
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-specific π-clamp-mediated radiosynthesis of 68Ga and 18F PET radiopharmaceuticals†
Thomas T. C.
Yue
 ab,
Jin Hui
Teh
ac,
Eric
Aboagye
c,
Michelle T.
Ma
b,
Truc T.
Pham
*b and
Nicholas J.
Long
*a
ab,
Jin Hui
Teh
ac,
Eric
Aboagye
c,
Michelle T.
Ma
b,
Truc T.
Pham
*b and
Nicholas J.
Long
*a
aDepartment of Chemistry, Molecular Sciences Research Hub, Imperial College London, White City Campus, Wood Lane, London W120BZ, UK. E-mail: n.long@imperial.ac.uk; Fax: +44 (0)20 7594 5804; Tel: +44 (0)20 7594 5781
bSchool of Biomedical Engineering and Imaging Sciences, King's College London, 4th Floor Lambeth Wing, St. Thomas’ Hospital, London SE17EH, UK. E-mail: truc.pham@kcl.ac.uk
cDepartment of Surgery & Cancer, Imperial Centre for Translational and Experimental Medicine, Imperial College London, Hammersmith Campus, UK
First published on 9th December 2024
Abstract
The π-clamp-mediated conjugation method, which enables site-specific modification of cysteine residues, is a promising strategy for developing well-defined radiolabelled biomolecules for positron emission tomography (PET) imaging. We have applied this method to site-specifically attach the macrocyclic chelators “NODA” and “NODAGA” to the somatostatin receptor 2-targeted peptide, octreotate. The resulting novel NODA-octreotate and NODAGA-octreotate compounds can be radiolabelled with either [18F]AlF− or [68Ga]Ga3+ respectively. In vivo PET imaging shows that the [68Ga]Ga3+-labelled derivative exhibits high stability and favourable pharmacokinetic properties.
Site-selective protein modification allows the precise installation of different functional entities such as drugs and radioactive labels onto biomolecules.1–3 This strategy produces well-defined bioconjugates, and enables fine-tuning of biomolecular properties.3,4 One significant application of this approach is in positron emission tomography (PET) imaging, where site-selective modification and radiolabelling can lead to improved imaging quality and accuracy, facilitating better diagnosis and monitoring of diseases.5,6
Cysteine (Cys) residues are amongst the most useful motifs for protein modification due to the high nucleophilicity of the thiol group. However, traditional Cys-based conjugation strategies utilising electrophilic reagents such as maleimides lack regioselectivity when multiple reactive Cys residues are available, resulting in ill-defined, heterogenous mixtures.7 Perfluoroaryl reagents are well known to undergo aromatic substitution reactions with Cys residues and the resulting conjugates are highly stable.8 Zhang et al. have demonstrated that regioselective Cys modification can be achieved using perfluoroaryl reagents through a “π-clamp” approach.9 This method utilises a four amino acid motif, Phe-Cys-Pro-Phe (FCPF), where the Cys thiol undergoes rapid and site-selective conjugation with perfluoroaryl reagents such as perfluorobiphenyl (PFBP) compounds in aqueous media. Notably, PFBP compounds do not react with other Cys residues, showcasing a high degree of regioselectivity and obviating the need for protecting group strategies or partial re-oxidation steps. This remarkable selectivity is facilitated by the hydrophobic interactions between the Phe side chains and the perfluoroaryl reagents, which creates a microenvironment that reduces the Cys thiol's pKa and decreases the enthalpy of activation for the SNAr reaction.10 Utilising the “π-clamp” strategy, perfluoroaryl-based reagents have been developed for the preparation of antibody–drug conjugates,9 protein–protein dimers,11 antibody-fluorophore conjugates,12,13 Re(I)- and Ir(III)-based peptide conjugates for photodynamic therapy.14,15 This method is yet to be deployed in radiopharmaceuticals for receptor-targeted molecular PET imaging, where it could have tremendous utility.
