
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsothiourea – catalyzed α-selective glycosylations†
Bhaswati
Ghosh‡
a,
Charles
Enlow‡
a,
Zhichen
Ma
a,
Ashley N.
Warden
a and
Abram J.
Axelrod
 *ab
*ab
aDepartment of Chemistry Purdue University, 720 Clinic Drive, West Lafayette, IN 47906, USA. E-mail: aaxelro@purdue.edu
bDepartment of Medicinal Chemistry and Molecular Pharmacology Purdue University, 575 Stadium Mall Drive, West Lafayette, IN 47907, USA
First published on 12th February 2025
Abstract
Herein, we present a catalytic strategy to efficiently form both α-1,2-cis and α-1,2-trans glycosyl linkages from either glycosyl bromide or chloride donors using the commercially available HyperBTM isothiourea in both good yields and selectivities.
Exerting stereocontrol in glycosylation is a fundamental goal within carbohydrate synthesis and is highly important for the efficient preparation of single-isoform oligosaccharides, enabling their interrogation in glycobiology and medicine. Considering the numerous variables inherent to glycosidic bond formation, the preparation of complex oligosaccharides remains a challenging endeavor.1 Accordingly, the development of methodologies that enable high degrees of stereocontrol have been an historical objective overall,2 and notably in the context of α-linked glycosides. While the synthesis of α-1,2-trans glycosides is often realized through anchimeric assistance with protecting groups, forming α-1,2-cis glycosides is less predictable due to stereoelectronic factors promoting a fluid SN1 and SN2 continuum.3 Historically, α-1,2-cis selectivity has been accomplished through the stoichiometric use of halide-ion,4 pyridine,5 amine,6 and phosphine7 nucleophiles, often requiring extended reaction times for full conversion. Numerous tactics have subsequently been advanced including the use of auxiliaries,8 directing groups,9 and exogenous modulators.10 Recently, α-1,2-cis glycosides have been prepared utilizing transition-metal catalysis11,12 or with main-group reagents.13,14 However, despite these advances, drawbacks exist regarding catalyst loadings, and specialized glycosyl donors. Additionally, requirements for high-dilution conditions and/or cryogenic temperatures are needed to ensure selectivity, and in the context of boron reagents, stoichiometric silver salts are necessary to activate the donor. Organocatalytic15 formation of α-linked glycans present an attractive alternative to metal-based methods, with Jacobsen disclosing a bio-inspired approach using anion-binding macrocycles,16 and Nguyen recently reporting C-2 symmetric phenanthrolines17 acting on glycosyl bromides as an evolution of pyridine nucleophiles.
Isothioureas, extensively studied as acyl transfer agents,18 represent an unexplored reagent in glycosylations and were identified as an attractive catalyst scaffold as they are strong Sigma donors.19 Herein, we disclose the discovery that isothioureas catalyze the formation of both α-1,2-cis glycosides and α-1,2-trans glycosides without the need for special directing groups or functionalization.
