
Activation of biochar by CO2 produced from the same pyrolysis process and captured in situ by CaO†
Mengjiao
Fan
a,
Yuewen
Shao
 a,
Chao
Li
a,
Yuchen
Jiang
a,
Yunyu
Guo
a,
Shu
Zhang
b,
Kai
Sun
c,
Yi
Wang
d and
Xun
Hu
*a
a,
Chao
Li
a,
Yuchen
Jiang
a,
Yunyu
Guo
a,
Shu
Zhang
b,
Kai
Sun
c,
Yi
Wang
d and
Xun
Hu
*a
aSchool of Material Science and Engineering, University of Jinan, Jinan, 250022, P. R. China. E-mail: Xun.Hu@outlook.com
bJoint International Research Laboratory of Biomass Energy and Materials, College of Materials Science and Engineering, Nanjing Forestry University, Nanjing, 210037, P. R. China
cSchool of Chemistry and Chemical Engineering, University of Jinan, 250022, P. R. China
dState Key Laboratory of Coal Combustion, Huazhong University of Science and Technology, P. R. China
First published on 6th December 2024
Abstract
Herein, recycled use of CO2 was proposed by using CaO to collect CO2in situ in the form of CaCO3 in the pyrolysis of willow branch (WB) at 600 °C. Subsequently, the formed CaCO3 and biochar together were heated to 900 °C to activate the biochar with both CO2 and CaO released.
CO2 is a gas that causes the greenhouse effect, which is mainly produced from the combustion of fossil fuels.1 Replacing fossil fuels with biomass can mitigate CO2 emission, as biomass is carbon-neutral.2,3 Nevertheless, the thermal conversion of biomass via processes like pyrolysis, gasification and activation also generate CO2 through intense deoxygenation reactions, due to the high O/C ratio in biomass.1 If the CO2 can be adsorbed in situ in pyrolysis, the heating value of pyrolytic gases could be enhanced.4 More importantly, negative CO2 emission could be achieved in the overall process. After adsorbing CO2, the next question would be how to further utilize it. It is well-known that CO2 is a reactive gas used in physical activation of biomass for the generation of activated carbon (AC).5,6 The adsorption of CO2 in the pyrolysis of biomass could also be regarded as a method for condensing or enriching CO2, which is highly desirable for the subsequent use of CO2 in activation.7–9 Thus, another challenge is finding a material to adsorb CO2 in pyrolysis at the typical temperature of ca. 600 °C, while releasing CO2 in the subsequent activation of biomass at ca. 800 °C.
CaO meets these criteria as it can absorb CO2 formed from pyrolysis to form CaCO3, which further decomposes to form CaO and CO2 at higher temperature.10,11 The released CO2 further etches carbonaceous materials, creating micropores.12,13 This process converts CaCO3 again into CaO, as demonstrated in Scheme 1.
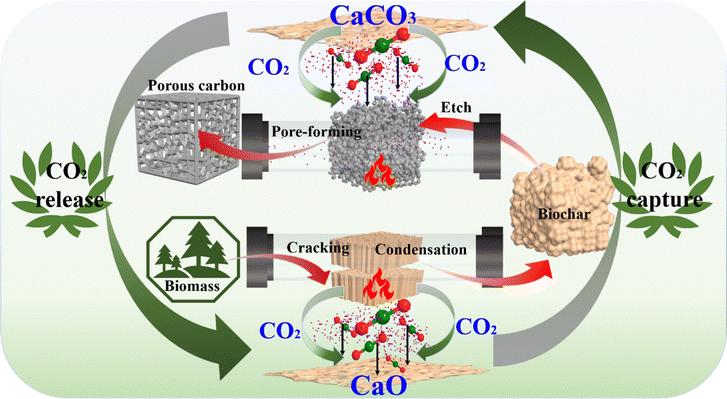 | ||
| Scheme 1 Schematic diagram of the concept of capturing CO2 from pyrolysis with CaO for subsequent use in the activation of biochar. | ||
To verify this hypothesis, pyrolysis of the mixture of CaO and willow branch (WB) was performed herein at 600 °C for 0.5 h. The obtained solid (CaCO3 from the reaction between CaO and CO2 together with biochar from pyrolysis) was further heated to 900 °C and held for 1 h, aiming to investigate the capability of the in situ formed CaCO3 for creating porous structures in the biochar through its further decomposition to form CO2 for gasification. The produced biochar/AC, bio-oil and gases in the first step of pyrolysis and second step of activation were analyzed in detail.
The synthesis process and supplementary characterization of the produced bio-oil and biochar/AC are shown in the ESI.†
The presence of CaO showed evident effects on the distribution of products from pyrolysis at 600 or 900 °C (Table 1). The yield of biochar from pyrolysis at 600 °C was obviously higher in the presence of CaO (21.3% vs. 20.8%). This could be attributed to three reasons: (1) the reaction between the CO2 derived from pyrolysis and CaO formed CaCO3, which will be verified later and contributed to increased weight of the samples even after pickling. (2) CaO as an alkaline oxide catalyzed the condensation of organics on the nascent biochar, resulting in retainment of more solid products.14,15 (3) CaO promoted deoxygenation of volatiles, during which some carbon deposits could form and increased formation of biochar at the expense of bio-oil. CaO also catalyzed gasification or cracking of the oxygen-containing organics in the bio-oil from its strong basicity, which reduced the yield of bio-oil. In terms of gases, the yields of CO2 were obviously lower in the pyrolysis in the presence of CaO, which was due to their reactions for the formation of CaCO3. The addition of CaO also enhanced the CO2 reforming,1 which together with the capture of CO2 jointly contributed to the lower CO2 yield. The low concentration of CO2 further facilitated the water–gas shift to form more H2.16 However, the promotion of the water–gas shift reaction would be expected to decrease the CO yield. Interestingly, such a decrease was not observed in the pyrolysis of WB. The reason for this might be the abundant lignin content in WB, as CaO could catalyze the decomposition of Ar–OH to increase the yield of CO.1 Intense decarbonylation reactions catalyzed by CaO also enhanced the formation of CO.4 Additionally, the presence of CaO promoted the fracture of the long chain C–C structure and demethylation reaction,17 forming more CH4 and C2H4 at 600 °C.
| Feedstock | Distribution of product (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Biochar/ACa | Bio-oil | Gas | CO | CH4 | CO2 | H2 | C2H4 | |
| a The yield of AC after pickling based on only biomass. | ||||||||
| WB-600 | 20.8 | 54.9 | 24.2 | 8.2 | 1.6 | 14.1 | 0.15 | 0.28 |
| WB + CaO-600 | 58.5 (21.3)a | 21.7 | 19.8 | 10.8 | 3.8 | 3.2 | 1.5 | 0.59 |
| Char-900 | 84.5 (17.6)a | 12.1 | 3.4 | 2.1 | 0.10 | 0.95 | 0.35 | 0 |
| Char + CaO-900 | 74.2 (6.6)a | 0.5 | 25.3 | 16.0 | 0.09 | 8.8 | 0.41 | 0 |
With the increase of the temperature to 900 °C for further pyrolysis of biochar, little bio-oil and gases were generated and carbonaceous solid was observed as the main product in the absence of CaO. This was related to the increased resistivity of biochar towards cracking from the enhanced aromatic degree via pyrolysis at 600 °C. In comparison, much more gases and little bio-oil were formed in the further pyrolysis of biochar containing CaO at 900 °C. The substantial increase in gas production was due to the formation of more CO2 from the decomposition of CaCO3 and also more CO from the cracking or gasification of carbonaceous intermediates with CO2. The high activity of CaO for cracking also led to the formation of a limited amount of bio-oil. Evidently, CaO catalyzed condensation reactions during pyrolysis at 600 °C, but cracking reactions together with CO2 release at 900 °C. The formation of bio-oil in the presence or absence of CaO was further analyzed.
The light compounds in the bio-oil from the pyrolysis at 600 °C were identified with GC-MS, and the relative abundances of grouped organics or specific compounds are shown in Fig. 1a and Table S1 (ESI†), respectively. As shown in Fig. 1a, the presence of CaO remarkably suppressed the generation of the majority of organics, including sugars and derived acids, esters, aldehydes, alcohols and ethers in bio-oil, but enhanced the production of some aromatics and alkanes. Hence, the increase of the yields of biochar resulted from not only the reactions between CaO and CO2, but also from many organics in bio-oil. Specifically, CaO could react with carboxyl-containing components of bio-oil, such as acids and esters, to form calcium carboxylate intermediates,1,18 which could decompose to produce ketones, as evidenced by the retainment of some ketones in the bio-oil. Furans were mainly formed from dehydration of hemicellulose and cellulose under especially acidic conditions.19 CaO and nascent CaCO3 inhibited the dehydration of sugars, due to their alkalinity. Similarly, CaO did not suppress the formation of nitrogen-containing compounds with alkalinity. The degradation of lignin to form phenolics was also interrupted by the presence of CaO. CaO could also react with phenolic compounds to form hydrocarbon salts that further decomposed,4,20 resulting in the decrease of the phenol content in the bio-oil. Besides, CaO could enhance aldol condensation to form new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, as evidenced by the emerge of aromatics and alkanes. The heavy component in bio-oil was further characterized by UV fluorescence spectroscopy.
C, as evidenced by the emerge of aromatics and alkanes. The heavy component in bio-oil was further characterized by UV fluorescence spectroscopy.
 | ||
| Fig. 1 (a) and (b) GC-MS and 2D UV analysis of bio-oil from the pyrolysis of willow branch with/without CaO at 600 °C; (c) and (d) 3D UV fluorescence analysis of the bio-oil produced at 600 °C; (e) and (f) 3D UV fluorescence analysis of the bio-oil produced at 900 °C. | ||
As shown in Fig. 1b, two main fluorescence peaks at 308 and 321 nm and one shoulder peak at 342 nm were observed in the pyrolysis of WB at 600 °C. The presence of CaO inhibited the formation of heavier organics with π-conjugated structures, reflected by the general decrease of the intensity of all the fluorescence peaks. This inhibition was achieved via direct reactions between CaO and both the light and heavier organics. For the bio-oil from further pyrolysis of the biochar at 900 °C, less aromatics with lower fluorescence intensity were observed. The Excitation–Emission-Matrix spectra of bio-oil shown in Fig. 1c–f exhibited similar results. The fluorescence peaks of the bio-oils mainly distributed in regions II and IV, which were ascribed to the organics with tyrosine-like structures and polycyclic aromatic structures.21,22 The presence of CaO in the further pyrolysis of biochar at 900 °C did lead to an increase of the fluorescence intensity in Region IV, but the increase was not significant. This was due to the combined effects of enhanced gasification of both organics on the surface of biochar and volatile matters.
In the feedstock mixed with CaO, CaO adsorbed moisture in WB to form Ca(OH)2. During the process of pyrolysis at 600 °C, Ca(OH)2 further decomposed back to CaO via dehydration, and CaO reacted with the CO2 generated from the pyrolysis of WB to form CaCO3. This formed CaCO3 further decomposed into CaO and CO2 at 900 °C, as evidenced by XRD and FTIR (Fig. 2a and b). This confirmed the concept of using CaO to collect CO2 from pyrolysis for further use of the CO2 for activation in the higher temperature region.
 | ||
| Fig. 2 (a) and (b) XRD patterns and FTIR spectra of the biochars and ACs of willow branch with/without CaO; (c) tetracycline (50 mg L−1) adsorption over the biochars and ACs of willow branch; SEM images of (d) and (e) biochars from the pyrolysis of willow branch with/without CaO at 600 °C and (f) and (g) biochars and ACs from subsequent activation of willow branch-derived char with/without CaO at 900 °C. | ||
The released CO2 etched the carbonaceous component, leading to the formation of micropores that contributed to an increase in the specific surface area of AC (136.2 m2 g−1 in blank test versus 469.6 m2 g−1), referred to as the “internal activation effect”.10,23 The continuous release of CO2 also accelerated the merging of micropores, resulting in the formation of hierarchical porous structure. Besides, the resulting AC had a relatively stable carbon skeleton with more mesoporous and even part of macropores, due to the template role of CaCO3 and CaO. This significantly enhanced the pore diameters (3.3 versus 6.7 nm) and also the pore volume (0.11 versus 0.79 cm3 g−1) drastically. This rendered superior performance of the AC derived from CaO activation for adsorption of tetracycline (Fig. 2c). The N2 physical adsorption isotherms of both biochar and AC from the activation of WB with/without CaO belonged to the combination of type I and IV isotherms, indicating the co-existence of micropores and mesopores (Fig. S1a, ESI†). This was further verified by the pore size distribution curve in Fig. S1b (ESI†).
Besides, the biochar produced by the pyrolysis of only WB at 600 °C retained the biological features of WB including the tubular and fold structures (Fig. 2d), while the further pyrolysis at 900 °C led to more broken structures due to the more intensive cracking reactions at higher temperature (Fig. 2e). The addition of CaO destroyed the biological structure of WB even in the biochar produced from pyrolysis at 600 °C (Fig. 2f). The catalytic effects of CaO for polymerization and the reactions with the majority of organics in volatiles further reshaped the morphology of the biochar (Fig. 1g).
As shown in Fig. 3, the in situ DRIFTS results showed that remarkable dehydration and dehydrogenation reactions above 400 °C reduced the abundance of –OH and aliphatic C–H during the pyrolysis of WB, forming more C–H in alkenes and CC.24 CO species could be formed at lower temperatures and the elevated temperature also resulted in its transformation into CC, C–O–C or adsorbed CO2. For the pyrolysis of WB in the presence of CaO, the CaO adsorbed moisture and existed in the form of Ca(OH)2. The Ca species also promoted the dehydrogenation and dehydration reactions, resulting in the formation of abundant C–H even at lower temperature (ca. 200 °C). The latency of the CO2 adsorption peak and the emergence of CaCO3 further verified the transformation of CaO. Additionally, the abundance of CO was much lower than the counterpart biochar, which was due to the reactions of the CO-containing organics with CaO, as evidenced by the decreased yields of ketones and carboxylic acids (Fig. 1a and Table S1, ESI†). As for the biochar, the pyrolysis at 600 °C formed stable structures and the intensity of the functionalities did not vary much in the further heating process during characterization. The decomposition of CaCO3 in the subsequent pyrolysis at 900 °C exhibited the reverse tendency for evolution of functionalities comparted to the pyrolysis of WB in the presence of CaO. CaCO3 further enhanced the fragmentation of the aromatic ring structures at high temperature, due to the etching reactions caused by the released CO2, resulting in negligible adsorption of C–H and C–O–C. A potential reaction mechanism of the whole process has been proposed and is shown in Scheme S1 (ESI†).
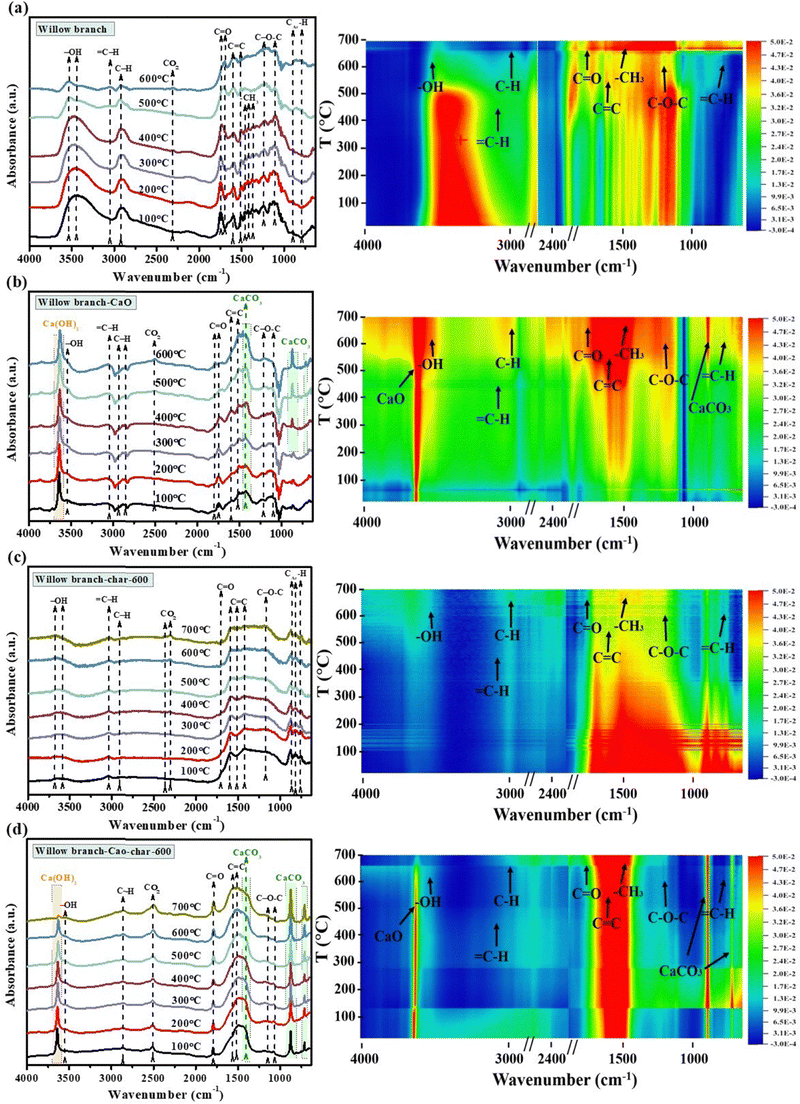 | ||
| Fig. 3 In situ DRIFTS characterization of the pyrolysis process. (a) Pyrolysis of willow branch; (b) pyrolysis of a mixture of willow branch and CaO; (c) subsequent activation of the willow branch-derived char; (d) subsequent activation of the mixture-derived char. | ||
During the process of preparing AC, the addition of CaO inevitably increased the environmental burden compared to pyrolysis of the feedstock only. Concrete manifestation could be demonstrated in the following aspects. Firstly, heating the device required the loading of more feedstock that contained CaO, which consumed more energy. Secondly, a pickling step was required as post-treatment of ACs. The use of acid for pickling and the formation of waste water in this process also had a negative impact on the environment. Nevertheless, the synthesis of AC from CaO activation generally followed the concept of green chemistry, reflected in the in situ activation of CaCO3 and the fact that CaO could be regarded as zero loss throughout the process.
The assessment of the potential environmental impacts of the activation of WB with CaO based on the TRACI 2.1 method is further shown in Fig. 4b. The results showed that the environmental effects of introducing CaO were mainly reflected in the ecotoxicity, but had little impact on acidification, eutrophication, and respiratory effects, non-carcinogenicity, ozone depletion and other aspects. CO2 was first absorbed by CaO and then released by subsequent decomposition of CaCO3, which together caused a moderate effect on global warming. By comparing the results of 20- and 100-year GHG assessments (Fig. 4c), it was found that the cumulative environmental damage from CaO activation increased with a small gradient. How to further reduce the environmental influence of CaO activation requires further exploration.
 | ||
| Fig. 4 (a) System boundaries for the preparation of ACs from CaO activation; (b) environmental impacts of the activation process; (c) effects of different activation processes on the greenhouse effect (20 years and 100 years). | ||
In summary, CaO could capture CO2 from WB pyrolysis to form CaCO3. CaCO3 further decomposed to CaO and CO2, which gasified biochar, generating the final AC. The enhanced gasification induced by both CaO and CO2 markedly increased the pore size, pore volume and specific surface area of AC with more mesopores and even macropores. CaO possessed high capability for creating macropores through merge of the micropores. The in situ IR characterization showed that the Ca species promoted dehydrogenation and cracking of the hydroxyl group, but consumed CO-containing organics and further enhanced the fragmentation of aromatic structures through gasification reactions. These destroyed the biological structure of the feedstock and transformed their morphologies through the reactions between CaO and organics in volatiles and on the surface of the biochar, resulting in the destruction of skeleton structure. The abundant mesopores and macropores created in these processes resulted in high performance for the adsorption of tetracycline. However, the high porosity of AC was obtained at the expense of high carbon yield. How to simultaneously achieve high specific surface area and high mass yield of AC could be further explored.
This work was supported by the National Natural Science Foundation of China (Grant No. 52276195), and Major Project of Hubei Province Science and Technology (Grant No. 2023BCA006).
Data availability
All data generated or analyzed during this study are included in this published article (and its ESI,† files).Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Zhang and J. Wang, et al. , Fuel Process. Technol., 2023, 247, 10775 Search PubMed.
- J. Yang and Y. Wu, et al. , Fuel, 2023, 331, 125994 CrossRef CAS.
- H. Li and Y. Wang, et al. , J. Cleaner Prod., 2021, 291, 125826 CrossRef CAS.
- X. Chen and Y. Chen, et al. , Bioresource Technol., 2017, 233, 15–20 CrossRef CAS PubMed.
- Y. Jiang and C. Ming, et al. , J. Anal. Appl. Pyrolysis, 2023, 176, 106242 CrossRef CAS.
- C. Jiang and G. A. Yakaboylu, et al. , Renewable Energy, 2020, 155, 38–52 CrossRef CAS.
- C. Li and S. Zhou, et al. , J. Anal. Appl. Pyrolysis, 2023, 171, 105968 CrossRef CAS.
- Z. Yi and C. Li, et al. , J. Anal. Appl. Pyrolysis, 2022, 167, 105689 CrossRef CAS.
- Y. Ding and Y. Li, et al. , Energy, 2021, 216, 119227 CrossRef CAS.
- J. Zhu and T. Huang, et al. , Green Chem., 2024, 26, 5441–5451 RSC.
- F. Gao and Y.-q Xie, et al. , New Carbon Mater., 2022, 37, 752–763 CrossRef CAS.
- J. Li and Q. Jiang, et al. , J. Mater. Chem. A, 2020, 8, 1469–1479 RSC.
- C. Zhao and L. Ge, et al. , Energy, 2023, 274, 127353 CrossRef CAS.
- H. Sun and K. Sun, et al. , Fuel, 2021, 285, 119192 CrossRef CAS.
- C. Cao and Y. Cheng, et al. , Waste Manage., 2022, 153, 367–375 CrossRef CAS PubMed.
- M. Li and Z. Sun, et al. , J. Mater. Chem. A, 2021, 9, 12495–12520 RSC.
- F. Chireshe and F.-X. Collard, et al. , J. Anal. Appl. Pyrolysis, 2020, 146, 104751 CrossRef CAS.
- X. Chen and S. Li, et al. , Bioresource Technol., 2019, 287, 121493 CrossRef CAS.
- J. Xu and Y. Guo, et al. , J. Anal. Appl. Pyrolysis, 2023, 173, 106102 CrossRef CAS.
- M. R. Khan, Fuel Sci. Technol. Int., 1987, 5, 185–231 CrossRef.
- W. Chen and P. Westerhoff, et al. , Environ. Sci. Technol., 2003, 37, 5701–5710 CrossRef CAS.
- Y. Shao and C. Li, et al. , Green Chem. Eng., 2024, 5, 222–235 CrossRef.
- G. Yang and H. Han, et al. , Carbon, 2012, 50, 3753–3765 CrossRef CAS.
- X. Li and L. Lu, et al. , China Powder Sci. Technol., 2023, 29, 49–60 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04931d |
| This journal is © The Royal Society of Chemistry 2025 |