Herein, we report a new class of 1,4,7-triazacyclononane-1,4-diacetate (NODA) and 1,4,7-triazacyclononane, 1-glutaric acid-4,7-acetic acid (NODAGA) based bifunctional chelates, PFBP-NODA (1) and PFBP-NODAGA (2) (Scheme 1), suitable for aluminium-[18F]fluoride ([18F]AlF) and [68Ga]Ga3+ radiolabelling respectively, while simultaneously facilitating site-specific conjugation via the “π-clamp” approach. Receptor-targeted peptide-based radiotracers have exhibited extraordinary clinical utility in cancer treatment.16 This includes octreotate (TATE) derivatives, [68Ga]Ga-DOTA-TATE and [177Lu]Lu-DOTA-TATE, which target the somatostatin receptor subtype 2 (SSTR2) and are now routinely clinically used to diagnose and treat patients with neuroendocrine cancers.17–19 Here, a TATE peptide modified with an N-terminal FCPF-tag was identified as a suitable target to demonstrate the utility of the bifunctional chelates. Importantly, TATE contains a pair of competing Cys residues which would serve as a proof of concept for site-specificity.
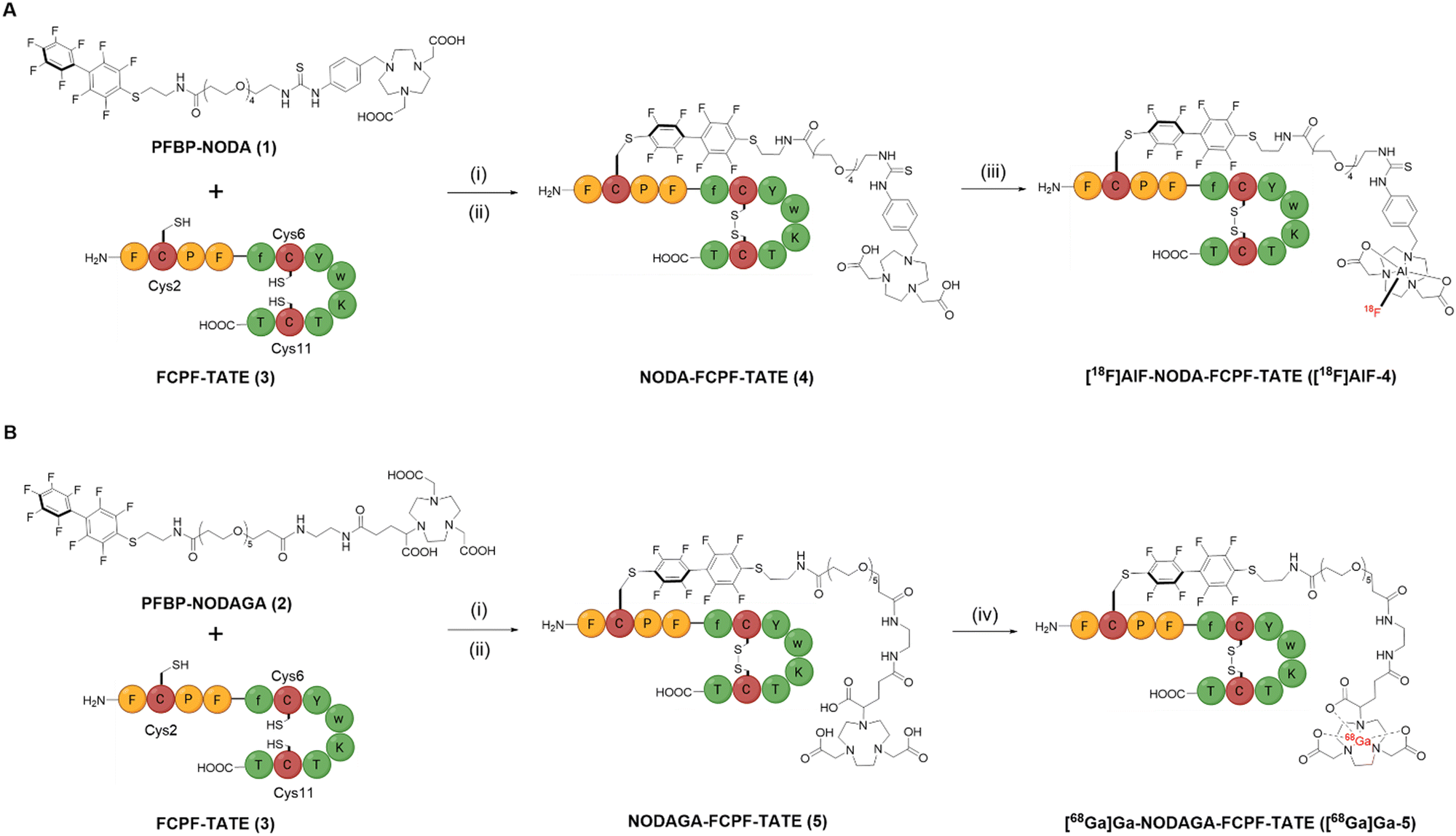 | ||
| Scheme 1 π-clamp mediated site-specific conjugation of FCPF-TATE (3) at Cys2 with (A) PFBP-NODA (1) and (B) PFBP-NODAGA (2), site-specificity confirmed by peptide mapping MS/MS experiment (Fig. S13, ESI†). Reaction conditions: (i) 20 mM TCEP, 0.2 M phosphate buffer, pH 8, 37 °C; (ii) 2,2′-dipyridyldisulphide, 2 M NH4OAc, pH 6, RT, 20 min; (iii) [18F]F−, AlCl3, 0.5 M NaOAc, pH 4.2, 100 °C, 20 min; (iv) [68Ga]Ga3+, 0.2 M NH4OAc, pH 6, 37 °C, 15 min. | ||
The di-acetate and tri-acetate N-substituted 1,4,7-triazacyclononane derivatives, NODA and NODAGA, were attached to a PFBP unit, via polyethylene glycol (PEG) linkers (Scheme 1). We selected PEG linkers so as to (i) enhance the solubility of the PFBP derivatives in aqueous environments, and (ii) potentially prolong circulation of any resultant radiotracer in blood circulation. Synthetic procedures and characterisation data for the new bifunctional chelators, 1 and 2, are included in the ESI.†
We then interrogated the reaction of 1 and 2 with the functionalised peptide, FCPF-TATE (3) (Scheme 1). Reaction of 3 (1 mM) with 5 mol eq. of 1 or 2 (0.2 M phosphate buffer, 20 mM TCEP, 37 °C) resulted in formation of the desired reduced conjugates after just 30 min (Fig. S9, ESI†). Importantly, peptide mapping MS/MS experiments (Fig. S13, ESI†) revealed preferential modification at the FCPF Cys2 residue.
Surprisingly, formation of a minor second species, which has a m/z signal corresponding to a dual-conjugated product, was also observed after 1 h, and in increasing amounts over time (Fig. S9, ESI†). Peptide mapping revealed this to be a Cys6-modified species (Fig. S14, ESI†). Cys6 is immediately flanked by aromatic D-Phe5 and a Tyr7 residues, alongside Phe4 and D-Trp8 adjacent to these. We postulate that π-stacking between the FfCYw sequence and PFBP motifs leads to secondary regioselectivity in conjugation. Whilst undesirable for our purposes, we envisage that this “secondary” selectivity could be deployed in future, for applications in which two different payloads are site-selectively attached to peptides.
We next optimised the reaction conditions, including reducing the equivalence of the bifunctional chelator used and lowering the reaction temperature, to minimise the formation of dual conjugated products (Fig. S10, ESI†). In this fashion, higher yields of the desired product were achieved, confirming that 1 and 2 each preferentially react with Cys2. The reduced TATE conjugates were subsequently oxidised using 2,2′-dipyridyldisulphide and the resulting conjugates 4 and 5 were isolated via preparative-HPLC and characterised by high-resolution MS (Fig. S11 and S12, ESI†).
Conjugate 5 was radiolabelled with generator-produced [68Ga]Ga3+, by reaction of [68Ga]Ga3+ with 5 in aqueous solution at 37 °C for 15 min, at pH 6. This resulted in 79.5 ± 6.8% radiochemical conversion (RCC) (n = 4), as determined by radio-HPLC. [68Ga]Ga-NODAGA-FCPF-TATE ([68Ga]Ga-5) was simply purified using a Sep-Pak® C18 cartridge, yielding the conjugate in >99% radiochemical purity (RCP) and 49.7 ± 8.5% decay corrected (d.c.) radiochemical yield (RCY) (n = 3) (Fig. S15c, ESI†). Additionally, [18F]AlF radiolabelling of 4 was achieved, by incubating 4 with AlCl3 (2 mM) and [18F]Fluoride in aqueous solution at 100 °C for 20 min, at pH 4.2. This resulted in 74% RCC as determined by radio-HPLC analysis. [18F]AlF-NODA-FCPF-TATE ([18F]AlF-4) was purified using a Sep-Pak® C18 cartridge, and was recovered in 60% d.c. RCY and 93% RCP (Fig. S16b, ESI†).
We then elected to take forward the novel [68Ga]Ga-5 radiotracer for biological evaluation. [68Ga]Ga-5 was stable in human serum, with only 2% dissociation of the radiotracer observed after incubation at 37 °C for 5 h (Fig. S15d, ESI†). The dissociated activity had a radio-HPLC retention time coinciding with unchelated [68Ga]Ga3+, indicating that any instability is likely a result of 68Ga decomplexation rather than instability at the conjugation site. [68Ga]Ga-5 also demonstrated excellent stability in human serum solution containing 5 mM glutathione (GSH) for up to 4 h, with no observed thiol exchange or 68Ga-decomplexation (Fig. S15e, ESI†).
To evaluate the binding of [68Ga]Ga-5 towards its target SSTR2 receptor, the radiotracer was incubated with SSTR2-expressing AR42J rat pancreatic cells. After 60 min incubation at 37 °C, 25.3 ± 3.0% of added [68Ga]Ga-5 radioactivity was associated with AR42J cells (Fig. 1). The tracer was found to internalise rapidly, with only 20% of cell-associated radioactivity remaining surface-bound after 15 min. These observations are consistent with reports of other radiolabelled TATE derivatives.20,21 When AR42J cells were co-incubated with [68Ga]Ga-5 and an excess of unmodified octreotide peptide (200-fold), to “block” SSTR2 receptors and assess specificity of radiotracer uptake, significantly reduced amounts of radioactivity were associated with cells: uptake measured ≤ 6% uptake at all time points (P < 0.001 at 15 min, P < 0.01 at 60 min). This indicates that [68Ga]Ga-5 uptake in AR42J cells is largely mediated by SSTR2 receptor binding.
 | ||
| Fig. 1 Uptake and internalisation of [68Ga]Ga-5 in SSTR2-positive AR42J cells (black), AR42J cells in the presence of 200-fold excess of octreotide (red) over time. Biological repeats were carried out in experimental triplicates (Mean ± SD, n = 3). See also Table S1 (ESI†). | ||
Next, the biodistribution of [68Ga]Ga-5 in healthy mice was studied using PET/CT imaging alongside ex vivo tissue counting experiments. [68Ga]Ga-5 (0.8–1.5 MBq) was administered intravenously (via tail vein) to healthy BALB/c mice (n = 4) and dynamic PET/CT images were obtained over 1 h (Fig. 2A). Quantitative PET imaging analysis indicated that [68Ga]Ga-5 was cleared from the blood pool predominantly hepatically, with uptake in the liver and kidney measuring 24.61 ± 4.79% ID per g and 8.81 ± 0.73% ID per g respectively. This is in contrast to reported radiolabelled TATE derivatives, where renal clearance was predominantly observed.20–23 This is likely due to the comparatively high lipophilic nature of the radiotracer: the logD7.4 of [68Ga]Ga-5 measured −0.40 ± 0.03. In addition, relatively high concentrations of 68Ga radioactivity remained in circulation at 1 h p.i. (13.81 ± 1.61% ID per g), again, contrasting the behaviour of previously reported radiolabelled TATE bioconjugates.20–23 The radiotracer's high lipophilicity alongside its PEG5 linker likely play a role in extending its in vivo plasma half-life.24 The π-clamp strategy, not only offers a facile method for site-specific radiolabelling, but may also serve as an effective strategy for extending the pharmacokinetic half-life of radiotracers when desired.25 Following PET/CT imaging, the mice were culled, and organs weighed and counted for radioactivity (Fig. 2B). Ex vivo biodistribution analysis at 1 h p.i. corroborated with the PET/CT data: 68Ga radioactivity concentrations in the liver and kidneys measured 54.59 ± 22.47% ID per g and 9.58 ± 5.81% ID per g respectively, while the concentration of 68Ga radioactivity in the blood measured 27.24 ± 3.52% ID per g. Crucially, minimal skeletal uptake was observed, with uptake in the bone measuring 0.95 ± 0.46% ID per g, indicating the high stability of the 68Ga-NODAGA complex in vivo.
 | ||
| Fig. 2 (A) Representative PET/CT maximum intensity projection (MIP) of healthy mice administered with [68Ga]Ga-5 at 1 h p.i. of radiotracer. (B) Ex vivo biodistribution of [68Ga]Ga-5 and [68Ga]Ga-5 co-administered with a blocking dose of octreotide (200-fold) in healthy mice at 1 h p.i. (Mean ± SD, n = 4). (C) RP-HPLC analysis of an ex vivo serum sample shows a single radioactive signal with retention time corresponding to [68Ga]Ga-5. See also Table S2 (ESI†). | ||
Healthy organs such as the adrenal glands, pancreas and the pituitary glands are known to express SSTR2, and previous studies have shown that radiolabelled TATE derivatives could visualise these organs in mice.21–23 In order to assess the specificity of the radiotracer in these SSTR2-expressing organs, a second group of mice (n = 4) were co-administered with [68Ga]Ga-5 and a blocking dose of octreotide peptide (200-fold) to saturate endogenous SSTR2 receptors (Fig. 2B). In mice administered only [68Ga]Ga-5, there was considerable uptake of the tracer in SSTR2-expressing healthy organs such as the pancreas (3.49 ± 1.06% ID per g), adrenal glands (6.45 ± 2.28% ID per g) and pituitary glands (6.04 ± 4.82% ID per g), and these values all decreased for animals co-administered a blocking dose (mean difference: pancreas 0.85 ± 0.65% ID per g; adrenal glands 1.15 ± 1.36% ID per g; pituitary glands 2.94 ± 2.76% ID per g). However, these differences were not statistically significant in this experiment. This could be due to the high amounts of activity remaining in circulation at 1 h p.i., preventing accurate determination of specific organ uptake. Future studies could evaluate the biodistribution at later time points where lower amounts of radioactivity remain in circulation, offering a clearer picture of specific SSTR2-mediated tissue retention. Serum fractions from these mice were also collected at 1 h p.i. of the radiotracer, and analysed by RP-HPLC (Fig. 2C), with the resulting radiochromatogram revealing a single signal, coinciding with that of [68Ga]Ga-5. This indicates the high metabolic stability of the radiotracer in vivo, with no evidence of 68Ga-decomplexation or thiol exchange observed.
In summary, we present the first application of the π-clamp method for developing well-defined radiopharmaceuticals. The novel bifunctional chelates PFBP-NODA and PFBP-NODAGA were shown to selectively conjugate to the FCPF tag amidst other cysteines without requiring protecting group strategies, using a modified octreotate derivative. In vitro studies revealed that [68Ga]Ga-NODAGA-FCPF-TATE exhibited high affinity toward SSTR2 receptors. In vivo, [68Ga]Ga-NODAGA-FCPF-TATE demonstrated excellent stability, primarily exhibiting hepatic clearance and an extended circulation half-life compared to other radiolabelled TATE derivatives. These promising results open up new possibilities for using the facile π-clamp method with other biomolecules and radionuclides.
This research was supported by a Cancer Research UK Career Establishment Award (C63178/A24959) (to M. T. M.), the EPSRC Programme for Next Generation Molecular Imaging and Therapy with Radionuclides (EP/S032789/1, ‘MITHRAS’), the Wellcome Multiuser Equipment Radioanalytical Facility (212885/Z/18/Z), and a Schrodinger Studentship from Imperial College London (to T. T. C. Y.).
Data availability
We can confirm that all the relevant research data is contained with the manuscript and electronic ESI.† No databases have been used and no references to such databases are contained in the manuscript or ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Farleigh, T. T. Pham, Z. Yu, J. Kim, K. Sunassee, G. Firth, N. Forte, V. Chudasama, J. R. Baker, N. J. Long, C. Rivas and M. T. Ma, Bioconjugate Chem., 2021, 32, 1214–1222 CrossRef CAS PubMed.
- T. T. C. Yue, Y. Ge, F. A. Aprile, M. T. Ma, T. T. Pham and N. J. Long, Bioconjugate Chem., 2023, 34, 1802–1810 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- C. Bai, E. E. Reid, A. Wilhelm, M. Shizuka, E. K. Maloney, R. Laleau, L. Harvey, K. E. Archer, D. Vitharana, S. Adams, Y. Kovtun, M. L. Miller, R. Chari, T. A. Keating and N. C. Yoder, Bioconjugate Chem., 2020, 31, 93–103 CrossRef CAS PubMed.
- L. K. Kristensen, C. Christensen, M. M. Jensen, B. J. Agnew, C. Schjöth-Frydendahl, A. Kjaer and C. H. Nielsen, Theranostics, 2019, 9, 4409–4420 CrossRef CAS PubMed.
- D. Vivier, K. Fung, C. Rodriguez, P. Adumeau, G. A. Ulaner, J. S. Lewis, S. K. Sharma and B. M. Zeglis, Theranostics, 2020, 10, 1746–1757 CrossRef CAS.
- K. J. Hamblett, P. D. Senter, D. F. Chace, M. M. C. Sun, J. Lenox, C. G. Cerveny, K. M. Kissler, S. X. Bernhardt, A. K. Kopcha, R. F. Zabinski, D. L. Meyer and J. A. Francisco, Clin. Cancer Res., 2004, 10, 7063–7070 CrossRef CAS.
- C. Zhang, E. V. Vinogradova, A. M. Spokoyny, S. L. Buchwald and B. L. Pentelute, Angew. Chem., Int. Ed., 2019, 58, 4810–4839 CrossRef CAS PubMed.
- C. Zhang, M. Welborn, T. Zhu, N. J. Yang, M. S. Santos, T. Van Voorhis and B. L. Pentelute, Nat. Chem., 2016, 8, 120–128 CrossRef CAS.
- P. Dai, J. K. Williams, C. Zhang, M. Welborn, J. J. Shepherd, T. Zhu, T. Van Voorhis, M. Hong and B. L. Pentelute, Sci. Rep., 2017, 7, 1–11 CrossRef.
- R. J. Taylor, M. Aguilar Rangel, M. B. Geeson, P. Sormanni, M. Vendruscolo and G. J. L. Bernardes, J. Am. Chem. Soc., 2022, 144, 13026–13031 CrossRef CAS PubMed.
- J. A. Baccile, P. J. Voorhees, A. J. Chillo, M. Berry, R. Morgenstern, T. J. Schwertfeger, F. M. Rossi and C. D. S. Nelson, ChemBioChem, 2021, 22, 3037–3041 CrossRef CAS.
- M. D. Lee, W. Y. Tong, T. Nebl, L. A. Pearce, T. M. Pham, A. Golbaz-Hagh, S. Puttick, S. Rose, T. E. Adams and C. C. Williams, Bioconjugate Chem., 2019, 30, 2539–2543 CrossRef CAS.
- L. C. C. Lee, A. W. Y. Tsang, H. W. Liu and K. K. W. Lo, Inorg. Chem., 2020, 59, 14796–14806 CrossRef CAS PubMed.
- P. K.-K. K. Leung, L. C.-C. C. Lee, T. K.-Y. Y. Ip, H.-W. W. Liu, S.-M. M. Yiu, N. P. Lee and K. K.-W. W. Lo, Chem. Commun., 2021, 57, 11256–11259 RSC.
- J. A. Jackson, I. N. Hungnes, M. T. Ma and C. Rivas, Bioconjugate Chem., 2020, 31, 483–491 CrossRef CAS.
- E. Pauwels, F. Cleeren, G. Bormans and C. M. Deroose, Am. J. Nucl. Med. Mol. Imaging, 2018, 8, 311–331 CAS.
- J. Strosberg, G. El-Haddad, E. Wolin, A. Hendifar, J. Yao, B. Chasen, E. Mittra, P. L. Kunz, M. H. Kulke, H. Jacene, D. Bushnell, T. M. O’Dorisio, R. P. Baum, H. R. Kulkarni, M. Caplin, R. Lebtahi, T. Hobday, E. Delpassand, E. Van Cutsem, A. Benson, R. Srirajaskanthan, M. Pavel, J. Mora, J. Berlin, E. Grande, N. Reed, E. Seregni, K. Öberg, M. Lopera Sierra, P. Santoro, T. Thevenet, J. L. Erion, P. Ruszniewski, D. Kwekkeboom and E. Krenning, N. Engl. J. Med., 2017, 376, 125–135 CrossRef CAS.
- M. S. Hofman, W. F. E. Lau and R. J. Hicks, RadioGraphics, 2015, 35, 500–516 CrossRef PubMed.
- M. T. Ma, C. Cullinane, K. Waldeck, P. Roselt, R. J. Hicks and P. J. Blower, EJNMMI Res., 2015, 5, 52 CrossRef PubMed.
- T. Maina, B. Nock, A. Nikolopoulou, P. Sotiriou, G. Loudos, D. Maintas, P. Cordopatis and E. Chiotellis, Eur. J. Nucl. Med., 2002, 29, 742–753 CrossRef CAS PubMed.
- M. Ginj, H. Zhang, B. Waser, R. Cescato, D. Wild, X. Wang, J. Erchegyi, J. Rivier, H. R. Mäcke and J. C. Reubi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16436–16441 CrossRef CAS PubMed.
- J. S. Lewis, M. R. Lewis, A. Srinivasan, M. A. Schmidt, J. Wang and C. J. Anderson, J. Med. Chem., 1999, 42, 1341–1347 CrossRef CAS.
- H. van de Waterbeemd, D. A. Smith, K. Beaumont and D. K. Walker, J. Med. Chem., 2001, 44, 1313–1333 CrossRef CAS.
- C. T. Mendler, T. Gehring, M. Schwaiger and A. Skerra, J. Nucl. Med., 2015, 56, 1112–1118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05223d |
| This journal is © The Royal Society of Chemistry 2025 |