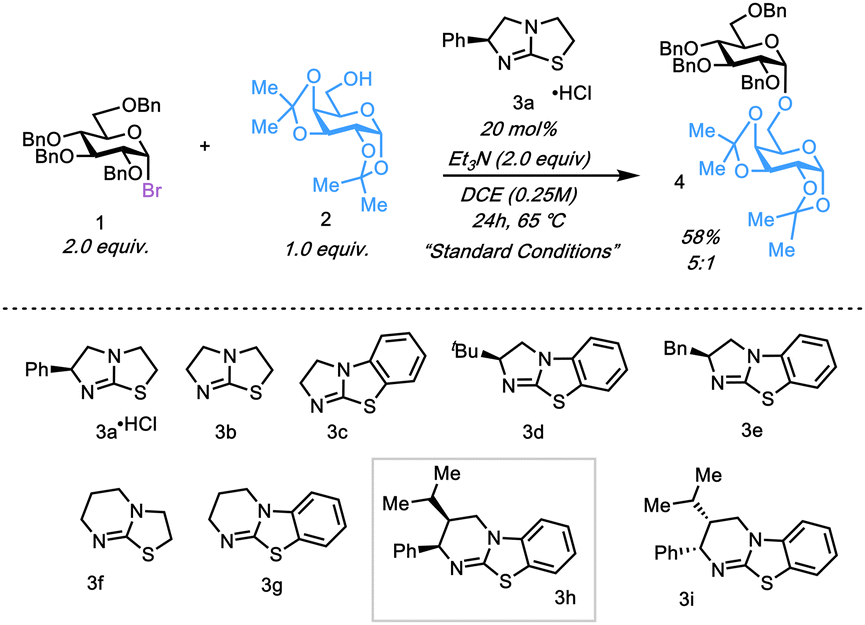
We began our investigation with glycosyl bromide 1 and galactose acceptor 2 as our study coupling partners with commercially available tetramisole hydrochloride 3a as the initial catalyst (Table 1). Glycosylation of 1 with 2 formed 4 in moderate yield (64%) but with low selectivity (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β) at 10 mol% catalyst loading in toluene at 50 °C in the presence of (iPr)2NEt (Fig. S1, ESI†). Systematic evaluation of reactant stoichiometries, catalyst loading, solvent, concentration, temperature, and choice of acid scavenger identified conditions that furnished 4 in 5:1 α:β selectivity in 58% yield. (Fig. S1, ESI†). A panel of other isothioureas was then screened for improved selectivity and yield. While catalyst 3b performed comparably to 3a, we observed that catalysts 3c–3e furnished disaccharide 4 in similar selectivities but in higher yields, suggesting the arene moiety augments reactivity. Catalyst 3f, a [6,5]-containing scaffold led to a lower yield, but with enhanced selectivity (6:1 α:β) relative to the [5,5]-scaffold isothiorureas, suggesting the [6,5] motif is important in conveying selectivity. Similarly, catalyst 3g, containing both an arene and the [6,5] scaffold mediated glycosylation with both higher selectivity and yield relative to 3a. Upon screening the commercially available HyperBTM isothioruea (3h) a significant increase in both yield and selectivity was observed, furnishing 4 in 83% yield at 9.5:1 α:β selectivity (Table 1). Further investigation showed that ent-HyperBTM (3i) provided 4 in both diminished selectivity and yield, suggesting the specific stereocenters of the phenyl and isopropyl groups within 3h play a crucial role to impart selectivity. Lowering the catalyst loading (entry 9, Table 1) of 3h to 10 mol% showed diminished yield, but with similar selectivity to entry 7.
:1 α:β) at 10 mol% catalyst loading in toluene at 50 °C in the presence of (iPr)2NEt (Fig. S1, ESI†). Systematic evaluation of reactant stoichiometries, catalyst loading, solvent, concentration, temperature, and choice of acid scavenger identified conditions that furnished 4 in 5:1 α:β selectivity in 58% yield. (Fig. S1, ESI†). A panel of other isothioureas was then screened for improved selectivity and yield. While catalyst 3b performed comparably to 3a, we observed that catalysts 3c–3e furnished disaccharide 4 in similar selectivities but in higher yields, suggesting the arene moiety augments reactivity. Catalyst 3f, a [6,5]-containing scaffold led to a lower yield, but with enhanced selectivity (6:1 α:β) relative to the [5,5]-scaffold isothiorureas, suggesting the [6,5] motif is important in conveying selectivity. Similarly, catalyst 3g, containing both an arene and the [6,5] scaffold mediated glycosylation with both higher selectivity and yield relative to 3a. Upon screening the commercially available HyperBTM isothioruea (3h) a significant increase in both yield and selectivity was observed, furnishing 4 in 83% yield at 9.5:1 α:β selectivity (Table 1). Further investigation showed that ent-HyperBTM (3i) provided 4 in both diminished selectivity and yield, suggesting the specific stereocenters of the phenyl and isopropyl groups within 3h play a crucial role to impart selectivity. Lowering the catalyst loading (entry 9, Table 1) of 3h to 10 mol% showed diminished yield, but with similar selectivity to entry 7.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield, αβ |
|
a All reactions were run at 0.1 mmol scale relative to 2. Yields refer to isolated, purified products, and (α:β) ratios were determined by 1H NMR analysis of unpurified reaction mixtures.
