1310 CrossRef.
- R. Salvio and M. D'Abramo, Eur. J. Org. Chem., 2020, 6004–6011 CrossRef CAS.
- Y. Matsumoto, Y. Taguchi, N. Yoshida, S. Tokai, T. Maruyama and T. Iwasawa, Supramol. Chem., 2021, 33, 1–13 CrossRef.
- R. Salvio, S. Volpi, T. Folcarelli, A. Casnati and R. Cacciapaglia, Org. Biomol. Chem., 2019, 17, 7482–7492 RSC.
- G. Persiani, D. Sokolova, R. Ivanov, S. Merget, T. Maintok, D. Häussinger and K. Tiefenbacher, Eur. J. Org. Chem., 2024, e202400923, DOI:10.1002/ejoc.202400923.
- D. Thompson and C. A. Nijhuis, Acc. Chem. Res., 2016, 49, 2061–2069 CrossRef CAS PubMed.
- P. Cyganik, A. Terfort and M. Zharnikov, Nano Res., 2023, 17, 4231–4243 CrossRef.
- J. Ji, W. Wu, X. Wei, M. Rao, D. Zhou, G. Cheng, Q. Gong, K. Luo and C. Yang, Chem. Commun., 2020, 56, 6197–6200 RSC.
- D. Lisi, C. A. Vezzoni, A. Casnati, F. Sansone and R. Salvio, Chem. – Eur. J., 2023, 29, e202203213 CrossRef CAS.
- Y. Chen, D. Cao, L. Wang, M. He, L. Zhou, D. Schollmeyer and H. Meier, Chem. – Eur. J., 2013, 19, 7064–7070 CrossRef CAS PubMed.
- S. Wang, N. Li, B. Zhu and W. Guan, ACS Catal., 2024, 14, 12793–12805 CrossRef CAS.
- P. Jin, Y. Rong, W. Liang, X. Liang, R. Lu, W. Wu, M. Gou, Y. Tang, C. Yang and Y. Inoue, J. Org. Chem., 2021, 87, 1679–1688 CrossRef.
- D. A. Poole, S. Mathew and J. N. H. Reek, J. Am. Chem. Soc., 2021, 143, 16419–16427 CrossRef.
- V. Martí-Centelles, R. L. Spicer and P. J. Lusby, Chem. Sci., 2020, 11, 3236–3240 RSC.
- J. Xie, G. Pan, Y. Li and L. Lai, J. Chem. Phys., 2023, 158, 105102 CrossRef CAS PubMed.
- L. Liu, L. Wang, C. Liu, Z. Fu, H. Meier and D. Cao, J. Org. Chem., 2012, 77, 9413–9417 CrossRef CAS PubMed.
- M. Kondo, K. Nakamura, C. G. Krishnan, S. Takizawa, T. Abe and H. Sasai, ACS Catal., 2021, 11, 1863–1867 CrossRef CAS.
- K. Endo, H. Ube and M. Shionoya, J. Am. Chem. Soc., 2019, 142, 407–416 CrossRef.
- G. Cera, D. Balestri, M. Bazzoni, L. Marchiò, A. Secchi and A. Arduini, Org. Biomol. Chem., 2020, 18, 6241–6246 RSC.
- D.-N. Yan, L.-X. Cai, P.-M. Cheng, S.-J. Hu, L.-P. Zhou and Q.-F. Sun, J. Am. Chem. Soc., 2021, 143, 16087–16094 CrossRef CAS PubMed.
- M.-Y. Li, S. Wang, X.-T. Li, B. Zhu and W. Guan, Inorg. Chem., 2024, 63, 15906–15914 CrossRef CAS PubMed.
- R. Varadharajan, A. Mohan Raj and V. Ramamurthy, Photochem. Photobiol. Sci., 2020, 19, 976–986 CrossRef CAS PubMed.
- H. Guo, L. W. Zhang, H. Zhou, W. Meng, Y. F. Ao, D. X. Wang and Q. Q. Wang, Angew. Chem., Int. Ed., 2020, 59, 2623–2627 CrossRef CAS.
- D. Jansen, J. Gramüller, F. Niemeyer, T. Schaller, M. C. Letzel, S. Grimme, H. Zhu, R. M. Gschwind and J. Niemeyer, Chem. Sci., 2020, 11, 4381–4390 RSC.
- R. G. DiNardi, S. Rasheed, S. S. Capomolla, M. H. Chak, I. A. Middleton, L. K. Macreadie, J. P. Violi, W. A. Donald, P. J. Lusby and J. E. Beves, J. Am. Chem. Soc., 2024, 146, 21196–21202 CrossRef PubMed.
- Y. Satoh, L. Catti, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2019, 141, 12268–12273 CrossRef CAS PubMed.
- Q. Wang, W. Liang, X. Wei, W. Wu, Y. Inoue, C. Yang and Y. Liu, Org. Lett., 2020, 22, 9757–9761 CrossRef CAS PubMed.
- N. Rad and V. Sashuk, Chem. Commun., 2022, 58, 5249–5252 RSC.
- K. Ono, M. Niibe and N. Iwasawa, Chem. Sci., 2019, 10, 7627–7632 RSC.
- X. Wei, A. M. Raj, J. Ji, W. Wu, G. B. Veerakanellore, C. Yang and V. Ramamurthy, Org. Lett., 2019, 21, 7868–7872 CrossRef CAS.
- K. Yang, Z. Ma, H.-X. Tong, X.-Q. Sun, X.-Y. Hu and Z.-Y. Li, Chin. Chem. Lett., 2020, 31, 3259–3262 CrossRef CAS.
- X. Hu, F. Liu, X. Zhang, Z. Zhao and S. Liu, Chem. Sci., 2020, 11, 4779–4785 RSC.
- D. Gentile, G. Floresta, V. Patamia, A. Nicosia, P. G. Mineo and A. Rescifina, Org. Biomol. Chem., 2020, 18, 1194–1203 RSC.
- G. Norjmaa, J.-D. Maréchal and G. Ujaque, J. Am. Chem. Soc., 2019, 141, 13114–13123 CrossRef CAS PubMed.
- G. Xu, S. Leloux, P. Zhang, J. Meijide Suárez, Y. Zhang, E. Derat, M. Ménand, O. Bistri-Aslanoff, S. Roland, T. Leyssens, O. Riant and M. Sollogoub, Angew. Chem., Int. Ed., 2020, 59, 7591–7597 CrossRef CAS PubMed.
- C. Tugny, N. del Rio, M. Koohgard, N. Vanthuyne, D. Lesage, K. Bijouard, P. Zhang, J. Meijide Suárez, S. Roland, E. Derat, O. Bistri-Aslanoff, M. Sollogoub, L. Fensterbank and V. Mouriès-Mansuy, ACS Catal., 2020, 10, 5964–5972 CrossRef CAS.
- G. Cera, G. Giovanardi, A. Secchi and A. Arduini, Chem. – Eur. J., 2021, 27, 10261–10266 CrossRef CAS PubMed.
- X. Zhu, G. Xu, L. M. Chamoreau, Y. Zhang, V. Mouriès-Mansuy, L. Fensterbank, O. Bistri-Aslanoff, S. Roland and M. Sollogoub, Chem. – Eur. J., 2020, 26, 15901–15909 CrossRef CAS PubMed.
- A. Berlanga-Vázquez, R. A. Talmazan, C. A. Reyes-Mata, E. G. Percástegui, M. Flores-Alamo, M. Podewitz and I. Castillo, Eur. J. Inorg. Chem., 2022, e202200596 Search PubMed.
- E.-K. Xiao, X.-T. Wu, F. Ma, X. Feng, P. Chen and Y.-J. Jiang, Org. Lett., 2020, 23, 449–453 CrossRef.
- R. A. Talmazan, J. Refugio Monroy, F. del Río-Portilla, I. Castillo and M. Podewitz, ChemCatChem, 2022, 14, e202200662 CrossRef CAS.
- V. Marcos, A. J. Stephens, J. Jaramillo-Garcia, A. L. Nussbaumer, S. L. Woltering, A. Valero, J.-F. Lemonnier, I. J. Vitorica-Yrezabal and D. A. Leigh, Science, 2016, 352, 1555–1559 CrossRef CAS PubMed.
- N. Basílio, M. Pessêgo, A. Acuña and L. García-Río, ChemCatChem, 2019, 11, 5397–5404 CrossRef.
- E. T. Luis, A. I. Day, B. König and J. E. Beves, Inorg. Chem., 2020, 59, 9135–9142 CrossRef CAS.
- L. J. Jongkind and J. N. H. Reek, Chem. – Eur. J., 2020, 15, 867–875 CAS.
- L. W. Zhang, X. D. Wang, Y. F. Ao, D. X. Wang and Q. Q. Wang, Chem. – Eur. J., 2024, 30, e202400498 CrossRef CAS.
- J. Wei, L. Zhao, C. He, S. Zheng, J. N. H. Reek and C. Duan, J. Am. Chem. Soc., 2019, 141, 12707–12716 CrossRef CAS.
- D. Fiedler, H. van Halbeek, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2006, 128, 10240–10252 CrossRef CAS.
- Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2007, 129, 7000–7001 CrossRef CAS PubMed.
- W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231–236 CrossRef CAS PubMed.
- R. Ning, H. Zhou, S. X. Nie, Y. F. Ao, D. X. Wang and Q. Q. Wang, Angew. Chem., Int. Ed., 2020, 132, 10986–10990 CrossRef.
- J. Li, X. Feng, W. Wang, M. Guo, J. Yang, X. Li, R. Yang and B. Yang, Appl. Catal., A, 2023, 663, 119309 CrossRef CAS.
- J. Tiwari, S. Singh, D. Jaiswal, A. K. Sharma, S. Singh, J. Singh and J. Singh, Curr. Catal., 2021, 9, 92–101 CrossRef.
- K. U. Künnemann, L. Schurm, D. Lange, T. Seidensticker, S. Tilloy, E. Monflier, D. Vogt and J. M. Dreimann, Green Chem., 2020, 22, 3809–3819 RSC.
- A. A. Husain and K. S. Bisht, J. Org. Chem., 2020, 85, 9928–9935 CrossRef CAS.
- K. K. Shrestha, M. A. Hilyard, I. Alahakoon and M. C. Young, Org. Biomol. Chem., 2022, 20, 6646–6653 RSC.
- X. Hao, T. R. Li, H. Chen, A. Gini, X. Zhang, S. Rosset, C. Mazet, K. Tiefenbacher and S. Matile, Chem. – Eur. J., 2021, 27, 12215–12223 CrossRef CAS.
- H. Chen, T.-R. Li, N. Sakai, C. Besnard, L. Guénée, M. Pupier, J. Viger-Gravel, K. Tiefenbacher and S. Matile, Chem. Sci., 2022, 13, 10273–10280 RSC.
- K. Li, K. Wu, Y.-Z. Fan, J. Guo, Y.-L. Lu, Y.-F. Wang, G. Maurin and C.-Y. Su, Natl. Sci. Rev., 2021, 9, nwab155 CrossRef.
- X.-B. Peng, D. He, G.-N. Sun, Y. Yu, Y.-H. Ma, S.-A. Tang, W.-L. Dong and S.-Y. Li, J. Org. Chem., 2021, 86, 3952–3959 CrossRef CAS.
- J. C. Ji, X. Q. Wei, W. H. Wu, C. Y. Fan, D. Y. Zhou, K. Kanagaraj, G. Cheng, K. Luo, X. G. Meng and C. Yang, J. Am. Chem. Soc., 2022, 144, 1455–1463 CrossRef CAS PubMed.
- L. Yang, L. Zhang, M. Quan, J. S. Ward, Y.-L. Ma, H. Zhou, K. Rissanen and W. Jiang, Nat. Commun., 2020, 11, 2740 CrossRef CAS.
- H. Zheng, L. Fu, R. Wang, J. Jiao, Y. Song, C. Shi, Y. Chen, J. Jiang, C. Lin, J. Ma and L. Wang, Nat. Commun., 2023, 14, 590 CrossRef CAS PubMed.
- S. S. Thomas, H. Tang and C. Bohne, J. Am. Chem. Soc., 2019, 141, 9645–9654 CrossRef CAS PubMed.
- Y. Sun, H. Fang, X. Lin, X. Wang, G. Chen, X. Wang, Z. Tian, L. Yang, M. Utz and X. Cao, CCS Chem., 2022, 4, 557–565 CrossRef CAS.
- R. Shusterman-Krush, L. Grimm, L. Avram, F. Biedermann and A. Bar-Shir, Chem. Sci., 2021, 12, 865–871 RSC.
- D. Zhang, J. Cheng, L. Wei, W. Song, L. Wang, H. Tang and D. Cao, J. Phys. Chem. Lett., 2020, 11, 2021–2026 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |