
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the selectivity of bimetallic Cu electrocatalysts for CO2 reduction using atomic layer deposition†
Si Young
Lee‡
 a,
Julia D.
Lenef‡
b,
Daniel O.
Delgado Cornejo
b,
Alondra M.
Ortiz-Ortiz
a,
Tao
Ma
c,
Timothy S.
Arthur
d,
Charles A.
Roberts
*d and
Neil P.
Dasgupta
*ab
a,
Julia D.
Lenef‡
b,
Daniel O.
Delgado Cornejo
b,
Alondra M.
Ortiz-Ortiz
a,
Tao
Ma
c,
Timothy S.
Arthur
d,
Charles A.
Roberts
*d and
Neil P.
Dasgupta
*ab
aDepartment of Mechanical Engineering, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: ndasupt@umich.edu
bDepartment of Materials Science & Engineering, University of Michigan, Ann Arbor, MI 48109, USA
cMichigan Center for Materials Characterization, University of Michigan, Ann Arbor, MI 48109, USA
dToyota Research Institute of North America, Ann Arbor, MI 48105, USA. E-mail: charles.roberts@toyota.com
First published on 17th December 2024
Abstract
Cu–Zn bimetallic catalysts were synthesized on 3-D gas diffusion electrodes using atomic layer deposition (ALD) techniques. Electrochemical CO2 reduction was evaluated, and a significant variation in the product selectivity was observed compared to unmodified Cu catalysts. As low as a single ALD cycle of ZnO resulted in a reduction of C2H4 production and shift towards CO selectivity, which is attributed to changes in the chemical state of the surface. Our findings demonstrate the impact of atomically-precise surface modifications on electrocatalyst selectivity.
The electrochemical CO2 reduction reaction (CO2RR) has garnered significant attention as a promising carbon utilization technology to convert CO2, a major contributor to the climate crisis, into valuable products using renewable energy sources.1 In particular, extensive research has been conducted on Cu-based catalysts to produce value-added C2+ compounds.2,3 Owing to the ability of these catalysts to generate more than 16 different products, control of selectivity is a critical issue for the commercialization of CO2RR technology.4 Recent studies have shown that enhanced C–C coupling can be achieved by (1) tuning the nanostructure morphology,5,6 (2) controlling crystal facets or shapes,7,8 (3) alloying or introducing secondary metals,9,10 and (4) adjusting the chemical state of the Cu catalyst surface or interfacial environment.11,12 Such studies indicate that further analysis of catalyst surface modification and interfacial environments is necessary.
Atomic layer deposition (ALD) is a powerful technique that enables programmable control of the surface composition and coating thickness with sub-nm precision.13 ALD offers an effective method for catalyst deposition by conformally depositing metal nanoparticles (NPs) on porous, high-aspect ratio structures.14,15 Beyond simple binary oxides, ALD enables the precise control of stoichiometry of solid solutions and alloy materials,16 which allows for tuning of the chemical state and lattice constants of the catalyst. In addition to synthesis of the catalyst material itself, ALD has also been explored for deposition of saturated, sub-monolayer “overcoats” on catalyst surfaces, which can modulate the adsorption energy of intermediates on the catalyst surface, and also has been shown to improve catalyst stability during operation.17–20
Despite these benefits of ALD for precise catalyst synthesis and modification, there have been few reports to date of ALD for Cu-based CO2RR catalysts.21 Recently, we reported a plasma-enhanced ALD (PEALD) process to incorporate Cu NP catalysts onto gas diffusion electrodes (GDEs), achieving a high Faradaic efficiency (FE) of over 75% for C2+ products.22 Additionally, ALD of CuSx electrocatalysts for CO2RR have been reported to be effective in improving the selectivity of formate.23 Furthermore, ALD overcoats of metal oxides have been applied to CuO nanowires to tune selectivity.17,18 This indicates the need for further studies to understand the factors that influence the product selectivity of Cu catalyst architectures using ALD.
In previous studies on Cu-based bimetallic catalysts for CO2RR using alternative synthetic strategies, elements such as Au, Ag, and Zn have been introduced to modify the selectivity of C2+ products such C2H4 and C2H5OH (EtOH), while Sn has been introduced to improve the selectivity of C1 products such as formate.24–26 The introduction of elements such as Ag and Au has been known to improve the selectivity towards EtOH due to the spillover effect of the generated CO,24,27 but the economic burden of precious metal introduction can be a limiting factor. Recent studies on Cu–Zn bimetallic catalysts have shown inconsistent trends in selectivity, with either C1 or C2 products increasing depending on the method and ratio of Zn introduction.25,28–30 However, there have been no reports to date of bimetallic Cu-based NP catalysts deposited by ALD.
In this study, we expand up on our previously developed PEALD method for Cu catalysts,22,31 and explore the deposition of Cu–Zn bimetallic alloys and Cu@ZnO core–shell catalysts with a ZnO overcoat on Cu (Scheme 1). Using these two model systems, we measured the effects of Zn introduction on product selectivity. The influences of these chemical and structural changes on the Cu catalyst are described below.
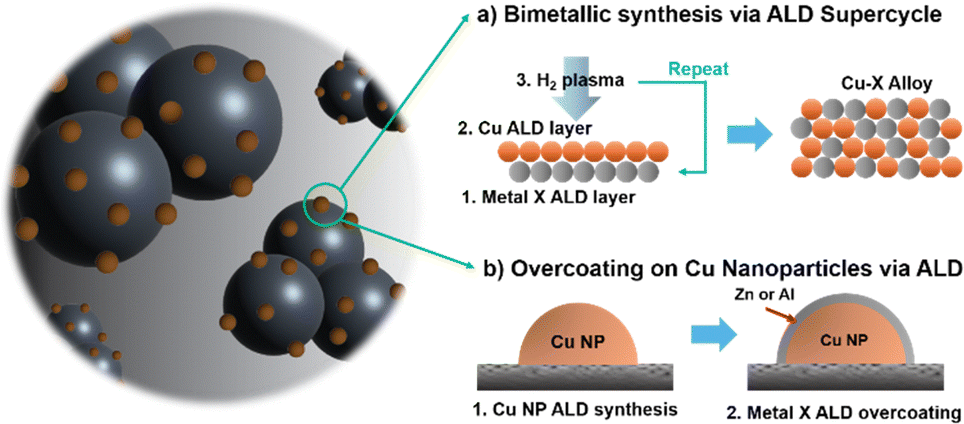 | ||
| Scheme 1 Cu bimetallic catalyst synthesis strategy that can be introduced through ALD. | ||
To fabricate Cu–Zn bimetallic alloy NPs, a supercycle approach was used alternating ALD Cu and ZnO cycles in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 1a). This is a common strategy to generate multi-element ALD films, where both elements are incorporated into the bulk material.32 Experimental details on the ALD process are provided in the ESI.†
:1 ratio (Scheme 1a). This is a common strategy to generate multi-element ALD films, where both elements are incorporated into the bulk material.32 Experimental details on the ALD process are provided in the ESI.†
Scanning electron microscopy (SEM) and scanning transmission electron microscopy (STEM) were performed to examine the morphology of the catalyst surfaces on carbon GDE substrates. Fig. 1a and Fig. S1 (ESI†) shows an SEM and TEM image of Cu NPs (4 ± 1 nm) after depositing 510 PEALD Cu cycles. The bimetallic alloy NPs had a slightly larger diameter 9 ± 2 nm, while maintaining a uniform distribution across the complex 3-D GDE structure (Fig. 1b and Fig. S2, ESI†). We have previously shown that varying the ALD Cu NP size in the nanometer range had a minimal effect on product selectivity, so these slight differences in particle diameter are not expected to have a strong effect. In both instances, the ability to conformally distribute NPs on a high aspect-ratio 3-D substrate demonstrates the power of the ALD method for electrocatalyst architectures.
 | ||
| Fig. 1 (a) SEM image of Cu PEALD catalyst on carbon GDE support. (b) SEM image of Cu–Zn (9:1 supercycle) bimetallic catalyst on GDE support. (c) XRD spectrum of Cu PEALD and Cu–Zn (9:1 supercycle) bimetallic catalyst on Si substrate (ICDS no: 15985). (d) Comparison of the Faradaic efficiency of the gaseous products for the Cu catalyst at −0.93 V vs. RHE and the Cu–Zn (9:1 supercycle) bimetallic catalyst at −0.90 V vs. RHE. (e) Cu 2p XPS spectra of Cu and Cu–Zn (9:1 supercycle) bimetallic catalysts on GDE substrate after 1 min Ar sputtering. | ||
The supercycle approach resulted in an approximately 1:1 ratio of Cu:Zn, as confirmed using X-ray photoelectron spectroscopy (XPS) (Table S1, ESI†). Further evidence of the formation of a bimetallic alloy was observed by STEM-energy dispersive X-ray spectroscopy (EDS) mapping (Fig. S3, ESI†) and the change in visible color, which appeared similar to brass (Fig. S4, ESI†). Grazing incidence X-ray diffraction (XRD) analysis revealed that the 2-theta value of (002) shifted to a lower angle by 0.96 degrees when Zn was introduced to the Cu catalyst, indicating an increase in d-spacing (Fig. 1c). This can be rationalized by the fact that Zn has a larger atomic radius than Cu.
The presence of Zn in the Cu catalyst significantly impacted overall product selectivity. Chronoamperometry measurements were performed for each system (Cu and Cu–Zn) at a potential of −0.93 V and −0.90 V vs. RHE, respectively, for 1 hour. The gas-phase products were quantified using gas chromatography. The pure Cu metal sample exhibited a ∼38% FE for C2H4 (Fig. 1d) which is consistent with the previously report.21 In contrast, the Cu–Zn bimetallic catalyst reduced this value to below 3%, and the CO content increased by about 31%. This represents an order-of-magnitude change in the selectivity for each of these products, illustrating the strong impact of Zn on the selectivity.
To examine the influence of Zn alloying on the chemical environment of Cu, XPS core scans were collected at the Cu 2p peak position. The presence of Zn resulted in a shift to lower binding energies for the Cu atoms (Cu PEALD: 933.3 eV, CuZn: 932.9 eV), indicating a shift to a more electron-rich environment (Fig. 1e).33 This shift is attributed to the lower electronegativity of the Zn atom compared to Cu. This result suggests that an electron-rich Cu state might be favorable for CO production.
To study the influence of the Zn concentration in the alloy on the chemical structure and corresponding product selectivity, bimetallic catalysts with a lower Zn content were synthesized. To accomplish this, the ALD cycle ratio of Cu:Zn in the supercycle recipe was increased from 9:1 to 81:1. XPS analysis showed a surface atomic ratio of about 3:1 (Table S1, ESI†). Examining a simple rule-of-mixtures analysis, these trends in stoichiometry suggest a nucleation delay of Cu growth on ZnO (Fig. S5, ESI†). The 81:1 supercycle catalyst had similar NP sizes to pure Cu PEALD (Fig. S6a, ESI†). In contrast to the 9:1 supercycle catalyst, which produces 30.7% CO at −0.90 V vs. RHE, the 81:1 supercycle catalyst with a lower Zn ratio suppresses CO production to around 10% FE across a wider potential range. The 81:1 supercycle sample also exhibits one order-of-magnitude lower FE for CH4 at potentials more negative than −0.90 V vs. RHE (Fig. S6b and c, ESI†), showing a higher suppression of C1 products compared to the 9:1 catalyst. These results suggest that in Cu–Zn bimetallic catalysts synthesized by ALD methods, reducing the amount of Zn introduced can enhance the total activity and C–C coupling selectivity of the catalyst. Further analysis and discussion of the 81:1 supercycle catalysts is provided in the ESI.†
To study the influence of how the Zn atoms were incorporated into the Cu NP catalysts during the ALD process, an overcoating strategy was also explored to form a core–shell structure, rather than mixing Zn into the bulk of the NP (Scheme 1b). For these samples, a series of ALD ZnO depositions were performed using 1, and 3 cycles on the surface of PEALD Cu catalysts. While the alloying method resulted in slight variations in NP morphology and size according to Cu–Zn ratios, the NP size and morphology remained similar across different cycles of ZnO ALD on Cu (Fig. 2a and Fig. S8, ESI†). This further illustrates the core–shell effect, as 1–3 cycles of ALD ZnO corresponds to only around 2–6 anstroms of nominal thickness. In other words, the NP nucleation and growth is determined by the PEALD Cu process, while the ALD ZnO overcoat conformally coats the NP topology.
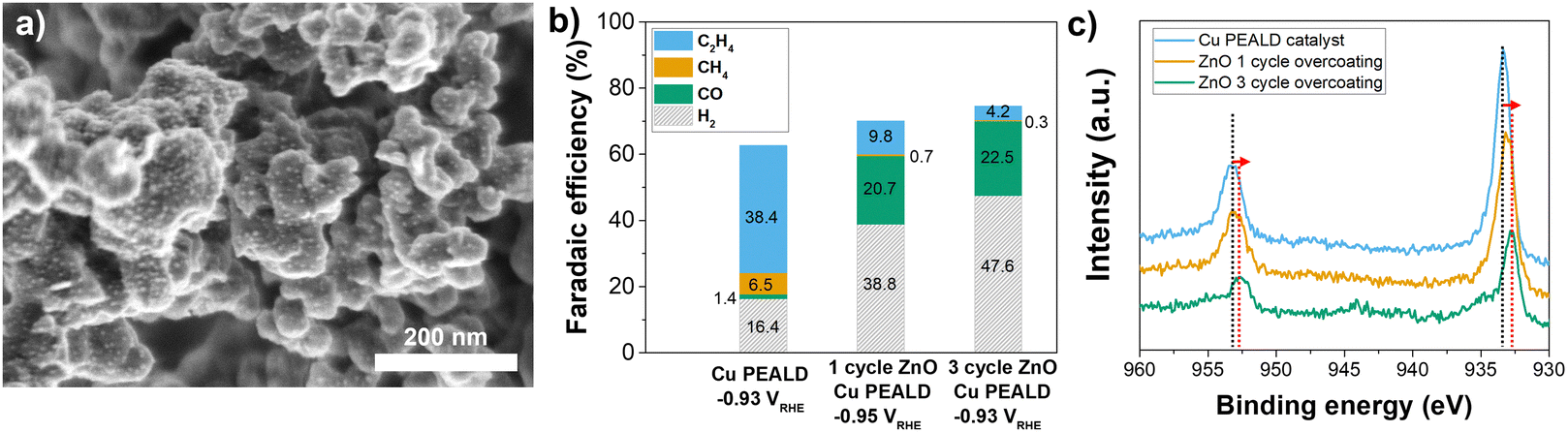 | ||
| Fig. 2 (a) SEM image after 1 cycle ZnO ALD overcoating on a bare Cu PEALD catalyst. (b) FE comparison of gaseous products of the catalyst Cu PEALD and after 1, and 3 cycles of ZnO ALD overcoats. (c) Comparison of Cu 2p XPS spectra of Cu PEALD catalyst and after 1, and 3 cycle ZnO ALD overcoating after 1 min Ar sputtering treatment. | ||
Fig. 2b displays the product analysis for the Cu PEALD and 1–3 ALD overcoat cycles on PEALD Cu catalysts. Notably, the selectivity for C2H4 drastically decreased to 9.8% at −0.95 V vs. RHE after only 1 cycle of ZnO ALD, compared to nearly 38.4% at −0.93 V vs. RHE for pristine Cu PEALD. Furthermore, the selectivity towards CO increased over an order of magnitude. This indicates that even minimal Zn introduction (a nominal overcoat thickness of ∼2 angstroms, which corresponds to less than a single unit cell of crystalline ZnO) can significantly alter catalyst selectivity. After additional ALD depositions of 3 cycles, the C2H4 selectivity dropped further to 4.2% at −0.93 V vs. RHE, with a slight increase in CO production (from 20.7% to 22.5%), These trends in FE were similar at various potentials for each catalyst (Fig. S9, ESI†), and the current density (activity) was also similar under each condition (Fig. S10, ESI†). This illustrates that the first ALD cycle of ZnO had the most profound effect on selectivity, while further increases in the surface Zn concentration had less significant effects.
The Cu 2p binding energy measured by XPS for pure Cu catalysts was 933.3 eV, and the Cu with a 1-cycle ZnO overcoat (CZ1) catalyst shows a nearly identical value at 933.2 eV (Fig. 2c). When a ZnO ALD overcoat is applied using 3 cycles, the Cu 2p binding energy shifts to 932.9 eV, similar to that of the CuZn alloy catalyst, and the FE also changes accordingly. This indicates that the surface properties and the chemical state of Cu can change significantly within a ZnO layer thickness range of around 2–6 angstroms. These findings suggest that in Cu–Zn systems, not only does the chemical state of Cu change due to the interaction between Cu and Zn, but even a few angstrom-thick hetero-structure ALD layer on the catalyst surface can significantly impact CO2RR performance (i.e., CO2 adsorption, intermediate surface diffusion, etc.).
To examine whether other metal oxide overcoats would also result in these significant shifts in CO2RR selectivity, another sample was fabricated coating Cu PEALD catalysts with 1 cycle of Al2O3 (CA1). SEM imaging of this catalyst showed a similar morphology to the pristine Cu PEALD catalysts (Fig. S11, ESI†). Unlike the CZ1 sample, which predominantly produced 9.8% C2H4 and 20.7% CO at −0.95 V vs. RHE, the CA1 sample had a similar C2H4 FE of 32.7% to Cu PEALD at −0.91 V vs. RHE (Fig. 3a). This tendency in CA1 remained similar across other potential ranges (Fig. S12, ESI†). Despite the 1:4 Cu:Al ratio revealed by XPS analysis (Table S2, ESI†), the CA1 catalyst exhibited similar C2H4 selectivity to that of pure Cu, implying different impacts on Cu selectivity depending on the metallic element used in the overcoat.
 | ||
| Fig. 3 (a) FE of gaseous products using a pristine Cu PEALD catalyst, 1 cycle ZnO ALD overcoat, and 1 cycle Al2O3 ALD overcoat. (b) Comparison of Cu 2p XPS spectra of each catalyst sample with no Ar sputtering used in the measurement. | ||
To study the influence of the Al2O3 and ZnO overcoats on the surface binding environment, Cu 2p XPS core scans were measured without any Ar sputtering (Fig. 3b). The trend in observed binding energies was Cu (933.3 eV) > CZ1 (933.2 eV) > CA1 (932.9 eV). After Ar sputtering, all three samples showed the same binding energy (Fig. S14, ESI†). This indicates that the impacts of single-cycle ALD overcoats on the Cu chemical state is highly localized to the surface.
We also observe a difference in the satellite peak near 944 eV, which is associated with the Cu2+ oxidation state.11,33 The ALD ZnO process reduces the presence of Cu2+ species on the Cu NP surface, unlike the ALD Al2O3 process, which preserved the Cu2+ surface oxide layer. Previous studies on copper oxide-based CO2RR catalysts indicated that reduction of Cu oxide layers could enhance C2H4 selectivity,11,34–36 which suggests that the ability to maintain surface oxides might contribute to the differences in C2H4 selectivity. These results suggest that ALD overcoating strategies for CO2RR catalysts must consider not only the composition of the coating material, but also changes in the surface oxidation of the underlying substrate.
To investigate the role of the Al2O3 overcoat on the electrocatalyst stability, chronoamperometry tests were conducted on the CA1 catalyst (Fig. S13, ESI†). The CA1 catalyst maintained a stable current density of ∼−28 mA cm−2 at −0.93 V vs. RHE for 24 hours. However, the FE for C2H4 increased from 28.5% to 35.2% in the first 3 hours before decreasing, while H2 selectivity increased continuously. This differs from previous observations where Cu PEALD showed a more monotonic trend in stability.22 These results suggest that ALD overcoats can also influence the surface reconstruction processes of Cu catalysts under CO2RR conditions, necessitating further research.
In conclusion, Zn was introduced into PEALD Cu NP catalysts using both alloying and overcoating methods. It was proposed that the electron-rich environment of Cu atoms induced by Zn may contribute to decreased C–C coupling selectivity. Additionally, ultrathin ALD overcoats altered the chemical state of Cu and consequently reduced the C2H4 selectivity. By changing the overcoat material from ZnO to Al2O3, the selectivity for C2 products remained similar to that of Cu. These differences suggest that altering the surface chemistry of Cu with overcoats can have a profound impact on selectivity. Overall, this work highlights the power of ALD to both tune and understand the mechanisms that determine CO2RR selectivity.
This research was supported by Toyota Research Institute of North America (TRINA) and by the National Science Foundation under Grant No. 2131709.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- O. S. Bushuyev, P. D. Luna, C. T. Dinh, L. Tao, G. Saur, J. V. D. Lagemaat, S. O. Kelley and E. H. Sargent, Joule, 2018, 2, 825–832 CrossRef CAS
.
- M. Jiang, H. Wang, M. Zhu, X. Luo, Y. He, M. Wang, C. Wu, L. Zhang, X. Liao, Z. Jiang and Z. Jin, Chem. Soc. Rev., 2024, 53, 5149–5189 RSC
- P. D. Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 350 CrossRef PubMed
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC
- K. D. Yang, W. R. Ko, J. H. Lee, S. J. Kim, H. Lee, M. H. Lee and K. T. Nam, Angew. Chem., Int. Ed., 2017, 56, 796–800 CrossRef CAS
- M. Jun, C. Kwak, S. Y. Lee, J. Joo, J. M. Kim, D. J. Im, M. K. Cho, H. Baik, Y. J. Hwang, H. Kim and K. Lee, Small Methods, 2022, 6, 2200074 CrossRef CAS
- G. L. D. Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef PubMed
- J. A. Gauthier, J. H. Stenlid, F. Abild-Pedersen, M. Head-Gordon and A. T. Bell, ACS Energy Lett., 2021, 6, 3252–3260 CrossRef CAS
- K. Tran and Z. W. Ulissi, Nat. Catal., 2018, 1, 696–703 CrossRef CAS
- L. Xie, J. Liang, C. Priest, T. Wang, D. Ding, G. Wu and Q. Li, Chem. Commun., 2021, 57, 1839–1854 RSC
- S. Y. Lee, H. Jung, N.-K. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2018, 140, 8681–8689 CrossRef CAS
- S. Y. Lee, J. Kim, G. Bak, E. Lee, D. Kim, S. Yoo, J. Kim, H. Yun and Y. J. Hwang, J. Am. Chem. Soc., 2023, 145, 23068–23075 CrossRef CAS
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed
- B. J. O’Neil, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef
- C. Detavernier, J. Dendooven, S. P. Sree, K. F. Ludwig and J. A. Matens, Chem. Soc. Rev., 2011, 40, 5242–5253 RSC
- J. Lu, K.-B. Low, Y. Lei, J. A. Libera, A. Nicholls, P. C. Stair and J. W. Elam, Nat. Commun., 2014, 5, 3264 CrossRef
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS
- D. Ren, J. Gao, L. Pan, Z. Wang, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Angew. Chem., Int. Ed., 2019, 58, 15036–15040 CrossRef CAS
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS
- N. Cheng, M. N. Banis, J. Liu, A. Riese, X. Li, R. Li, S. Ye, S. Knights and X. Sun, Adv. Mater., 2015, 27, 277–281 CrossRef CAS PubMed
- J. O. Olowoyo, V. S. Gharahshiran, Y. Zeng and Y. Zheng, Chem. Soc. Rev., 2024, 53, 5428–5488 RSC
- J. D. Lenef, S. Y. Lee, K. M. Fuelling, K. E. Rivera Cruz, A. Prajapati, D. O. Delgado Cornejo, T. H. Cho, K. Sun, E. Alvarado, T. S. Arthur, C. A. Roberts, C. Hahn, C. C. L. McCrory and N. P. Dasgupta, Nano Lett., 2023, 23, 10779–10787 CrossRef CAS
- M. Suominen, M. Mäntymäki, M. Mattinen, J. Sainio, M. Putkonen and T. Kallio, Mater. Today Sustainability, 2023, 24, 100575 CrossRef
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC
- J. Zhang, C. Guo, S. Fang, X. Zhao, L. Li, H. Jiang, Z. Liu, Z. Fan, W. Xu, J. Xiao and M. Zhong, Nat. Commun., 2023, 14, 1298 CrossRef CAS
- M. Zhang, Z. Zhang, Z. Zhao, H. Huang, D. H. Anjum, D. Wang, J.-H. He and K.-W. Huang, ACS Catal., 2021, 11, 11103–11108 CrossRef CAS
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS
- Y. Feng, Z. Li, H. Liu, C. Dong, J. Wang, S. A. Kulinich and X. Du, Langmuir, 2018, 34, 13544–13549 CrossRef CAS PubMed
- H. S. Jeon, J. Timoshenko, F. Scholten, I. Sinev, A. Herzog, F. T. Haase and B. R. Cuenya, J. Am. Chem. Soc., 2019, 141, 19879–19887 CrossRef CAS
- L. Wan, X. Zhang, J. Cheng, R. Chen, L. Wu, J. Shi and J. Luo, ACS Catal., 2022, 12, 2741–2748 CrossRef CAS
- J. D. Lenef, J. Jo, O. Trejo, D. J. Mandia, R. L. Peterson and N. P. Dasgupta, J. Phys. Chem. C, 2021, 125, 9383–9390 CrossRef CAS
- A. R. Bielinski, S. Lee, J. J. Brancho, S. L. Esarey, A. J. Gayle, E. Kazyak, K. Sun, B. M. Bartlett and N. P. Dasgupta, Chem. Mater., 2019, 31, 3221–3227 CrossRef CAS
- J. Conradie and E. Erasmus, J. Electron. Spectrosc. Relat. Phenom., 2022, 259, 147241 CrossRef CAS
- S. Y. Lee, S. Y. Chae, H. Jung, C. W. Lee, D. L. T. Nguyen, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 6210–6218 RSC
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. R. Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef
- X. Li, L. Li, L. Wang, Q. Xia, L. Hao, X. Zhan, A. W. Robertson and Z. Sun, Chem. Commun., 2022, 58, 7412–7415 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, SEM, images, XRD, XPS data and electrochemical results. See DOI: https://doi.org/10.1039/d4cc04820b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |