
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign of a stapled peptide that binds to the Ebola virus matrix protein dimer interface†
Roopashi
Saxena‡
a,
Madison M.
Wright‡
b,
Benjamin M.
Rathman
b,
Ukesh
Karki
c,
Prem P.
Chapagain
 cd,
Juan R.
Del Valle
*b and
Robert V.
Stahelin
*a
cd,
Juan R.
Del Valle
*b and
Robert V.
Stahelin
*a
aBorch Department of Medicinal Chemistry and Molecular Pharmacology and the Purdue Institute of Inflammation, Immunology and Infectious Disease, Purdue University, West Lafayette, IN 47907, USA. E-mail: rstaheli@purdue.edu
bDepartment of Chemistry & Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: jdelvalle@nd.edu
cDepartment of Physics, Florida International University, Miami, FL 33199, USA
dBiomolecular Sciences Institute, Florida International University, Miami, FL 33199, USA
First published on 25th April 2025
Abstract
The Ebola virus (EBOV) is a filamentous lipid-enveloped RNA virus that can cause viral hemmorhagic fever and has a high fatility rate. EBOV encodes seven genes including the lipid-binding matrix protein, VP40, which lies beneath the lipid-envelope. VP40 is a 326 amino acid protein with a N-terminal domain (NTD) harboring a high affinity dimer interface and a C-terminal domain (CTD) critical to plasma membrane lipid interactions. Disruption of VP40 dimer formation via mutagenesis inhibits assembly and budding of VP40. A series of conformationally constrained mimics of the VP40 α2 helix were designed based on the crystal structures of the VP40 dimer. A thermal shift assay was used to screen constrained and native peptides for significant alterations in VP40 stability. The most meritorious peptides were then confirmed to directly bind VP40 using microscale thermophoresis and isothermal titration calorimetry. A constrained VP40 peptide mimetic with a di-cysteine staple emerged with micromolar affinity for the VP40 dimer. This peptide was able to shift the VP40 dimer–monomer equilibrium as evidenced by size exclusion chromatography and bound near the NTD α-helix dimer interface. This study provides the first evidence of a designed small molecule induced disruption of VP40 dimer–monomer equilibrium.
Introduction
Ebolavirus (EBOV) is a lipid-enveloped filamentous virus harbouring a negative sense single stranded RNA genome. The first outbreak of EBOV was reported in 19761 and since then multiple outbreaks of different strains of virus have led to infections in humans often with fatality rates greater than 50%.2 EBOV consists of a 19 kb genome encoding 7 genes including nucleoprotein (NP), viral protein 35 kDa (VP35), viral protein 40 kDa (VP40), glycoprotein (GP), viral protein 30 kDa (VP30), viral protein 24 kDa (VP24) and polymerase (L).3 The virus ultrastructure has an outer lipid envelope containing GP, which are trimeric spikes expressed on the viral surface and are required for viral entry into the host cell. Below the viral surface, VP40 forms a matrix layer that provides stability and shape to the virus and plays important roles in viral assembly and budding.4–6 Below the matrix layer is the ribonucleoprotein (RNP) complex formed by viral genome RNA bound to NP, VP24, VP30, VP35, and L.3VP40 is an abundantly expressed viral protein, which performs multiple functions during the viral life cycle. VP40 achieves its multifunctionality by existing as a transformer protein, with conformational changes that lead to different oligomerization states.5,7 VP40 predominantly forms a dimer, which binds to the host cell plasma membrane inner leaflet and serves as a building block for matrix layer formation.5,6 The dimer further oligomerizes at the plasma membrane to form hexamers for cylindrical matrix layer formation.4 VP40 also exists as a ring-shaped octamer which binds viral RNA to downregulate viral replication and transcription.5,8 Further, expression of VP40 in mammalian cells in the absence of other EBOV proteins is sufficient to form virus-like particles (VLPs),9–11 highlighting the importance of VP40–host interactions for viral assembly and budding.11–13
The VP40 crystal structure demonstrated formation of a high affinity butterfly-shaped dimer with a distinct N-terminal domain (NTD) and C-terminal domain (CTD) connected by a flexible linker.5 The NTD has a critical role in forming NTD–NTD interactions at the dimer interface5,7,14,15 and formation of the octameric ring through a different NTD interface.6,8 Meanwhile, the CTD is required for binding to host cell plasma membrane lipids and formation of dimer–dimer interactions.7,13,16,17 The NTD forms a β-sandwich with two α-helices while the CTD features two β-sheets and two α-helices connected by a disordered loop structure.6,18 The dimerization interface is formed by an NTD α-helix along with an adjacent loop region, constituting residues 106–120 and 52–65,5,14,15 respectively. These regions bring the two protomers together through an array of hydrophobic interactions.5,14,15 Mutations at critical residues in both these regions of the dimerization interface have been reported to disrupt dimer formation, strongly favouring formation of the octameric ring.5,7,14,15 This indicates that interdomain contacts regulate conformational dynamics, dimer stability, and eventually formation of VP40 filaments.
Despite broad knowledge of VP40 structure and dimer formation, few chemical tools are available to probe VP40 structure, function, and the equilibrium between different VP40 oligomeric forms. To address this gap, we designed constrained peptides to probe the VP40 dimerization interface. The peptidomimetics were designed to mimic the NTD α-helical region, to either prevent the NTD interaction of two protomers or disrupt the dimerization interface. Di-cysteine sidechain stapling was employed to restrict accessible backbone conformations at distinct sites along the helix. Biophysical analysis along with site directed mutagenesis revealed a VP40 mimic capable of binding the VP40 dimer and shifting the dimer–monomer equilibrium.
Results and discussion
Design and synthesis of stapled VP40 peptides
We designed a series of conformationally constrained mimics of the VP40 α2 helix based on the crystal structures of the VP40 dimer.5 The α2 helix engages in several key hydrophobic interactions with the opposing VP40 protomer across residues 112–116, in addition to polar contacts involving D109 and S110 (Fig. 1(A)).15 The backbone torsions of residues 116–120 depart from canonical α-helical geometry, with Y120 engaging in a sidechain-to-backbone H-bond to the carbonyl oxygen of I115. We selected positions Y106, T111, I115, and Y120 as candidate sites for covalent tethering since these sidechains reside opposite to the dimer interface (Fig. 1(B)). To introduce constraints at these positions, we substituted pairs of native residues with Cys to enable stapling of 3 linear substrates (Fig. 1(C)). Two linear substrates feature i → i + 5 spacing of the Cys residues and the other i → i + 4 spacing. Since Met can participate in alkylation reactions with bis-electrophiles we also introduced an M116X substitution, where X is norleucine.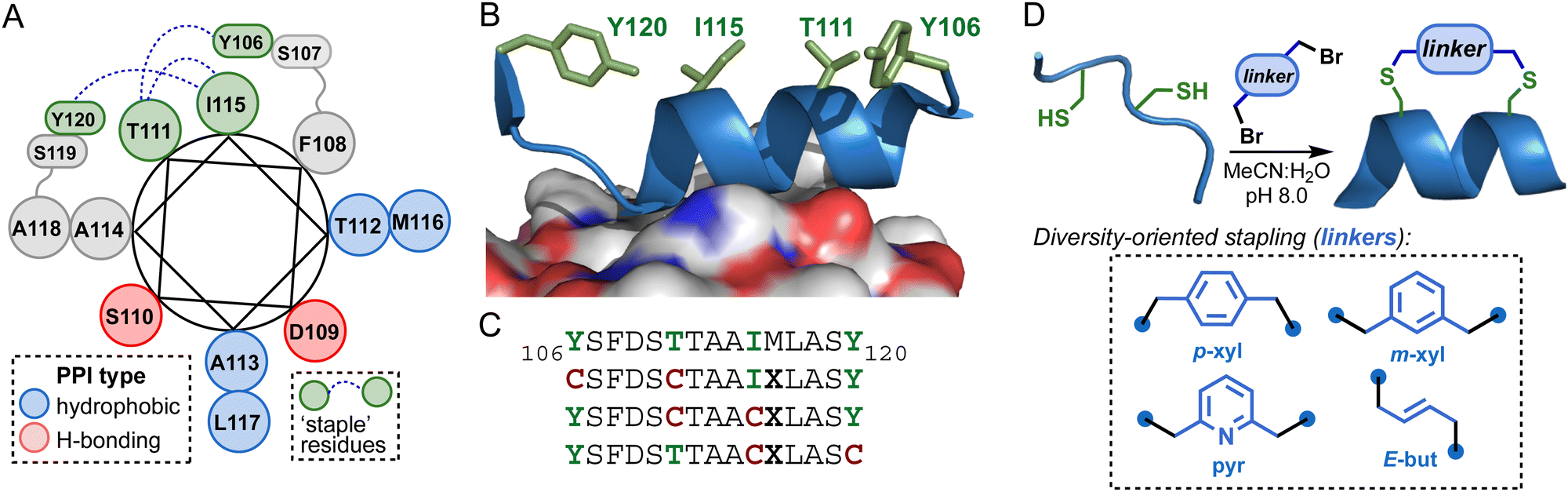 | ||
| Fig. 1 (A) Helical wheel diagram of VP40106–120 derived from the X-ray crystal structure of the VP40 dimer (PDB 4LDB). (B) Non-interface residue sidechains (green) in the structure of VP40106–120 bound to the opposing dimer protomer. (C) VP40106–120 lead sequence and designed linear di-Cys substrates for stapling. (D) Diversity-oriented stapling with 4 dibromide electrophiles. | ||
Linear di-Cys peptides were synthesized on Rink amide resin using standard Fmoc solid-phase protocols (HATU/NMM activation). Following cleavage from the resin, the crude unprotected peptides were each reacted with 4 different dibromide electrophiles to generate 12 unique macrocycles. The stapled peptides were purified to homogeneity by RP-HPLC and characterized by HRMS (Table 1 and Fig. S1, ESI†).
| Peptide | Sequencea | Linker | Yieldb (%) | HRMS (m/zobs) |
|---|---|---|---|---|
| a Di-Cys macrocycles indicated by brackets (X = norleucine). b Overall isolated yield following RP-HPLC purification, based on initial resin loading. | ||||
| 1a | [CSFDSC]TAAIXLASY | m-xyl | 9 | 1665.7640 [M + H]+ |
| 1b | [CSFDSC]TAAIXLASY | p-xyl | 7 | 1665.7687 [M + H]+ |
| 1c | [CSFDSC]TAAIXLASY | m-pyr | 11 | 1666.7601 [M + H]+ |
| 1d | [CSFDSC]TAAIXLASY | E-but | 8 | 1615.7465 [M + H]+ |
| 2a | YSFDS[CTAAC]XLASY | m-xyl | 14 | 1715.7454 [M + H]+ |
| 2b | YSFDS[CTAAC]XLASY | p-xyl | 4 | 1715.7439 [M + H]+ |
| 2c | YSFDS[CTAAC]XLASY | m-pyr | 10 | 1716.7402 [M + H]+ |
| 2d | YSFDS[CTAAC]XLASY | E-but | 11 | 1665.7269 [M + H]+ |
| 3a | YSFDSTTAA[CXLASC] | m-xyl | 10 | 1653.7258 [M + H]+ |
| 3b | YSFDSTTAA[CXLASC] | p-xyl | 5 | 1653.7290 [M + H]+ |
| 3c | YSFDSTTAA[CXLASC] | m-pyr | 11 | 1654.7240 [M + H]+ |
| 3d | YSFDSTTAA[CXLASC] | E-but | 9 | 1603.7121 [M + H]+ |
Screening of peptides for VP40 dimer interactions using a thermal shift assay
To identify compounds that can interact with the VP40 dimer, we screened our library of stapled peptides using a thermal shift assay.19,20 VP40 dimer was purified and isolated from the octamer fraction (Fig. S2, ESI†) and subjected to temperatures from 35–95 °C. The intrinsic fluorescence of VP40 contributed by 2 Trp and 4 Tyr residues was measured at both 330 nm and 350 nm for each degree change in temperature. The VP40 dimer exhibits minimal fluorescence in the folded state, and we observe an increase in the fluorescence with higher temperatures as the protein unfolds and aromatic residues become more exposed. Once protein is completely unfolded maximum fluorescence is recorded, which stays constant with further increases in temperature. This gives a sigmoidal melting profile of protein as shown in Fig. 2(A). The transition between the folded and unfolded states of a protein is determined by inflection temperatures, which indicates changes in structural integrity of the protein.19,20 VP40 dimer displays two inflection temperatures, one at 55 °C (Ti1) and the second at 70 °C (Ti2), respectively (Fig. 2(A)). Each peptide from the library was then mixed with the VP40 dimer and the melting profiles of VP40 in the presence of peptide was measured. In peptide series 1 with a chemical staple between position 106 and 111, we observed that m-pyridyl linked 1c decreased both inflection temperatures. In peptide series 2 with staples between residues 111 and 115, p-xylyl and m-pyridyl derivatives 2b and 2c changed the melting profile of VP40 dimer. Lastly, for series 3 with staples between positions 115 and 120, p-xylyl stapled peptide 3b significantly decreased both inflection temperatures of VP40 as compared to the vehicle control DMSO (Fig. 2(B)–(D)). A decrease in inflection temperature indicates that these peptides are destabilizing the VP40 dimer. Other stapled peptides tested did not shift the melting profiles (Fig. S3, ESI†) while peptide 3c only decreased Ti2 (Fig. 2(D)). | ||
| Fig. 2 A library of compounds was screened based on changes in melting profile of the VP40 dimer. (A) Ebolavirus VP40 dimer was purified and subjected to thermal melting with absorbance measured at 330 nm and 350 nm. The ratio of absorbance (350 nm/330 nm) was plotted for every degree change in temperature giving sigmoidal melting profile for VP40 with two inflection temperatures (Ti1 – 55 °C, Ti2 – 70 °C). (B) The library of compounds was individually assessed for altering the VP40 melting profile by addition of individual peptides to VP40 dimer. Inflection temperature 1 (C) and inflection temperature 2 (D) of VP40 with each of the peptides was compared to vehicle control DMSO. Statistics represent one way ANOVA with multiple comparisons to DMSO ****p < 0.0001. Each experiment was repeated three times. | ||
To verify that the impact of 3b on inflection temperatures is staple-dependent, we synthesized linear peptides in which the Cys residues at positions 115 and 120 were replaced with Ile and Tyr from the parent sequence (4) or 2 Ala residues (5). These peptides did not change Ti1 and only slightly decreased Ti2 (Fig. S4, ESI†). These results highlight the requirement of macrocyclic constraint between residues 115 and 120 for destabilization of VP40 dimer. In addition, peptides 3a and 3b are constitutional isomers and differ only in the regiochemistry of xylene linker substitution. Their distinct effects on inflection temperature suggests that interactions with the VP40 dimer are sensitive to subtle changes in linker geometry and macrocycle conformation.
Microscale thermophoresis (MST)21 was then used to assess VP40 interactions comparing peptides that decreased both inflection temperatures. Peptides were added to cell lysate containing GFP tagged VP40 for a range of concentrations and fluorescence signal at each concentration was determined. The greater the change in fluorescence readout with varying peptide concentrations indicates stronger binding of peptide to GFP tagged VP40. It was observed that 3b showed the most pronounced concentration dependent change in fluorescence (Fig. S5, ESI†) and was pursued for further studies.
To investigate the impact of sidechain stapling on conformation, we obtained circular dichroic (CD) spectra of wild-type VP40106–120 (4) and selected macrocyclic variants in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 MeCN:H2O (Fig. 3(A)). Peptide 4 exhibited a canonical α-helical CD signature characterized by ellipticity minima at 208 and 222 nm and a 208/222 nm band strength ratio of 1.3. Our lead peptide 3b, featuring a C-terminal p-xylyl staple between residues 115 and 120, exhibited a significant shift in 208/222 nm band strength ratio indicating potential disruption of canonical α-helical structure. Peptides 1b and 2b, which harbour the same p-xylyl staple positioned at the N-terminus or in the center of the sequence, showed CD signatures similar to that of the wild-type peptide. The i → i + 4 staple in 2b also resulted in enhanced helicity relative to 3b and 4 based on band intensity at 222 nm. The incorporation of other i → i + 5 staples at the C-terminus significantly altered the conformation of the peptide relative to 3b (Fig. 3(B)). Taken together, these results suggest that the precise nature and position of the p-xylyl staple are important for stabilizing a conformation of 3b that interacts with the VP40 dimer. However, the stronger binding of 3b to VP40 did not appear to stem from enhanced helicity. The i → i + 5 staple in 3b may instead serve to mimic the non-helical C-terminal capping motif observed in the X-ray structure of the VP40 dimer.5
:1 MeCN:H2O (Fig. 3(A)). Peptide 4 exhibited a canonical α-helical CD signature characterized by ellipticity minima at 208 and 222 nm and a 208/222 nm band strength ratio of 1.3. Our lead peptide 3b, featuring a C-terminal p-xylyl staple between residues 115 and 120, exhibited a significant shift in 208/222 nm band strength ratio indicating potential disruption of canonical α-helical structure. Peptides 1b and 2b, which harbour the same p-xylyl staple positioned at the N-terminus or in the center of the sequence, showed CD signatures similar to that of the wild-type peptide. The i → i + 4 staple in 2b also resulted in enhanced helicity relative to 3b and 4 based on band intensity at 222 nm. The incorporation of other i → i + 5 staples at the C-terminus significantly altered the conformation of the peptide relative to 3b (Fig. 3(B)). Taken together, these results suggest that the precise nature and position of the p-xylyl staple are important for stabilizing a conformation of 3b that interacts with the VP40 dimer. However, the stronger binding of 3b to VP40 did not appear to stem from enhanced helicity. The i → i + 5 staple in 3b may instead serve to mimic the non-helical C-terminal capping motif observed in the X-ray structure of the VP40 dimer.5
 | ||
| Fig. 3 Circular dichroic (CD) spectra of select peptides. (A) Selected macrocyclic variants (1b, 2b, and 3b) with a p-xyl staple and the WT peptide (4) were used to collect CD spectra in 1:1 MeCN:H2O. (B) Different CD spectra were detected for peptides of the same sequence with different stapling chemistry (3a–3d). | ||
To examine the peptide secondary structure of both WT (unstapled) and 3b in solution, we performed three replicates of 500-ns simulations of each system. As shown in Fig. S6 (ESI†), the unstapled WT sequence maintains the helical structure (Fig. S6A–C, ESI†), whereas the stapled 3b significantly loses the helicity during the simulations. Interestingly, the helical structure is retained for 3b when it is interacting with VP40 (see Molecular dynamics section).
Interactions of 3b with VP40 are independent of non-specific disulfide interactions
Since di-Cys stapling was used to constrain 3b, we wanted to rule out the possibility of non-specific cysteine residue interactions causing destabilization of VP40. We utilized a di-cysteine mutant of VP40 where VP40 cysteines at positions 311 and 314 were mutated to alanine. Previous studies demonstrated that these mutations do not affect VP40 dimer stability.22 We purified the di-cysteine VP40 mutant (C311A/314A) dimer and added different concentrations of 3b for the thermal shift assay. When different concentrations of 3b were added to WT VP40 dimer, it was observed that a higher concentration of peptide led to a greater shift in the melting profiles (Fig. 4(A) and (B)). Similarly, 3b was able to decrease the inflection temperatures of di-cysteine mutant VP40 in a concentration dependent manner, suggesting that destabilization is independent of cysteines present in VP40 (Fig. 2(D) and 4(C)). We also added a reducing agent, TCEP (tris(2-carboxyethyl)phosphine), to the WT VP40 reaction mixture to eliminate disulfide interactions. Although VP40 dimer gave a single inflection temperature (Ti1) at 55 °C in reducing buffer conditions, 3b was able to decrease Ti1 like native buffer conditions. Overall, these results indicate that 3b-mediated destabilization of VP40 dimer is independent of cysteines present in VP40 and disulfide interactions between protein and peptide. | ||
| Fig. 4 Peptide 3b destabilizes both wild type and di-cysteine mutant of VP40 as thermal titration of 3b shows similar results for wild type and di-cysteine double mutant of VP40. (A) Peptide 3b was titrated with wild type VP40 and melting profiles for each concentration is plotted. (B) The inflection temperature 1 (Ti1) and inflection temperature 2 (Ti2) of wild type VP40 with each concentration is shown by bar graphs. (C) Peptide 3b was titrated with C311A/314A dimer and melting profiles of di-cysteine double mutant are plotted. (D) The inflection temperature 1 (Ti1) and inflection temperature 2 (Ti2) of C311A/314A with each concentration of peptide is shown by bar graphs. Each experiment was repeated three times. | ||
Xylene staple in 3b is required for binding of peptide to the VP40 dimer
Next, we quantitatively assessed 3b binding to the VP40 dimer using isothermal titration calorimetry (ITC). 3b was titrated into the vessel harbouring the target VP40 dimer and monitored for binding by measuring the heat change with injections of 3b. We observed heat of titration changes with each 3b injection that became saturated at higher 3b concentrations, showing sigmoidal change in heat of titrations (Fig. 5(A)). The binding curve depicts that 3b binds to VP40 dimer with an apparent affinity of 60 μM (Fig. 5(B)). The linear peptide where both the cysteines used for stapling are changed to alanine (5) was used as a control. The control peptide with the same sequence but no chemical staple (5) did not show significant binding to the VP40 dimer (Fig. 5(C) and (D)). The heat of titration did not change with initial injections of 5 and only showed changes at higher concentration. This trend of heat of titration was like the heat of solvent, therefore indicating little to no interaction between 5 and VP40. These studies reveal that a p-xylene staple between residues 115–120 is crucial for 3b–VP40 interactions. | ||
| Fig. 5 Peptide 3b binds VP40 dimer in ITC studies. (A) Peptide with xylene linker (3b) was titrated into VP40 dimer for 20 injections and heat readings for each injection was plotted. Heat of titration indicates binding and saturation of peptide. Heat of dilution of peptide was calculated by titrating equivalent amount of DMSO with VP40 dimer over 20 injections. (B) Enthalpy of binding of peptide (3b) was calculated by subtracting enthalpy of DMSO from enthalpy of peptide and plotted with mole ratio of peptide to the dimer. Each experiment was repeated three times, giving an apparent KD of 60 ± 2 μM. Error bars indicate standard deviation of separate replicate experiments. (C) Peptide with no linker (5) was titrated into VP40 dimer for 20 injections and heat readings for each injection was plotted. Heat of titration indicate no changes in heat in initial injections and heat is reduced only at high concentrations. This pattern of heat titration is similar to that of DMSO. (D) Corresponding enthalpy of DMSO was subtracted from enthalpy of peptide 5 and plotted to give a binding curve. Each experiment was repeated three times and subtracted enthalpy indicate little to no binding of peptide 5 to VP40 dimer. Error bars indicate standard deviation from replicate experiments where each experiment was repeated three times. | ||
Compound 3b binds in proximity to the VP40 N-terminal domain helical dimer interface
These findings prompted us to understand the binding mechanism and probable binding sites of 3b on VP40. The NTD α-helical dimer interface is also exposed in the VP40 octamer structure5 (Fig. 6(A)) and therefore we investigated the binding of 3b to the VP40 octamer. ITC studies revealed that 3b binds to octamer with an apparent KD of 51 μM (Fig. 6(C)). This enabled us to investigate the binding of 3b with different VP40 mutants as we and others have previously observed that mutations at critical residues causes a shift in the oligomerization profile of VP40 from dimer to octamer.5,7,14,15 We assessed the binding of 3b to a mutant in which the isoleucine at position 307 is mutated to arginine (I307R) (Fig. 6(D)). I307R mutation prevents CTD–CTD interaction between the dimers5 and has been reported to favourably form the octamer (Fig. 6(E)).5,7 We observed that 3b can bind the I307R octamer mutant with an affinity of 29 μM (Fig. 6(F)). We next examined the mutation L117A, which is present in the NTD α-helix and is essential for maintaining inter-protomer interactions to retain VP40 dimer stability (Fig. 6(G)).5,7,15 We observed that 3b is unable to bind to the octamer formed by the L117A mutation (Fig. 6(H) and (I)). This suggests that 3b binding to VP40 is dependent on the presence of a bulky hydrophobic residue at position 117 where mutation to Ala disrupted 3b interactions. Altogether, these studies demonstrate that 3b was able to bind to VP40 when the NTD α-helical dimer interface is intact or exposed in the VP40 octamer structure. This is supported by similar affinity of 3b for WT VP40 octamer and the I307R mutant whereas binding of 3b is disrupted to the L117A mutant, indicating 3b likely interacts near residue L117. | ||
| Fig. 6 Peptide 3b binds to intact VP40 dimer. (A) Representative figure of VP40 octamer with residues 106–120 highlighted in red for each protomer (shown in blue and cyan). (B) Wild type VP40 was recombinantly expressed and purified using Ni-NTA chromatography and subjected to a size exclusion column. Wild type VP40 shows predominant dimer and octamer peaks. Octamer fractions were collected and concentrated for ITC studies. (C) Peptide 3b was titrated with wild type VP40 octamer for over 20 injections and heat of titration for each concentration were measured. As described previously, heat of dilution was measured by titrating an equivalent amount of DMSO with VP40 octamer. Enthalpy of binding of peptide was plotted by subtracting enthalpy of DMSO from enthalpy of peptide 3b with mole ratio of peptide to VP40 octamer. Experiments were repeated three times with error bars representing standard deviation and binding affinity KD of 51 ± 3 μM. (D) Representative image indicates mutation of isoleucine to arginine at position 307 abrogates CTD–CTD interactions. (E) VP40 with I307R mutation was purified as above and subjected to size exclusion chromatography indicating the mutated protein predominantly forms octamer. (F) Peptide 3b was titrated with I307R octamer and binding curve was plotted by subtracting enthalpy of DMSO from the enthalpy of peptide. Each experiment was replicated three times giving KD of 29 ± 7 μM. Error bars indicate standard deviation of replicate experiments. (G) Representative image indicates mutation of leucine to alanine at position 117 disrupts the interprotomer hydrogen bonding required for stabilization of dimer. (H) Recombinant expression and purification of L117A mutant shows predominant octamer formation. (I) Peptide 3b was titrated for 20 injections with L117A mutant octamer and enthalpy of binding was determined by subtracting enthalpy of DMSO from the enthalpy of peptide and plotted with mole ratio of peptide to L117A mutant. Experiments were repeated three times with error bars indicating standard deviation. No binding was observed for peptide 3b with L117A mutant octamer. Each experiment was repeated three times. | ||
Compound 3b binds to VP40 of different Ebolavirus strains
There is high sequence similarity in the VP40 NTD α-helical region of VP40 between different strains of Ebolavirus (Fig. 7(A)). Residues 101–125 are identical between Zaire and Sudan Ebolavirus. As the results above suggested 3b binding is near L117 in the NTD α-helical interface, we tested binding of 3b to the VP40 dimer of Sudan Ebolavirus (Fig. 7).23 We observed that 3b binds to Sudan VP40 dimer with an apparent KD of 43 μM (Fig. 7(B)). Further, the control peptide 5 did not show binding to Sudan VP40 like results obtained with the Zaire strain (Fig. 7(C)). This suggests that 3b can interact with the conserved NTD dimer interface and can be used to probe the VP40 dimerization in different strains of Ebolavirus. | ||
| Fig. 7 Peptide 3b binds to Sudan Ebolavirus VP40 dimer. (A) Conserved sequence of dimerization interface between different Ebolavirus strains. (B) Sudan VP40 dimer was recombinantly expressed and purified and peptide 3b was titrated for 30 injections with the dimer. Heat of dilution was calculated by titrating equivalent amount of DMSO for 30 injections. Enthalpy was plotted by subtracting enthalpy of DMSO from enthalpy of peptide. Experiment was repeated three times giving a KD of 43 ± 5 μM with error bars indicating standard deviation between experiments. (C) Similarly, peptide 5 was titrated with Sudan VP40 dimer for 20 injections and subtracted enthalpy from DMSO was plotted. Each experiment was repeated three times, with error bars indicating standard deviation and the experiments showed no binding of peptide 5 to the Sudan VP40 dimer. Each experiment was repeated three times. | ||
Compound 3b alters the oligomerization equilibrium of VP40
While the studies above indicate the specific binding of 3b to the VP40 dimer, the effect of peptide on the VP40 dimer remains unknown. To gain insights in this regard, we incubated VP40 dimer with 3b and performed size exclusion chromatography to separate different oligomeric forms of VP40. We observed that there is a dominant dimer peak in the peptide and vehicle-treated samples. In addition to the dimer peak, we observe a small peak around 22 mL in the peptide-treated fraction corresponding to VP40 monomer (Fig. 8). Immunoblotting revealed that fractions collected from the peptide-treated samples elute VP40 in later fractions. This indicates that 3b can cause a slight shift in the equilibrium of VP40 dimer towards formation of the monomeric form of VP40. | ||
| Fig. 8 Peptide 3b slightly shifts equilibrium of VP40 dimer. (A) Peptide 3b was added to purified VP40 dimer and incubated at 4 °C overnight. The sample was injected in a Superdex 200 10/300 GL column to separate the oligomeric forms of VP40. Elution profile of VP40 with peptide 3b is shown in red and it indicates a dimer peak and a smaller peak at 22 mL elution volume. As a vehicle control, VP40 dimer was treated with equivalent DMSO concentration and subjected to size exclusion chromatography (in black) showing a dimer peak. (B) 1 mL fractions were collected and immunoblotted for VP40 using anti-6xHistidine antibody. VP40 was detected in all the fractions from 11–24 mL for the peptide while DMSO samples show presence of VP40 from 12–21 mL and less amounts in 23 and 24 mL fractions. There was no protein detected in the 22 mL fraction in case of DMSO while there is presence of protein in case of peptide 3b indicating peptide induced formation of smaller order oligomers of VP40 which are eluted at higher volumes. | ||
Computational studies of 3b interactions with VP40
To explore 3b interactions with VP40 at the dimer interface, we modelled the complex of VP40 and 3b by taking the VP40 structure from residues 106 to 120 and connecting the cysteine sidechains of residues 115 and 120 with a p-xylyl staple using a custom patch (included in the ESI†). We performed a 1.0 μs all-atom molecular dynamics simulation of the complex using NAMD 3.0.24 Analysis of the trajectory showed that 3b remains bound for the duration of the simulation. As shown in the Movie S1 (ESI†), the peptide is stably bound with significant VP40-3b hydrophobic contacts (Fig. 9(B)) and hydrogen bond interactions (Fig. 9(C)–(E)) to hold the peptide in the interface. The most notable VP40–3b hydrogen bonding occurs between S81–D109, D80–S107, and D80–S110. Notably, the N-terminal tail that is aligned with a β-strand (indicated by a small arrow in Fig. 9(A)) reorients and wraps the peptide, adding to the stability of the complex. A transient reorientation of the peptide occurs around 65 ns but snaps back to its former position by 200 ns. A similar event is observed between 780–930 ns, suggesting a possibility of further optimization to enhance the interactions. Additional two 1.0 μs replicas runs of the system show similar interfacial interactions of S81–D109 or D80–S110 but we also see additional contacts for D27 interacting with L117 and S119 in one of the runs. The slight variations seem to be due to the loop orientations of the N-terminal tail wrapping the peptide. | ||
| Fig. 9 Structure and dynamics of VP40–3b complex. (A) Structure of the VP40–3b complex at 0 ns. The N-terminal tail (indicated by a small arrow) is pointing downward and not interacting with 3b. (B) As in the VP40 dimer structure, 3b has significant hydrophobic contacts with VP40 at the dimer interface. The hydrophobic side chains involved in the interactions are shown in stick whereas the hydrophobic surface is shown as a transparent white surface. The N-terminal tail (indicated by a small arrow) wraps the peptide during the simulation. (C) Residues involved in hydrogen bond interactions at the VP40–3b interface. (D) The total number of hydrogen bonds between VP40 and 3b during the simulation. (E) Specific VP40–3b hydrogen bond pairs that are observed to form at least 10% of the time during the 1 μs simulation. These simulations were run three different times. | ||
Conclusions
The EBOV VP40 dimer is necessary for formation of virus particles from the host cell plasma membrane. VP40 dimer formation is mediated by a NTD α-helix and adjacent loop region, mutation of which prompts VP40 octamer formation in vitro5,7,14,15 and stalls VP40's ability to reach the inner leaflet of the host cell plasma membrane.5,7,15 We identified several constrained peptides that could associate with VP40, altering the VP40 melting temperature, and bind directly to VP40 as evidenced by MST. We pursued 3b as it demonstrated the most significant binding affinity with MST and lowered Ti1 and Ti2 of VP40. We demonstrated that binding of 3b to VP40 was specific to the constrained peptide sequence and stapling chemistry using both modified peptides and mutations of VP40. Binding of 3b to VP40 was shown to be independent of VP40 Cys residues and 3b binding occurred to both VP40 dimer and octamer with micromolar affinity. Notably, the NTD α-helix that mediates VP40 dimerization is exposed in the VP40 octamer structure. Mutagenesis studies demonstrated 3b bound near the NTD α-helix that mediates dimerization as mutation of L117 to Ala abrogated binding of 3b to VP40. 3b was also able to associate with Sudan VP40 with similar affinity to that of Zaire VP40, further demonstrating the ability of 3b to interact near the VP40 dimer interface and be a viable future probe of VP40 oligomer equilibria. VP40 is highly conserved among EBOV strains and almost completely conserved across strains between residues 101 and 120. Thus, 3b and future analogues should be useful for examining VP40 biophysical properties from all known EBOV strains.The current study should inform further structure-based design efforts to target VP40 and alter dimer–monomer equilibria. Since the VP40 dimer is necessary for proper virus assembly and budding,5,7 shifting VP40 towards the monomer or octamer form would inhibit spread of virions from cell to cell. Further, inhibiting octamer formation or repressing octamer function could also be detrimental to the EBOV lifecycle. VP40 has been hypothesized to serve as a regulator of viral RNA transcription.5,25,26 While it is somewhat understood how VP40 binds RNA it is not yet known how to allosterically inhibit VP40 octamer RNA binding. Thus, since 3b bound the VP40 octamer, 3b and future analogues could be used to probe VP40 octamer activity and how best to alter VP40 RNA binding affinity. Recently, a pocket was found in Sudan VP40 near residues L158 and R214 that was able to accommodate salicylic acid27 while targeting VP40 host interactions with small molecule inhibitors has also shown efficacy.28,29 Thus, multiple sites are emerging as prime targets to alter VP40 oligomer equilibria to inhibit virus assembly and spread.
Experimental
Protein expression and purification
The pET46 plasmids encoding wild type Zaire Ebolavirus VP40 and Sudan Ebolavirus VP40 (deletion of N-terminal 43 residues) with 6x-His-tags were kind gifts from Dr Erica Ollmann Saphire (La Jolla Institute for Immunology).5,23 The C311A/C314A plasmid was previously made in the lab.22 The I307R and L117A mutations were made in Zaire Ebolavirus VP40 plasmid by Gene Universal (Newark, DE). Each of these plasmids were transformed into Rosetta™ BL21 DE3 cells (Merck Millipore Billerica, MA) and purified using Ni-NTA affinity chromatography as previously described.16 The eluted protein was further subjected to size exclusion chromatography on a HiLoad 16/600 Superdex 200 pg column using an AKTA PURE Protein Purification System) (Cytiva Life Sciences, Marlborough, MA) to separate oligomeric forms of VP40. The dimeric and octameric fractions of protein were concentrated and stored in buffer containing 10 mM HEPES, 150 mM NaCl, pH 8.0. Protein concentration was determined using Pierce™ BCA Protein Assay.Solid-phase synthesis of linear peptide substrates
Automated solid-phase peptide synthesis was carried out on NovaPEG Rink amide resin (35–100 mesh, 0.45 mmol g−1) on a 0.1 mmol scale. Fmoc deprotection steps were carried out by treating the resin with a solution of 20% piperidine/DMF once at room temperature (5 min), then at 75 °C (2 min). After Fmoc deprotection the resin was washed 4× with DMF. Coupling of Fmoc-protected amino acids was achieved using 5 equiv. HATU (0.25 M in DMF), 10 equiv. NMM (1 M in DMF), and 5 equiv. of Fmoc-protected amino acid (0.2 M in DMF) at 50 °C (10 min × 2). Deprotection and coupling steps were repeated until peptide synthesis was complete and then a final Fmoc deprotection was run to remove Fmoc from the N-terminus. The resin was transferred to a suitable vessel, washed with DCM (5 mL × 4) and dried under vacuum. Peptides were cleaved from the solid support and globally deprotected by incubating the dried resin in 4 mL of TFA:TIPS:H2O:DODT (92.5:2.5:2.5:2.5) for 2.5 h. The resin was filtered, and the filtrate was collected in a 50 mL centrifuge tube. The resin was washed with DCM (10 mL), filtered, and crude peptides were precipitated from the combined filtrate by the addition of cold Et2O (40 mL). The mixture was centrifuged, and the supernatant decanted. The pellet was washed with Et2O (25 mL × 2) and dried thoroughly under vacuum.
Synthesis of stapled peptides
For dithiol bis-alkylation, 1.5 equiv. of 1,3-bis(bromomethyl)benzene, 1,4-bis(bromomethyl)benzene, 2,6-bis(bromomethyl)pyridine, or E-1,4-dibromo-2-butene was added to a 1 mM solution of linear precursor peptide in 1:1 MeCN:H2O buffered with NH4HCO3 (20 mM) and the pH was adjusted to 8.0 using 2 M aq. NaOH. Reaction progress was monitored by analytical HPLC. The reaction was stirred for 2 h before evaporating the MeCN under a stream of N2, freezing, and lyophilization. All peptides were purified by preparative RP-HPLC (C12, 250 mm × 21.2 mm, 4 μm, 90 Å) using linear gradients of MeCN in H2O (mobile phases modified with 0.1% formic acid) over 30 minutes. RP-HPLC samples for all purified peptides were acquired (C12, 150 mm × 4.6 mm, 4 μm, 90 Å) using linear gradients of MeCN in H2O (mobile phases modified with 0.1% formic acid) over 20 minutes and LC spectra are provided for λ = 220 nm. HRMS of purified peptides were acquired using a Bruker Impact II ESI-QTOF. Lyophilized peptides were dissolved in DMSO to make 10 mM stock solutions and were stored at −20 °C until further use.
Thermal shift assay
Samples were prepared by adding peptides at a final concentration of 125 mM (1.25% DMSO) to VP40 dimer at concentration of 1 mg ml−1 in 10 mM HEPES, 150 mM NaCl, pH 8.0 buffer. Melting profiles of VP40 were measured using a Tycho NT.6 (NanoTemper Technologies, München, Germany) and the data was directly exported from the instrument. The inflection temperatures of different samples, as calculated by the instrument, were compared using a one-way ANOVA in GraphPad Prism.Microscale thermophoresis
GFP-VP40 plasmid was prepared as previously.12 HEK293 cells from ATCC were cultured in DMEM (Corning) media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. GFP-VP40 was transfected using Lipofectamine 2000 and cells were collected 24 hours post transfection in Dulbecco's phosphate buffer saline (DPBS no calcium, no magnesium). Cells were then lysed in DPBS containing 1% TritonX-100 (PBS-T) and the lysate was clarified by low-speed centrifugation. Samples for assay were prepared by serially diluting peptides in the cell lysate and fluorescence was measured using a Monolith NT.115 (NanoTemper Technologies). Fluorescence readout was analysed using Monolith analysis software and binding curves plotted in GraphPad Prism.Circular dichroism
All peptides were analysed at a concentration of 75 μM in 1:1 MeCN:H2O. CD spectra were acquired using a JASCO J-1700 CD spectrometer in a 1 mm path length quartz cell with 2 s digital integration time, 1 nm bandwidth, 0.5 nm datapitch, and a scan speed of 100 nm min−1 at 20 °C. Mean residue ellipticity at a given wavelength (MRE; deg cm2 dmol−1 residue−1) was calculated based on the equation MRE = θ/(10 × b × M × n), where θ is ellipticity (mdeg), b is the pathlength (cm), M is the peptide concentration (mol L−1), and n is the total number of residues.
Isothermal titration calorimetry (ITC)
ITC studies were performed with 50 μL of peptide in the ITC syringe and 300 μL of purified VP40 or mutant in the ITC cell using a Nano ITC (TA instruments, New Castle, DE, USA). Measurements were made at 20 °C at 200 rpm stir speed. In each experiment, 0.5 mM of peptide was titrated into the cell containing 1 mg mL−1 of protein. Data was exported from the instrument and corresponding heats of DMSO were subtracted from heat of peptide to obtain heat of binding of peptide. All thermograms were fit to specific binding with a Hill slope model (equation: Y = Bmax × X^h/(Kd^h + X^h) using GraphPad Prism to obtain apparent binding affinity.Size exclusion chromatography studies
Purified VP40 dimer at a concentration of 1 mg mL−1 was incubated overnight with 3b at a final concentration of 125 μM. The samples were then subjected to size exclusion chromatography using a Superdex 200 10/300 GL column (AKTA Pure, GE Healthcare). 1 mL fractions were collected and immunoblotted using a previously described protocol for VP40 detection.16 Briefly, fractions were loaded on a gel (10% SDS-PAGE), run, and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% milk-TBS-T and VP40 was identified using an anti-his tag polyclonal antibody (Sigma-Aldrich, Inc., St. Louis, MO). The antibody was detected using an AP Conjugate Substrate Kit (Bio-Rad, Hercules, CA) and imaged on an Amersham™ Imager 600 (Cytiva Life Sciences). Precision Plus Protein™ Kaleidoscope Ladder (Bio-Rad) was used to confirm the size of the VP40 band (40 kDa).Molecular dynamics simulations
The VP40 dimer structure available in the protein data bank (PDB ID 4LDB)5 was used to model the VP40 monomer-3b complex. The missing residues were added using Modeller.30 The simulation system was prepared with CHARMM-GUI31,32 web server using the CHARMM36m force field.33 The complex was solvated with TIP3 water and ionized/neutralized with 0.15 M KCl in a cubic box of size 89 × 89 × 89 Å3. A custom patch for stapling the p-xylyl to the cysteines was defined by generating a force field file using CGenFF34 parameters. The prepared system contained a total of 65970 atoms, with 6816 water molecules, 58 potassium ions, and 59 chloride ions. Molecular dynamics simulation was performed with NAMD 3.024 using 2 fs timesteps. The system was first minimized for 10000 steps, followed by 1 ns equilibration at 303.15 K with all protein and peptide heavy atoms restrained. An unrestrained production simulation was run for 1.0 microseconds at constant temperature and pressure (NPT). Temperature was controlled using Langevin dynamics with damping of 1 ps−1 and constant pressure was maintained at 1 atm with the Nose–Hoover Langevin piston35 pressure control with piston period of 0.05 ps and decay 25. The long-range electrostatic interactions were calculated with the Particle Mesh Ewald method (PME)36 method with periodic boundary conditions and a non-bonded cut-on and cut-off set at 10 Å and 12 Å respectively. The covalent bonds of hydrogen atoms were restricted using ShakeH.37 Visual molecular dynamics (VMD)38 was used to analyze the trajectories and create images. Hydrogen bonds between VP40 and the peptide were defined with a 3.5 Å distance and 30° angle cutoff.
Author contributions
Conceptualization: R. S., J. R. D., P. P. C., and R. V. S.; methodology: R. S., M. M. W., B. M. R., U. K., and P. P. C.; software: P. P. C.; validation: R. S., M. M. W., B. M. R., and U. K.; formal analysis: R. S., M. M. W., B. M. R., and U. K.; investigation: R. S., M. M. W., B. M. R., U.K.; resources: P. P. C.; writing – original draft: R. S. and R. V. S.; writing – review & editing: R. S., M. W., B. R., U. K., P. P. C., J. R. D., and R. V. S.; visualization: R. S., M. M. W., B.M. R., and U.K.; supervision: J. R. D., P. P. C., and R. V. S.; project administration: J. R. D., P. P. C., and R. V. S.; funding acquisition: J. R. D., P. P. C., and R. V. S.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number R01AI158220 (to R. V. S., J. R. D., and P. P. C.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- R. T. Emond, B. Evans, E. T. Bowen and G. Lloyd, A case of Ebola virus infection, Br. Med. J., 1977, 2, 541–544, DOI:10.1136/bmj.2.6086.541.
- G. Ameni, A. Zewude, B. Tulu, M. Derara, B. Bayissa, T. Mohammed, B. A. Degefa, M. E. Hamad, M. Tibbo and R. Barigye, J. Epidemiol. Glob. Health, 2024, 14, 1397–1412, DOI:10.1007/s44197-024-00304-7.
- T. Hoenen, A. Groseth and H. Feldmann, Therapeutic strategies to target the Ebola virus life cycle, Nat. Rev. Microbiol., 2019, 17, 593–606, DOI:10.1038/s41579-019-0233-2.
- T. A. Bharat, T. Noda, J. D. Riches, V. Kraehling, L. Kolesnikova, S. Becker, Y. Kawoaka and J. A. G. Briggs, Structural dissection of Ebola virus and its assembly determinants using cryo-electron tomography, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4275–4280, DOI:10.1073/pnas.1120453109.
- Z. A. Bornholdt, T. Noda, D. M. Abelson, P. Halfmann, M. R. Wood, Y. Kawaoka and E. Ollmann Saphire, Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle, Cell, 2013, 154, 763–774, DOI:10.1016/j.cell.2013.07.015.
- W. Wan, M. Clarke, M. J. Norris, L. Kolesnikova, A. Koehler, Z. A. Bornholdt, S. Becker, E. Ollmann Saphire and J. A. Briggs, Ebola and Marburg virus matrix layers are locally ordered assemblies of VP40 dimers, eLife, 2020, 9, e59225, DOI:10.7554/eLife.59225.
- K. Del Vecchio, C. T. Frick, J. B. Gc, S.-I. Oda, B. S. Gerstman, E. Ollmann Saphire, P. P. Chapagain and R. V. Stahelin, A cationic, C-terminal patch and structural rearrangements in Ebola virus matrix protein VP40 protein control its interactions with phosphatidylserine, J. Biol. Chem., 2018, 293, 3335–3349, DOI:10.1074/jbc.M117.816280.
- F. X. Gomis-Ruth, A. Dessen, J. Timmins, A. Bracher, L. Kolesnikova, S. Becker, H. Dieter Klenk and W. Weissenhorn, The matrix protein VP40 from Ebola virus octamerizes into pore-like structures with specific RNA binding properties, Structure, 2003, 11, 423–433, DOI:10.1016/s0969-2126(03)00050-9.
- L. D. Jasenosky, G. Neumann, I. Lukashevich and Y. Kawoaka, Ebola virus VP40-induced particle formation and association with the lipid bilayer, J. Virol., 2001, 75, 5205–5214, DOI:10.1128/JVI.75.11.5205-5214.2001.
- T. Noda, H. Sagara, E. Suzuki, A. Takada, H. Kida and Y. Kawaoka, Ebola virus VP40 drives formation of virus-like filamentous particles along with GP, J. Virol., 2002, 76, 4855–4865, DOI:10.1128/jvi.10.4855-4865.2002.
- J. M. Licata, M. Simpson-Holley, N. T. Wright, Z. Han, J. Paragas and R. N. Harty, Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: involvement of host proteins TSG101 and VPS-4, J. Virol., 2003, 77, 1812–1819, DOI:10.1128/jvi.77.3.1812-1819.2003.
- E. Adu-Gyamfi, K. A. Johnson, M. E. Fraser, J. L. Scott, S. P. Soni, K. R. Jones, M. A. Digman, E. Gratton, C. R. Tessier and R. V. Stahelin, Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus, J. Virol., 2015, 89, 9440–9453, DOI:10.1128/JVI.01087-15.
- K. A. Johnson, M. R. Budicini, N. Bhattarai, T. Sharma, S. Urata, B. S. Gerstman, P. P. Chapagain, S. Li and R. V. Stahelin, PI(4,5)P2 binding sites in the Ebola virus matrix protein VP40 modulate assembly and budding, J. Lipid Res., 2024, 65, 100512, DOI:10.1016/j.jlr.2024.100512.
- Y. B. Narkhede, A. Bhardwaj, B. B. Motsa, R. Saxena, T. Sharma, P. P. Chapagain, R. V. Stahelin and O. Wiest, Elucidating residue-level determinants affecting dimerization of Ebola virus matrix protein using high-throughput site saturation mutagenesis and biophysical approaches, J. Phys. Chem. B, 2023, 127, 6449–6461, DOI:10.1021/acs.jpcb.3c01759.
- Y. B. Narkhede, R. Saxena, T. Sharma, J. P. Conarty, V. Toro Ramirez, B. B. Motsa, S. Amiar, S. Li and P. P. Chapagain, Computational and experimental identification of keystone interactions in Ebola virus matrix protein VP40 dimer formation, Protein Sci., 2024, 33, e4978, DOI:10.1002/pro.4978.
- B. B. Motsa, T. Sharma, M. D. Cioffi, P. P. Chapagain and R. V. Stahelin, Minor electrostatic changes robustly increase VP40 membrane binding, assembly, and budding of Ebola virus matrix protein derived virus-like particles, J. Biol. Chem., 2024, 300, 107213, DOI:10.1016/j.jbc.2024.107213.
- M. D. Cioffi, T. Sharma, B. B. Motsa, N. Bhattarai, B. S. Gerstman, R. V. Stahelin and P. P. Chapagain, Ebola virus matrix protein VP40 single mutations G198R and G201R significantly enhance plasma membrane localization, J. Phys. Chem. B, 2024, 128, 11335–11344, DOI:10.1021/acs.jpcb.4c02700.
- A. Dessen, V. Volchkov, O. Dolnik, H. D. Klenk and W. Weissenhorn, Crystal structure of the matrix protein VP40 from Ebola virus, EMBO J., 2000, 19, 4228–4236, DOI:10.1093/emboj/19.16.4228.
- K. Huynh and C. L. Partch, Analysis of protein stability and ligand interactions by thermal shift assay, Curr. Protoc. Protein Sci., 2015, 79, 28.9.1–28.9.14, DOI:10.1002/0471140864.ps2809s79.
- J. A. Bhayani and M. A. Ballicora, Determination of dissociation constants of protein ligands by thermal shift assay, Biochem. Biophys. Res. Commun., 2022, 590, 1–6, DOI:10.1016/j.bbrc.2021.12.041.
- M. Gonneau, S. Schoenaers, C. Broyart, K. Vissenberg, J. Santiago and H. Höfte, Microscale Thermophoresis (MST) to study rapid alkanization factor (RALF)-receptor interactions, Methods Mol. Biol., 2024, 2731, 279–293, DOI:10.1007/978-1-0716-3511-7_21.
- K. A. Johnson, N. Bhattarai, M. R. Budicini, C. M. LaBonia, S. C. B. Baker, B. S. Gerstman, P. P. Chapagain and R. V. Stahelin, Cysteine mutations in the Ebolavirus matrix protein VP40 promote phosphatidylserine binding by increasing the flexibility of a lipid-binding loop, Viruses, 2021, 13, 1375, DOI:10.3390/v13071375.
- M. C. Clifton, J. F. Bruhn, K. Atkins, T. L. Webb, R. O. Baydo, A. Raymond, D. D. Lorimer, T. E. Edwards, P. J. Myler and E. Ollmann Saphire, High resolution crystal structure of dimeric VP40 from Sudan ebolavirus, J. Infect. Dis., 2015, 212(Suppl 2), S167–S171, DOI:10.1093/infdis/jiv090.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernadi, R. Buch, G. Fiorin, J. Henin and W. Jiang, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130, DOI:10.1063/5.0014475.
- T. Hoenen, N. Biedenkopf, F. Zielecki, S. Jung, A. Groseth, H. Feldmann and S. Becker, Oligomerization of Ebola virus VP40 is essential for particle morphogenesis and regulation of viral transcription, J. Virol., 2010, 84, 7053–7063, DOI:10.1128/JVI.00737-10.
- T. Hoenen, S. Jung, A. Herwig, A. Groseth and S. Becker, Both matrix proteins of Ebola virus contribute to the regulation of viral genome replication and transcription, Virology, 2010, 403, 56–66, DOI:10.1016/j.virol.2010.04.002.
- A. D. Werner, N. Krapoth, M. J. Norris, A. Heine, G. Klebe, E. Ollmann Saphire and S. Becker, Development of a crystallographic screening to identify Sudan Virus VP40 ligands, ACS Omega, 2024, 9, 33193–33203, DOI:10.1021/acsomega.4c04829.
- H. M. Loughran, Z. Han, J. E. Wrobel, S. E. Decker, G. Ruthel, B. D. Freeman, R. N. Harty and A. B. Reitz, Quinoxaline-based inhibitors of Ebola and Marburg VP40 egress, Bioorg. Med. Chem. Lett., 2016, 26, 3429–3435, DOI:10.1016/j.bmcl.2016.06.053.
- Z. Han, J. Lu, Y. Liu, B. Davis, M. S. Lee, M. A. Olson, G. Ruthel, B. D. Freeman, M. J. Schnell, J. E. Wrobel, A. B. Reitz and R. N. Harty, Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses, J. Virol., 2014, 88, 7294–7306, DOI:10.1128/JVI.00591-14.
- B. Webb and A. Sali, Comparative protein structure modeling using MODELLER, Curr. Protoc. Bioinformatics, 2016, 54, 5.6.1–5.6.37, DOI:10.1002/cpbi.3.
- S. Jo, T. Kim, V. G. Iyer and W. Im, CHARMM-GUI: a web-based graphical user interface for CHARMM, J. Comput. Chem., 2008, 29, 1859–1865, DOI:10.1002/jcc.20945.
- B. R. Brooks, C. L. Brooks 3rd, A. D. Mackerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels and S. Boresch, et al. , J. Comput. Chem., 2009, 30, 1545–1614, DOI:10.1002/jcc.21287.
- J. Huang and A. D. MacKerell Jr., CHARM36 all-atom additive protein force field: validation based on comparison to NMR data, J. Comput. Chem., 2013, 34, 2135–2145, DOI:10.1002/jcc.23354.
- K. Vanommeslaeghe, E. Prabhu Raman and A. D. MacKerell Jr., Automation of the CHARMM general force field (CGenFF) II: Assignment of bonded parameters and partial atomic charges, J. Chem. Inf. Model., 2012, 52, 3155–3168, DOI:10.1021/ci3003649.
- M. M. Brooks, A. Hallstrom and M. Peckova, A simulation study used to design the sequential monitoring plan for a clinical trial, Stat. Med., 1995, 14, 2227–2237, DOI:10.1002/sim.4780142006.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593, DOI:10.1063/1.470117.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical integration of the Cartesian equations of proteins and nucleic acids, J. Comput. Phys., 1997, 23, 327–341, DOI:10.1016/0021-9991(77)90098-5.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14, 33–38, DOI:10.1016/0263-7855(96)000180-5.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00048c |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2025 |