|
||
| 1 | catalyst 3b | 57%, 4.5:1 |
| 2 | catalyst 3c | 67%, 5.2:1 |
| 3 | catalyst 3d | 75%, 5.2:1 |
| 4 | catalyst 3e | 64%, 4.8:1 |
| 5 | catalyst 3f | 55%, 6:1 |
| 6 | catalyst 3g | 71%, 6:1 |
| 7 | catalyst 3h |
83%, 9.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
| 8 | catalyst 3i | 74%, 4.5:1 |
| 9 | catalyst 3h, 10 mol% | 65%, 10:1 |
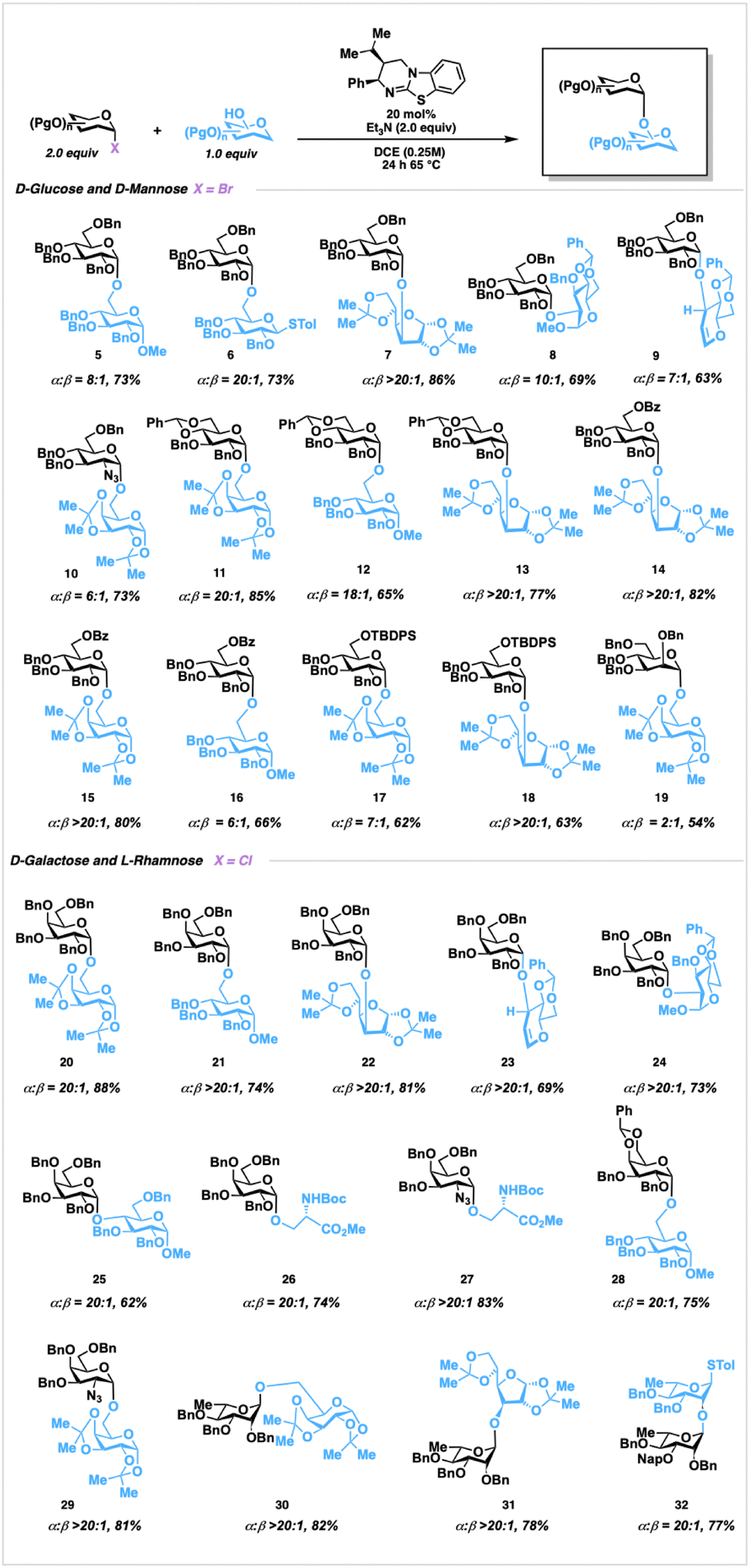
Experimentally, this transformation gives high – to complete α-selectivity and tolerates common sets of protecting groups, including benzyl, benzylidene, silyl, and benzoate groups. While use of chloride donors was found to be competent, reaction times were extended relative to the analogous bromide donors with slightly reduced selectivities (Fig. S3, ESI†). Exposure of 1 to primary alcohol acceptors furnished disaccharides 5 and 6 in good yields, and in good to outstanding alpha selectivity, compared with Lewis-acidic conditions (Fig. 1).20 Notably, in the formation of 6 no aglycone transfer was observed.21 Reactions of secondary hydroxyl acceptors, including challenging C-2 and C-3 hydroxyls, occurred in excellent selectivities forming 7 and 8. Glycals, typically coupled through alkylation conditions,22 reacted smoothly to form 9 with no hydrochlorination by-products. Additionally, 2-deoxy-2-azido sugars are tolerated, forming 10 in good yield, albeit at more modest levels of selection, potentially due to the electronic impact of the azido moiety influencing an SN1 pathway shift.23 We then explored benzylidene-protected donors, which can modify conformational plasticity24 of the donor, potentially impacting selectivity. To this end, benzylidene incorporation demonstrated augmented stereoselection, with both primary and secondary hydroxyl acceptors, furnishing disaccharides 11–13 in augmented chemical yields and α-selectivity relative to previous organocatalytic25 approaches and comparable with photoredox strategies, resepectively.11b Glycosylation with C-6 O-benzoyl and C-6 O-silyl protected donors are tolerated, as evidenced by the formation of 14–16 in both good yields and anomeric ratios. The presence of the sterically demanding C-6 O-TBDPS group attenuated reactivity, as reflected in lower yields of 17 and 18. Interestingly, the formation of 19 occurred with very low selectivity, suggesting that catalyst-glyca conformation is ineffective at promoting selectivity compared to the glucosyl donor-catalyst adducts.
 | ||
| Fig. 1
a
Reaction scope. aReactions run on 0.1 mmol scale relative to acceptor. Yields refer to isolated, purified products, (α:β) ratios were determined by 1H NMR analysis of unpurified reaction mixtures. | ||
Next, we examined galactosyl donors which were found to react with higher yields (Fig. S3, ESI†), selectivities, and were more stable relative to the bromides.
Disaccharides 20 and 21, for example, were formed with augmented selectivity in comparison to previous reports.25 Glycosylation with secondary acceptors formed the sterically encumbered adducts 22–24 with complete α-selectivity. In addition, disaccharide 25 containing a highly challenging α-1,4 linkage,26 was obtained in 20:1 α-selectivity. Serine nucleophiles are well tolerated to form O-glycan-type structures2726 and. Benzylidene protection of the chloride donor was well tolerated in furnishing 28, and azido disaccharide 29 was prepared in outstanding selectivity. Following this, we explored α-1,2-trans glycoside formation on rhamnoside donors without utilizing the influence of directing or protecting groups for selectivity. Rhamnosylation proceeded through use of the chloride donor and led to the formation of disaccharides 30 and 31 in both good yields and outstanding selectivities from the corresponding primary and secondary hydroxyl acceptors, respectively. Disaccharide 32 was obtained through glycosylation with an unreactive axial O-2 hydroxyl nucleophile. Expanding this reaction to catalyst-controlled oligosaccharide synthesis, we identified 43, a structural component of the of the Group B Streptococcus agalactiae cell wall as a suitable objective (Fig. 2).28 As oligorhamnans are found in bacterial pathogens, homogeneous access to specific glycoforms could enable investigation into their biological properties and potential as therapeutic targets. Proceeding, glycosylation of acceptor 33 with donor 35, furnished disaccharide 36 in 76% yield as a single α-anomer at gram scale. Removal of the tert-butyldimethylsilyl protecting group with TBAF furnished acceptor 37 in 91% yield, which was then reacted with 35 utilizing catalyst 3h to provide trisaccharide 38 in 78% yield as a single anomer. Following silyl deprotection, and isolation of trisaccharide acceptor 39, glycosylation with 35 generated tetrasaccharide 40 as a single anomer in 70% yield over two steps. Deprotection, followed by glycosylation of 41 with 42, furnished pentasaccharide 43 as a single anomer in 85% yield over two steps, and confirmed through both 1D and 2D NMR experiments.
 | ||
| Fig. 2 Synthesis of rhamnan pentasaccharide 42. | ||
Based on literature precedents of amines and heterocycles engaging glycosyl halides to form glycosyl ammoniums, we hypothesize that this reaction proceeds through a double SN2 reaction where in the first displacement the catalyst reacts with the glycosyl halide to form an equatorial ammonium species,17,29 In the second displacement, the glycosyl acceptor reacts to form a new α-linked glycosyl bond and releasing the catalyst (Scheme 1a). We were able to detect this ammonium species through 1H NMR spectroscopy by reacting 1 with catalyst 3h and within 1 hour identified two anomeric signals at δ5.92 ppm and δ5.73 ppm both in agreement based on previously observed glycosyl ammoniums,29 and the respective H1–H2 coupling constants are 4.05 Hz and 3.75 Hz, which are smaller than chair and suggestive of a different type of conformation (Fig. S4, ESI†). Additionally, we were able to isolate the catalyst-glycan adduct and characterize it through mass spectrometry, and subsequently react it with 2 to form 4 albeit in modest yield due to the moisture-sensitive adduct (Scheme 1b and Fig. S5, ESI†).
 | ||
| Scheme 1 (a) Proposed reaction mechanism, (b) mass spectrometry study. | ||
In conclusion, we have identified isothioureas as nucleophilic glycosylation catalysts and specifically the HyperBTM as highly effective for the selective formation of both α-1,2-cis and α-1,2-trans glycosidic linkages. This reaction proceeds without the need for assistance through protecting or directing groups and is operationally direct and mild. It is amenable to both glycosyl chloride and bromide donors with successful application to sterically encumbered linkages and an oligosaccharide. We are currently investigating both the structure of the putative ammonium intermediate, and the overall mechanism through both experimental and computational approaches. Simultaneously, we are currently expanding this platform to other glycan types and more complex oligosaccharides.
B. G., C. E. Z. M. and A. N. W. designed and conducted experiments, and collected and analyzed the data with A. J. A. A. J. A. supervised the research, conceived the project and wrote the manuscript with author input.
This research was supported through generous start-up funding from Purdue University. NIH P30 CA023168 is acknowledged for supporting shared NMR resources to the Purdue Institute for Cancer Research.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. Ling and C. S. Bennett, Asian J. Chem., 2019, 8, 802–813 CAS; (b) C. J. Crawford and P. H. Seeberger, Chem. Soc. Rev., 2023, 52, 7773–7801 RSC.
- (a) M. Liu, X. Qin and X.-S. Ye, Chem. Rec., 2021, 21, 3256–3277 CrossRef CAS PubMed; (b) L. Bohé and D. Crich, Carbohydrate Res., 2015, 403, 48–59 CrossRef PubMed.
- (a) S. S. Nigudkar and A. V. Demchencko, Chem. Sci., 2015, 6, 3859–3863 RSC; (b) Y. Fu, L. Bernasconi and P. Liu, J. Am. Chem. Soc., 2021, 143, 1577–1589 CrossRef CAS PubMed; (c) A. Ishiwata, K. Tanaka, J. Ao, F. Ding and Y. Ito, Front. Chem., 2022 DOI:10.3389/fchem.2022.972429.
- R. U. Lemieux, K. B. Hendriks, R. V. Stick and K. James, J. Am. Chem. Soc., 1975, 97, 4056–4062 CrossRef CAS.
- R. U. Lemieux and A. R. Morgan, J. Am. Chem. Soc., 1963, 85, 1889–1890 CrossRef CAS.
- A. C. West and C. Schuerch, J. Am. Chem. Soc., 1973, 95, 1333–1335 Search PubMed.
- L. Sun, X. Wu, D.-C. Xiong and X.-S. Ye, Angew. Chem., Int. Ed., 2016, 55, 8041–8044 CrossRef CAS PubMed.
- (a) T. J. Boltje, J.-H. Kim and G.-J. Boons, Nat. Chem., 2010, 2, 552–557 Search PubMed; (b) R. A. Mensink and T. J. Boltje, Chem. Eur. J., 2017, 23, 17637–17653 CrossRef CAS PubMed.
- (a) J. P. Yasomanee and A. V. Demchenko, Angew. Chem., Int. Ed., 2014, 53, 10453–10456 Search PubMed; (b) X. Liu, Y. Song, A. Liu, Y. Zhou, Q. Zhu, Y. Lin, H. Sun, K. Zhu, W. Liu, N. Ding, W. Xie, H. Sun, B. Yu, P. Xu and W. Li, Angew. Chem., Int. Ed., 2022, 61, e202201510 CrossRef CAS PubMed; (c) J. T. Walk, Z. A. Buchan and J. Montgomery, Chem. Sci., 2015, 6, 3448–3453 RSC.
- (a) Y. Kobayashi and T. Mukaiyama, Bull. Chem. Soc. Jpn., 2005, 78, 910–916 CrossRef; (b) S. K. Mulani, W.-C. Hung, A. B. Ingle, K.-S. Shiau and K.-K. T. Mong, Org. Biomol. Chem., 2014, 12, 1184–1197 RSC; (c) L. Wang, H. S. Overkleeft, G. A. Van Der Marel and J. D. C. Codée, J. Am. Chem. Soc., 2018, 140, 4632–4638 CrossRef CAS PubMed; (d) J. Park, S. Kawatkar, J.-H. Kim and G.-J. Boons, Org. Lett., 2007, 9, 1959–1962 CrossRef CAS PubMed.
- (a) Y. Feng, T. Guo, H. Yang, G. Liu, Q. Zhang, S. Zhang and Y. Chai, Org. Lett., 2022, 24, 6282–6287 CrossRef CAS PubMed; (b) C. Zhang, H. Zuo, G. Y. Lee, Y. Zou, Q.-D. Dang, K. N. Houk and D. Niu, Nat. Chem., 2022, 14, 686–694 CrossRef CAS PubMed; (c) S. Zhou, X. Zhong, A. Guo, Q. Xiao, J. Ao, W. Zhu, H. Cai, A. Ishiwata, Y. Ito, X.-W. Liu and F. Ding, Org. Lett., 2021, 23, 6841–6845 CrossRef CAS PubMed.
- X. Ma, Z. Zheng, Y. Fu, X. Zhu, P. Liu and L. A. Zhang, J. Am. Chem. Soc., 2021, 143, 11908–11913 CrossRef CAS PubMed.
- M. Tanaka, A. Nakagawa, N. Nishi, K. Iijima, R. Sawa, D. Takahashi and K. Toshima, J. Am. Chem. Soc., 2018, 140, 3644–3651 CrossRef CAS PubMed.
- K. A. D’Angelo and M. S. Taylor, J. Am. Chem. Soc., 2016, 138, 11058–11066 CrossRef PubMed.
- M. M. Nielsen, T. Holmstrøm and C. M. Pedersen, Angew. Chem., Int. Ed., 2021, 61, e202115394 CrossRef PubMed.
- Y. Park, K. C. Harper, N. Kuhl, E. E. Kwan, R. Y. Liu and E. N. Jacobsen, Science, 2017, 355, 162–166 CrossRef CAS PubMed.
- F. Yu, J. Li, P. M. DeMent, Y.-J. Tu, H. B. Schlegel and H. M. Nguyen, Angew. Chem., Int. Ed., 2019, 58, 6957–6961 CrossRef CAS PubMed.
- (a) V. B. Birman and X. Li, Org. Lett., 2006, 8, 1351–1354 CrossRef CAS PubMed; (b) G. Xiao, G. A. Cintron-Rosado, D. A. Glazer, B.-M. Xi, C. Liu, P. Liu and W. Tang, J. Am. Chem. Soc., 2017, 139, 4346–4349 CrossRef CAS PubMed; (c) J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109–2121 RSC; (d) A. J. Nimmo, J. Bitai, C. M. Young, C. McLaughlin, A. M. Z. Slawin, D. B. Cordes and A. D. Smith, Chem. Sci., 2023, 14, 7537–7544 RSC.
- B. Maji, C. Joannesse, T. A. Nigst, A. D. Smith and H. Mayr, J. Org. Chem., 2011, 76, 5104–5112 CrossRef CAS PubMed.
- S. S. Nigudkar, K. J. Stine and A. V. Demchenko, J. Am. Chem. Soc., 2014, 140, 11942–11953 Search PubMed.
- Z. Li and J. C. Gildersleeve, J. Am. Chem. Soc., 2006, 128, 11612–11619 CrossRef CAS PubMed.
- P. H. Seeberger, M. Eckhardt, C. E. Gutteridge and S. J. Danishefsky, J. Am. Chem. Soc., 1997, 119, 10064–10072 CrossRef CAS.
- P. R. Andreana and D. Crich, ACS Cent. Sci., 2021, 7, 1454–1462 CrossRef CAS PubMed.
- (a) D. Crich and L. Li, J. Org. Chem., 2007, 72, 1681–1690 CrossRef CAS PubMed; (b) R. A. Jeanneret, S. E. Johnson and M. C. Galan, J. Org. Chem., 2020, 85, 15801–15826 CrossRef CAS PubMed.
- M. Shaw, Y. Kumar, R. Thakur and A. Kumar, Beilstein J. Org. Chem., 2017, 13, 2385–2395 CrossRef CAS PubMed.
- F. Yu, J. L. Dickson, R. S. Loka, H. Xu, R. N. Schaugaard, H. B. Schlegel, L. Luo and H. M. Nguyen, ACS Catal., 2020, 10, 5990–6001 CrossRef CAS PubMed.
- (a) D. M. Beckwith and M. Cudic, Sem. Immunol., 2020, 47, 101389 CrossRef CAS PubMed; (b) S. D. Kuduk, J. B. Schwarz, X.-T. Chen, P. W. Glunz, D. Sames, G. Ragupathi, P. O. Livingston and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 12474–12485 CrossRef CAS.
- H. Guérin, S. Kulakauskas and M.-P. Chapot-Chartier, J. Biol. Chem., 2022, 298, 102488–102504 CrossRef PubMed.
- R. U. Lemieux and A. R. Morgan, Can. J. Chem., 1965, 43, 2205–2213 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05456c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |