
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRaPID discovery of cell-permeable helical peptide inhibitors con-taining cyclic β-amino acids against SARS-CoV-2 main protease†
Marina
Kawai
a,
Tika R.
Malla
b,
H. T. Henry
Chan
c,
Anthony
Tumber
b,
Lennart
Brewitz
 b,
Eidarus
Salah
b,
Naohiro
Terasaka
a,
Takayuki
Katoh
a,
Akane
Kawamura
bd,
Christopher J.
Schofield
b,
Fernanda
Duarte
c and
Hiroaki
Suga
*a
b,
Eidarus
Salah
b,
Naohiro
Terasaka
a,
Takayuki
Katoh
a,
Akane
Kawamura
bd,
Christopher J.
Schofield
b,
Fernanda
Duarte
c and
Hiroaki
Suga
*a
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, Tokyo, Japan
bDepartment of Chemistry and the Ineos Oxford Institute for Antimicrobial Research, Chemistry Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
cPhysical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK
dChemistry – School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, UK
First published on 21st May 2025
Abstract
Structurally constrained cyclic β-amino acids are attractive building blocks for peptide drugs because they induce unique and stable conformations. Introduction of (1S,2S)-2-aminocyclopentanecarboxylic acid [(1S,2S)-2-ACPC] into peptides stabilizes helical conformations, so improving proteolytic stability and cell membrane permeability. We report on the ribosomal synthesis of a helical peptide library incorporating (1S,2S)-2-ACPC at every third position and its application for the discovery of SARS-CoV-2 main protease (Mpro) inhibitors. We identified two peptide sequences containing multiple (1S,2S)-2-ACPC residues, which exhibit helical conformations and superior proteolytic stability compared with their α-Ala or β-Ala counterparts. Studies using the chloroalkane cell-penetration assay showed that their cell permeability values (CP50) are comparable with or even slightly better than that of the cell-penetrating nona-arginine (R9) peptide. The new approach is thus a highly efficient method that combines a helical peptide library containing structurally constrained cyclic β-amino acids with the classical RaPID discovery method, enabling de novo discovery of proteolytically stable and cell-penetrating bioactive peptides that target intracellular proteins.
Introduction
Helical structures are often observed in bioactive cell-penetrating peptides (CPPs). For example, melittin, found in the genus Apis, interacts with cell membranes and forms pores in lipid bilayers owing to its amphipathic α-helical structure.1,2 The α-helicity of melittin enables it to form a rigid binding interface that interacts with its target protein, calmodulin, resulting in potent inhibition of tumor cell proliferation.3–5 Therefore, helical peptides are an attractive class of peptides for developing probes against intracellular protein targets.In general, short peptides comprised solely of α-amino acids (α-peptides) are unable to fold into stable helical structures due to their flexible backbones; >20 residues are typically required for formation of stable helices.6 Various strategies for stabilizing helical peptide structures have been developed. For example, side-chain-to-side-chain covalent bridges can be introduced for stabilization of helices to develop α-helix-mediated protein–protein interaction inhibitors based on rational or semi-rational designs.7
By contrast with α-amino acids, structurally constrained cyclic β2,3-amino acids (cβAAs) can form well-defined, stable secondary structures in short peptides,8 which are often referred to as foldamers.9 (1S,2S)-2-Aminocyclopentanecarboxylic acid [(1S,2S)-2-ACPC] is a cβAA which strongly enhances the helical propensity of peptides when combined with α-amino acids (α/β-peptides),10,11 in a largely side chain independent manner. In particular, introduction of (1S,2S)-2-ACPC at every third residue gives rise to unique helical structures such as 10/11/11-helix and 14-helix conformations (Fig. 1).12,13 10/11/11-helix is characterized by an i → i + 3 hydrogen bonding pattern, whereas 14-helix adopts an i → i + 4 hydrogen bonding pattern, which is similar to that of the α-helix (also referred to as a 13-helix). Such α/β-helical peptides are anticipated to have high rigidity and compactness, properties that can be advantageous both for cell membrane permeation and strong binding to target molecules. Importantly, such cβAAs also potentially decrease susceptibility to proteolysis owing to their unnatural backbones.14 Thus, cβAAs are attractive building blocks for the development of structurally and biologically stable bioactive peptides targeting intracellular proteins, including for drug development. To date, reported helical α/β-peptides have been the result of rational or semi-rational designs, based on knowledge of how the parental α-peptides interact with the target proteins.15,16
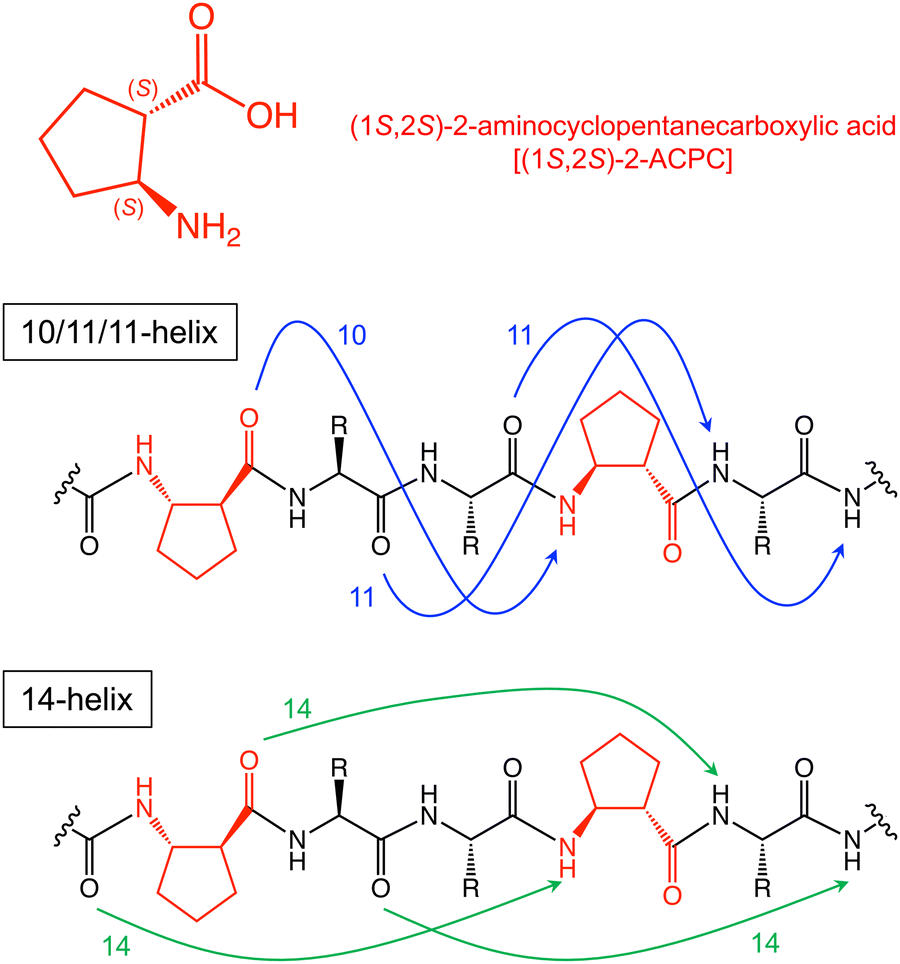 | ||
Fig. 1 Structure of (1S,2S)-2-aminocyclopentanecarboxylic acid and models of hydrogen-bond patterns that define 10/11/11-helix and 14-helix. Reported i, i + 3 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N hydrogen bonds are in blue (10/11/11-helix), and i, i + 4 CO⋯H–N hydrogen bonds are in green (14-helix). O⋯H–N hydrogen bonds are in blue (10/11/11-helix), and i, i + 4 CO⋯H–N hydrogen bonds are in green (14-helix). | ||
Ribosomal translation of non-proteinogenic α-amino acids has been achieved by means of a custom-made Escherichia coli translation system in the combination with the flexizyme technology, referred to as the flexible in vitro translation system (FIT system).17,18 Although ribosomally mediated multiple incorporation of β-amino acids was once extremely difficult due to the slow accommodation of β-aminoacyl-tRNA to the ribosomal A-site and slow peptidyl transfer, this limitation has been recently overcome by devising a new class of engineered tRNA, named tRNAPro1E2.19,20 This tRNA has a chimeric structure consisting of T-stem derived from tRNAGlu possessing a higher binding affinity to EF-Tu,21,22 and D-arm derived from tRNAPro1 capable of binding to EF-P,23 respectively. The use of β-aminoacyl-tRNAPro1E2 enhances its accommodation efficiency to the A site mediated by EF-Tu and peptidyl transfer reaction of peptidyl-tRNAPro1E2 mediated by EF-P, resulting in higher incorporation efficiency of β-amino acids. As a result, multiple incorporations of cβAA repertoires have become possible,24–26 prompting us to construct diverse peptide libraries containing multiple cβAAs.
The application of such libraries to the Random non-standard Peptides Integrated Discovery (RaPID) system, built based on mRNA display,27 has led to the discovery of de novo macrocycle inhibitors, F3 and F4, against human FXIIa.25 A co-crystal structure of FXIIa with F3 containing two residues of (1S,2S)-2-aminocyclohexanecarboxylic acid (ACHC) revealed that it has a unique β-sheet-like macrocyclic structure, wherein the ACHC residues enable significant structural stabilization of the macrocycle. Remarkably, the cyclohexane rings of the ACHCs also interact directly with the specific hydrophobic residues of FXIIa, contributing to not only high resistance against serum proteases (with a half-life of 60 h) but also to the strong binding ability of F3.
Encouraged by the above success, it occurred to us that a de novo α/β-helical peptide library can be built if the placement of (1S,2S)-2-ACPC is achieved at periodic positions in peptide sequences under the reprogrammed genetic code. Here we report ribosomal construction of (1S,2S)-2-ACPC-containing helical peptide libraries, which were used for the RaPID selection against the SARS-CoV-2 main protease (Mpro) to discover de novo α/β-helical peptide inhibitors with favorable serum peptidase resistance and membrane permeability properties.
Results
Ribosomal synthesis of (1S,2S)-2-ACPC-containing helical peptide libraries
The flexizyme technology afforded essential aminoacyl-tRNAs for construction of α/β-helical libraries, where the initiator fMet (AUG) was reprogrammed to N-acetyl-L-tyrosine (Ac-L-Tyr) using an Ac-L-Tyr-tRNAiniCAU while (1S,2S)-2-ACPC was assigned to the elongator Met (AUG) codon using a (1S,2S)-2-ACPC-tRNAPro1E2CAU (Fig. S1A and B, ESI†). The mRNA libraries encoding α/β-helical peptides with lengths of 7–15 residues were designed to have periodic AUG codons at every third position, introducing 2–5 residues of (1S,2S)-2-ACPC (Fig. 2A). Fifteen canonical amino acids (A, C, D, F, G, H, I, L, N, P, R, S, T, V, and Y) were assigned by the NNU codons, whereas unused amino acids (E, K, M, Q, and W) and the corresponding aminoacyl-tRNA synthetases (ARSs) were omitted to construct a FIT system for this particular setup.17,18 A C-terminal TGVTNG sequence was introduced as a spacer connected to the 3′-end of the corresponding mRNA. | ||
| Fig. 2 Library designs of the (1S,2S)-2-ACPC-containing helical peptides. (A) Sequences of the mRNA libraries, I–III, coding α/β-helical peptide libraries containing random 4–10 NNU codons and the corresponding peptide sequences. The puromycin linker covalently links the respective mRNA to the translated peptide by ribosome catalysis. These mRNA libraries were combined into one mRNA library for the expression of a peptide library containing (1S,2S)-2-ACPC at the designated positions using the elongator AUG codon. (B) A model peptide, Pcβ5, and its mRNA template, mRcβ5. Pcβ5 was designed to contain five (1S,2S)-2-ACPCs at the designated positions assigned by the elongator AUG codon. (C) MALDI-TOF MS analysis of Pcβ5 expressed in the custom-made FIT system. Purple arrows indicate monovalent ([M + H]+, and [M + K]+) and divalent ([M + 2H]2+) ions of Pcβ5, respectively. “Obsd.” and “Calcd” denote observed and calculated m/z values of monovalent ion of Pcβ5, respectively. Asterisks (*) show unknown impurities, inherently present in the FIT system. | ||
We previously observed that the multiple consecutive incorporations of β-amino acids suffered from poor translation efficiency depending on the type of β-amino acids.20,25,28 Hence we first investigated if the translation efficiency and fidelity of the designed library would be sufficient to construct it. We designed a model peptide containing five (1S,2S)-2-ACPCs at every third position (Pcβ5) and attempted to express it using the custom-made FIT system (Fig. 2B). Analysis of the product using matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS) (Fig. 2C) showed only the expected monovalent ([M + H]+) and divalent ([M + 2H]2+) ions for Pcβ5. Based on experience, we considered that this level of quality of translation should be sufficient for the expression of (1S,2S)-2-ACPC-containing peptides in a library format.
De novo discovery of (1S,2S)-2-ACPC-containing helical peptide ligands
Encouraged by the clean expression of the Pcβ5 peptide, we proceeded with a selection campaign using the (1S,2S)-2-ACPC-containing helical peptide libraries (Fig. S2A, ESI†). As a biomedicinally important intracellular target, we selected the SARS-CoV-2 main protease (Mpro), which is involved in the replication of the coronavirus.29 The initial mRNA libraries I–III (Fig. 2A) were combined into a single mRNA library, which was then ligated to a puromycin-linked oligonucleotide, then subject to in vitro translation using the FIT system and reverse transcription to produce an mRNA/cDNA-tagged peptide library. The peptide library was then applied to naked streptavidin-coated magnetic beads to remove bead-binding peptides using three rigorous washes. The supernatant was then applied to biotinylated Mpro-immobilized beads for affinity-based Mpro screening. The recovered cDNA was amplified by PCR and transcribed for the next round of selection.After the sixth and seventh rounds of the selection, the amount of recovered mRNA/cDNA-tagged peptides bound to Mpro substantially increased (Fig. S2B, ESI†). The recovered DNA sequences after the sixth and seventh rounds of the selection were analyzed by deep sequencing, leading to the identification of several families of enriched peptides (Table S2, ESI†).
Although the initial library was designed to have elongator AUG codons encoding (1S,2S)-2-ACPC at every third position, in the top 100 peptide sequences we found that the enriched peptides contained Ile-, Thr-, or Val-residues at the intended positions of (1S,2S)-2-ACPC (Table S2, ESI†). Such mutations could be introduced by PCR mutations, with the resulting peptides being translated more efficiently than the parental (1S,2S)-2-ACPC encoding peptides. The deep sequencing result suggests the plausible peptide sequences derived from the respective mRNA libraries as designed, except for Mph2 (Table 1); Mph2 has an extra α-amino acid at the N-terminus probably due to PCR mutations. It should be noted that no peptide was found from mRNA library III, probably because of its poorer expression level compared with mRNA library I and II, reducing the translation of the full-length peptides from mRNA library III. Based on the observed high read frequencies and the high numbers of (1S,2S)-2-ACPC appearing in their sequences, we chose peptides Mph1–Mph6 for further analysis (Table 1).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 fitting
:1 fitting
| Originated library | Peptide | Sequence | Read (%) at 6th round | k a (106 M−1 s−1) | k d (10−2 s−1) | K D (nM) |
|---|---|---|---|---|---|---|
| a The origin of library of Mph2 was not determined from the cDNA sequence. | ||||||
| I | Mph1 |

|
14 | — | ||
| Mph2 |

|
2.6 | — | |||
| II | Mph3 |

|
0.94 | 80.0 | 1.21 | 15.1 |
| II | Mph4 |

|
0.76 | — | ||
| I | Mph5 |

|
0.21 | 12.6 | 0.373 | 29.5 |
| I | Mph6 |
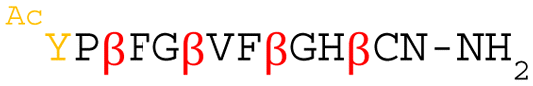
|
0.02 | — | ||
Binding kinetics and inhibitory activity of selected peptides
Peptides Mph1–6 were chemically synthesized (Fig. S3, ESI†) to evaluate binding affinity by surface plasmon resonance (SPR) (Table 1 and Fig. S4, ESI†). Mph1, Mph2, Mph4, and Mph6 did not show reliable SPR fitting data (note that Mph6 is identical to Mph1 except for Ile6). On the other hand, Mph3 and Mph5 exhibited reliable fitting data; their KD values were determined to be 15.1 and 29.5 nM, respectively. Therefore, we chose Mph3 and Mph5 for mutation studies to evaluate the contribution of (1S,2S)-2-ACPC to the binding affinity (Table 2 and Fig. S5, ESI†). Neither Mph3A nor Mph3B with quadruple substitutions manifested reliable binding kinetics with Mpro, indicating that the (1S,2S)-2-ACPC residues in Mph3 are crucial for its excellent binding affinity. Mph5A, which contains the double α-Ala substitutions, also lost binding ability (KD > 10 μM), whereas Mph5B, which contains the double β-Ala substitutions, exhibited a dissociation constant (KD = 16.4 nM) comparable to Mph5. Thus, although the cyclopentane ring of (1S,2S)-2-ACPC in Mph5 is not essential, it is critical to have β-amino acid residues at the relevant positions.| Peptide | Sequence | k a (106 M−1 s−1) | k d (10−2 s−1) | K D (nM) | IC50 (μM) | Serum half-life t1/2 (h) | CP50 (nM) |
|---|---|---|---|---|---|---|---|
| Mph3 |

|
80.0 | 1.21 | 15.1 | 0.62 | 2.4 | 340 |
| Mph3A |

|
— | — | — | 2.9 | <10 min | 1030 |
| Mph3B |

|
— | — | — | >17 | 0.59 | 770 |
| Mph3y |

|
65.2 | 0.576 | 8.83 | 0.96 | 11 | 240 |
| Mph3-tr2 |

|
— | — | — | 5.2 | >24 h | NT |
| Mph5 |
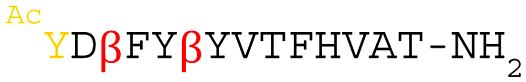
|
12.6 | 0.373 | 29.5 | 0.64 | 2.6 | NT |
| Mph5A |

|
— | — | — | >16 | <10 min | NT |
| Mph5B |

|
23.1 | 0.379 | 16.4 | 0.77 | 0.53 | NT |
The Mpro inhibitory activities of Mph3 and Mph5 were measured by solid-phase extraction coupled with mass spectrometry (Table 2, Fig. S6, ESI†).30 Variable concentrations of each peptide were co-incubated with an 11-mer Mpro substrate (TSAVLQ/SGFRK) and Mpro, and then both hydrolysis product and full-length substrate were quantified by the rapid-fire mass spectrometry. Inhibition of Mpro activity was evaluated by the percentage of substrate cleavage. Both peptides showed potent inhibitory activity with the IC50 values of 0.62 μM and 0.64 μM, respectively. On the other hand, Mph3A showed a ∼5-fold reduced potency (2.9 μM), while Mph3B was only weakly active (>17 μM) (Table 2 and Fig. S6A, ESI†), observations which are consistent with the KD measurement described above. As expected, Mph5B was able to inhibit with comparable potency (0.77 μM) to the parental Mph5, whereas Mph5A did not show inhibitory activity (Table 2 and Fig. S6B, ESI†).
Secondary structural analysis of Mph3 and Mph5
To evaluate the contribution of secondary structure elements of the peptides to binding affinity and inhibitory activity against Mpro, the peptides were analyzed by far-UV circular dichroism (CD) spectroscopy. Gellman and coworkers have reported that helical peptides containing cβAAs, such as 10/11/11-helix and 14-helix, are characterized by a single CD minimum at ∼206 nm and similar to a mixture of α-helix and random coil,31 while common α-helices have CD minima at 222 and 208 nm.32Mph3 has a CD minimum at ∼206 nm in 50%v/v trifluoroethanol (TFE) in phosphate buffer (pH 8.0), corresponding to the reported α/β-peptides with an ααβ pattern (Fig. 3). On the other hand, Mph5, that has an ααβ pattern at its N-terminus, showed a strong CD minimum at 208 nm and a weak shoulder at 222 nm, indicating that it likely has mixed structural features involving both α/β-helix and weak α-helix conformations (Fig. 3). Note that TFE was used to improve the peptide solubility for the CD analysis. As TFE is known to stabilize helical structures,33 the population of helix structures in 50%v/v TFE could be higher than that in water. Interestingly, Mph1, Mph2, Mph4, and Mph6 all exhibited a single intense CD minimum at ∼206 nm (Fig. S7, ESI†), indicating that their preferred secondary structures are α/β-helix regardless of poor binding ability to Mpro. These results imply that our library design enables production of the desired α/β-helical peptides. | ||
| Fig. 3 Far-UV circular dichroism spectra of Mph3, Mph3A, Mph3B (top), and Mph5, Mph5A, Mph5B (bottom). Measurements were performed using 75 μM peptide in 50%v/v trifluoroethanol in 10 mM phosphate buffer (pH 8.0) at room temperature. Red lines, peptides obtained from the RaPID selection; green lines, α-Ala mutants; and blue lines, β-Ala mutants. | ||
We next analyzed the secondary structures of α-Ala and β-Ala mutants of Mph3 and Mph5 (Fig. 3). Mph3A showed a weak CD minimum at 208 nm and a weak shoulder at 222 nm, suggesting that a fraction of Mph3A is folded with an α-helical structure. Mph5A also showed a pronounced α-helical CD signature. This observation is consistent with the data that neither Mph3A nor Mph3B could retain the activity of their (1S,2S)-2-ACPC-containing parent (Table 2). Although Mph3B showed no signature of helicity, Mph5B showed a profile of helicity similar to the parental Mph5. Among the α-Ala/β-Ala-substituted analogs of Mph3 and Mph5, only Mph5B exhibited a comparable inhibitory activity against Mpro to the parent peptide (Mph5), indicating that the α/β-helicity plays a critical role in efficient Mpro inhibition.
Proteolytic stability of the Mph3, Mph5, and mutants
We evaluated the proteolytic stability of Mph3 and Mph5, as well as their α-Ala or β-Ala mutants, Mph3A, Mph5A, Mph3B, and Mph5B (Fig. 4). Each sample was co-incubated with an internal standard peptide in human serum at 37 °C. The quantity of remaining full-length peptides was estimated at each time point (0, 0.5, 1, 2, 4, 6, and 20 or 24 h) by liquid chromatography/mass spectrometry (LC–MS). The results reveal that Mph3 and Mph5 exhibited protease resistance with half-lives (t1/2) of 2.4 and 2.6 h, respectively. Although these half-lives are not comparable to the reported α/β-peptides obtained from RaPID selection,25 these t1/2 values in serum were substantially higher than those of their α-Ala/β-Ala mutants (t1/2 = <10 min for Mph3A and Mph5A, and 0.59 h and 0.53 h for Mph3B and Mph5B, respectively). This result well agrees with the earlier notion that the greater helical propensity owing to the cβAA residues could give rise to greater protease resistance in serum.34,35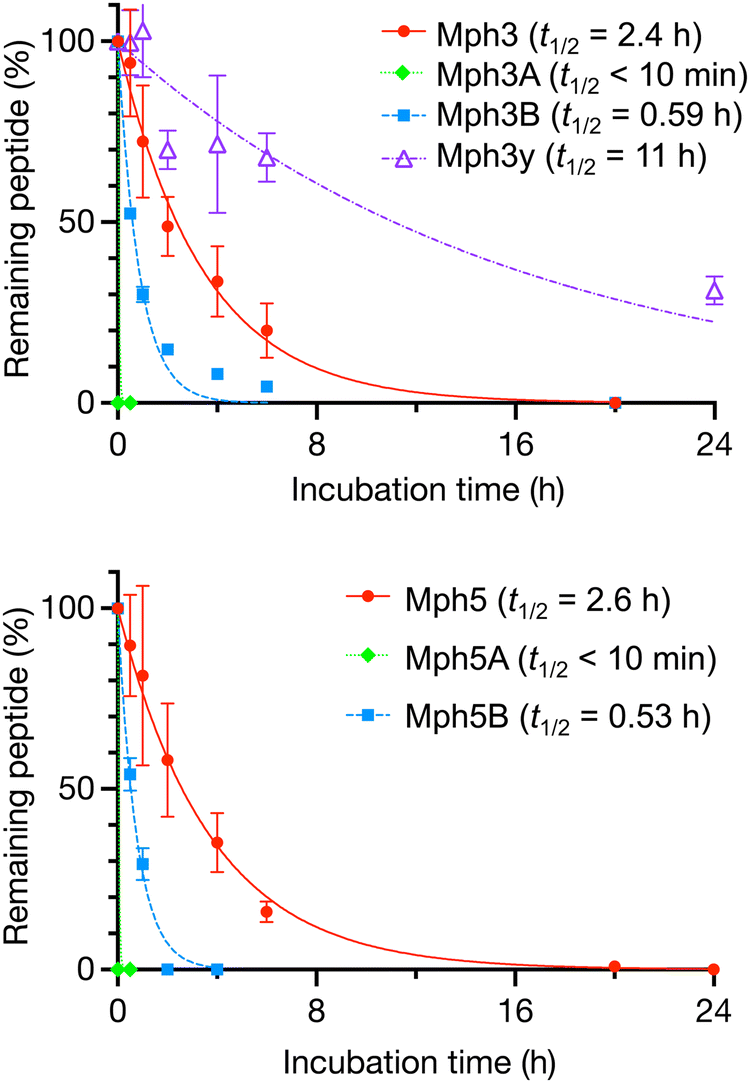 | ||
| Fig. 4 Proteolytic stability of Mph3, Mph3A, Mph3B, Mph3y (top), and Mph5, Mph5A, Mph5B (bottom). The peptides were co-incubated with an internal standard peptide (NH2-PEG5-DWDSDTDNDDDWDSDTDNDD-PEG5-CONH2) in human serum for up to 20 h for (1S,2S)-2-ACPC mutants, and 24 h for Mph3y. The percentage of remaining peptides were estimated by LC/MS. Half-life (t1/2) values were calculated using Prism8 GraphPad software. n = 3. | ||
The peptide bond cleavage sites of Mph3 were analyzed by MALDI-TOF MS and LC–MS (Fig. S8, ESI†). After 20 h incubation in serum, 13-mer and 12-mer fragments, lacking the N-terminal AcY-R (Mph3-tr1) or AcY-R-F (Mph3-tr2) of Mph3, were observed by MALDI-TOF MS (Fig. S8A and B, ESI†). This observation suggested that the N-terminus of Mph3 might be susceptible to peptidases present in serum. To validate the proteolytic stability of these fragments, Mph3-tr1 and Mph3-tr2 were chemically synthesized and tested for serum stability. Surprisingly, the half-life of Mph3-tr1 was even shorter (t1/2 = 0.38 h, Fig. S8C, ESI†) than that of Mph3 (t1/2 = 2.4 h), indicating that the Phe residue was rapidly removed from the N-terminus. On the other hand, Mph3-tr2 was apparently immune to peptidases over 24 h (Fig. S8C, ESI†), but a relatively large fluctuation of data and error range suggested that it might bind to serum proteins, such as albumin, possibly prohibiting peptidase digestions. To design an peptidase-resistant Mph3 analog, we prepared Mph3y, whose N-terminal L-Tyr residue was substituted with the chiral counterpart D-Tyr (y). Notably, the Y1y substitution of Mph3y enhanced resistance to serum peptidases (t1/2 = 11 h) by 4.6-fold compared with the parental Mph3 (t1/2 = 2.4 h).
We also evaluated the Mpro inhibitory activities of Mph3-tr2 and Mph3y (Table 2 and Fig. S6A, S9, ESI†). Mph3-tr2 showed a ∼8-fold lower inhibitory activity (IC50 = 5.2 μM) than the full-length Mph3 (IC50 = 0.62 μM). By contrast to Mph3-tr2, Mph3y exhibited similar levels of inhibition as Mph3 (IC50 = 0.96 μM).
In summary, these studies reveal that the N-terminal AcY-R-F residues of Mph3 play a role in exhibiting its Mpro inhibition activity, but these residues are susceptible to serum peptidases. Complete deletion of these residues increased their serum stability of the helical peptides but in turn caused detrimental loss of inhibitory activity. Alternatively, the Y1y substitution (Mph3y) improved the serum resistance without loss of inhibitory activity.
Cellular uptake of chloroalkane-labeled Mph3
Finally, we examined the cell permeability of the helical peptides by means of the chloroalkane penetration assay (CAPA) developed by the Kritzer group.36 CAPA utilizes covalent bond formation between HaloTag protein and chloroalkane-tag for quantification of the cytosolic delivery of peptides. HEK293H cells expressing the HaloTag on the mitochondrial surface were pulsed with chloroalkane-tagged peptides (ct-peptides), followed by treatment with a chloroalkane-tagged fluorescent dye (ct-TAMRA). If the chloroalkane-tagged molecules permeate cells, they covalently bind to the HaloTag inside the cells via their chloroalkane-tag. Quantification of cell-permeated peptides was conducted by measuring fluorescence of ct-TAMRA that competes with ct-peptides. The chloroalkane-tag was conjugated to the C-terminus of peptides to minimize the false positive caused by peptidase degradation products (Fig. S10, ESI†). The conformations of C-terminal chloroalkane-tagged Mph3 and its mutants (Mph3-ct, Mph3A-ct, Mph3B-ct, and Mph3y-ct, respectively) were similar to those of their parental peptides based on circular dichroism measurements (Fig. S11, ESI†). The potential cytotoxicity of Mph3-ct was also assessed using the Cell Counting Kit-8, showing no significant cytotoxicity even at 20 μM (Fig. S12, ESI†). For CAPA, cells were treated with 10, 40, 156, 625, 2500, and 10000 nM of each ct-peptide for 22 h under 10% FBS conditions to determine a concentration at 50% cell permeation (CP50). As a positive control, a well-studied polycationic CPP with an N-terminal chloroalkane-tag (ct-R9) was used in parallel at concentrations of 4, 15, 62.5, 250, 1000, and 4000 nM.
Remarkably, Mph3-ct exhibited a substantial cell membrane permeability with a CP50 values of 340 nM (Fig. 5), comparable to that of a positive control peptide, ct-R9 (380 nM). Notably, the peptidase-resistant Mph3y-ct showed even better cell permeability with a CP50 value of 240 nM. In contrast, the CP50 values of Mph3A-ct and Mph3B-ct were approximately 3-fold and 2-fold higher (CP50 = 1030 nM and 770 nM, respectively) than that of Mph3-ct.
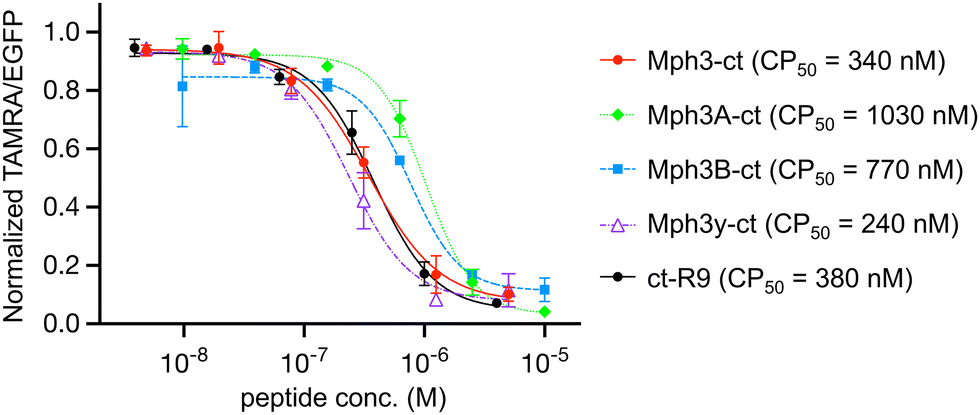 | ||
| Fig. 5 Measurement of cell penetration using the chloroalkane penetration assay (CAPA). Each peptide with chloroalkane-tag (ct) was incubated with HaloTag-GFP-Mito cells in Opti-MEM for 24 h. The fluorescence of living cells and ct-TAMRA was detected and qualified by FACS. The data were normalized using peptide(−) control as 100% signal and peptide(−), ct-TAMRA(−) as 0% signal. CP50 values were calculated using Prism8 GraphPad software. n = 3. | ||
A possible reason for the observed better cell membrane permeability of Mph3/3y-ct over Mph3A/3B-ct is due to the pronounced helicity of Mph3 observed by CD spectroscopy (Fig. 3), likely contributing to the compactness and low water coordination to the backbone. To substantiate this hypothesis, we performed molecular dynamics (MD) simulations to model the tertiary structure of Mph3, Mph3A, and Mph3B. The results imply that the most enriched (or abundant) structures of Mph3 seems to be a 14-helix-like structure characterized by 11 polar contacts (Fig. 6A), with a cluster population of 47% (Fig. 6B). This suggests that Mph3 may have a rigid helical structure and little chance of polar contacts with aqueous solution, both of which should be advantageous for improved cell permeation. In addition, hydrophobic side chains of (1S,2S)-2-ACPCs aligned along one face of the helical conformation, representing another structural feature that might have contributed to enhanced membrane permeability. In contrast, the most enriched structure of Mph3A was calculated as β-sheet-like structure characterized by 4 polar contacts, the cluster population of which was 14% (Fig. 6B). For Mph3B, the maximum cluster population was extremely low (4%), suggesting that Mph3B does not fold into any specific structures.
 | ||
| Fig. 6 Structural calculation of the conformation of (1S,2S)-2-ACPC-containing peptides Mph3, Mph3A, and Mph3B. (A) A MD-generated structure of Mph3. (1S,2S)-2-ACPC is shown in pink and the initiator AcTyr in orange. Polar contacts are shown as dotted lines. (B) Comparison of structural clusters and enriched structures of Mph3, Mph3A, and Mph3B. | ||
Conclusions
The results presented here reveal the power of a modified RaPID system for the ribosomal synthesis of an α/β-helical peptide library and its application for the discovery of SARS-CoV-2 main protease (Mpro) inhibitors. One of the peptides, Mph3, containing four (1S,2S)-2-ACPC residues at designated periodical positions, possesses both Mpro affinity (KD = 15.1 nM) and Mpro inhibitory activity (IC50 = 620 nM, Table 2). Far-UV CD spectroscopy and MD simulation analyses imply that Mph3 very likely adopt 14-helix-like structure characterized by 11 polar intramolecular contacts (Fig. 6A). Importantly, chloroalkane-tagged Mph3-ct exhibits notable cell membrane permeability with a CP50 value of 340 nM, comparable to a known membrane permeable peptide, ct-R9, which has a CP50 of 380 nM (Fig. 5). Despite these favorable properties, Mph3 is susceptible to peptidase degradation in serum with half-lives (t1/2) of only 2.4 h, resulting in fragmentation at the first two N-terminal residues (Table 2). To address this, we substituted its N-acetyl-D-Tyr residues for its L-counterpart, giving Mph3y, an alteration which successfully elevated the serum stability, i.e. from t1/2 = 2.4 to 11 h. Importantly, Mph3y also maintains the favorable KD/IC50 values, as well as cell membrane permeability, of the parental Mph3, identifying it as the most promising peptide inhibitor in our study (Table 2 and Fig. 5).Recently, several studies have identified macrocyclic peptide Mpro inhibitors using the RaPID selection.37–40 Our group reported a novel thioether-macrocyclic peptide Mpro inhibitor containing a cyclic γ-amino acid, referred to as GM4 (IC50 = 50 nM).37 Johnsen-Leete et al. also reported a thioether-macrocyclic peptide Mpro inhibitor (IC50 = 70 nM), whose pharmacophore sequence consist of proteinogenic amino acids.38 However, neither of these macrocyclic peptides showed significant cell membrane permeability. To overcome this issue, the latter group prepared CPP conjugates of the peptide, resulting in antiviral activity in the 10 μM range.38 There is, however, a discrepancy between the in vitro data and cellular data of greater than two orders of magnitude. Harrison et al. developed a hydrophobic cyclic peptide Mpro inhibitor, M1-5X, with the antiviral activity (EC50) of 13.3 μM. However, conjugation with the CPP resulted in cytotoxicity, thereby preventing further improvement in antiviral activity.39 These findings indicates that cell membrane permeability remains a major issue.
The α/β-helical peptides developed in this study, Mph3 and Mph3y, contain the backbone hydrogen bond donors and acceptors in the context of their stable helix structures, enabling them to manifest cell permeability in the 300 nM range. Importantly, our work shows that the de novo α/β-helical peptides freshly isolated by the RaPID selection can exhibit the desired properties against Mpro, giving a potential route for the development of in vivo active peptide inhibitors in the future.
Before progressing to check the intracellular inhibitory activities of obtained peptides, two critical issues must be addressed. Firstly, although the Mph3y variant of Mph3 has an improved anti-peptidase/protease stability, it is yet probably insufficiently stable with only a t1/2 of 11 h. Though Mph3-tr2 is near immune to peptidase digestion, its non-specific interaction with serum proteins and poor binding ability to Mpro prohibited us from advancing it further. The results with Mph3y suggests that a programming for incorporation of unnatural initiators, such as N-acetyl-D-Tyr, will generate an initial library that contains active species with better serum stability; we are thus working to redesign the library and selection campaign. Secondly, despite the positive outcomes of this study, Mph3 shows tight binding to Mpro with 15 nM of KD but its inhibitory activity is limited to an IC50 of 620 nM, i.e., there is a ∼40-fold of difference in KD and IC50 values. GM4 shows a ∼8-fold difference between its KD and IC50 values.37 It is possible that the dimeric nature of Mpro relates to the differences in KD and IC50 values. An X-ray structure of the complex of Mpro and macrocycle GM4 reveals that the certain critical residues of GM4 bind in a deep pocket at the active site; no helical motif was formed in the Mpro complexed GM4 structure. As yet, we do not have an X-ray structure of Mph3 complexed with Mpro which will inform on its binding mode, in particular how deeply the helical motif accesses to the active site and whether or not induced fit occurs on binding. It is possible that the α/β-helical scaffold may be unsuited for potent inhibitors accessing deeply in the active site pocket of Mpro. We envision that a library similar to the helix library described in this work can be applied to other targets to test if potent molecules can be obtained for other proteins, with one focus being on induced fit, particularly in targets with known helical binders.41,42 Nevertheless, the results presented here provide insight that will help in the design of improved α/β-helical peptide libraries.
In conclusion, we have designed and generated an α/β-helical peptide library via genetic code reprogramming that enables installation of up to five cyclic β-amino acids at designated positions. The RaPID selection using this library against SARS-CoV-2 Mpro yielded peptides containing up to four (1S,2S)-2-ACPC residues, one of which exhibited both Mpro inhibitory activity (IC50 = 620 nM) and cell membrane permeability (CP50 = 340 nM). These results show for the first time that de novo α/β-helical peptides can be discovered from such a library against a protein target of choice, opening an avenue for simultaneously identifying drug-like peptides with target binding/inhibitory ability and cell membrane permeability/proteolytic stability properties. This is an important advance because optimizing cell membrane permeability for (cyclic) α-peptides, even when they have very high target affinity, is often time-consuming, if possible at all.
Experimental
Preparation of flexizymes and tRNAs
Flexizymes (dFx, eFx) and tRNAs (tRNAiniCAU, tRNAPro1E2CAU) were prepared by in vitro transcription using the T7 RNA polymerase. Template DNAs with an upstream T7 promoter sequence were prepared by the extension of forward and reverse extension primers followed by PCR amplification using forward and reverser PCR primers [RNA and primer sequences are shown in Table S1(a), ESI†]. PCR products were purified by phenol-chloroform extraction and ethanol precipitation. The purified DNAs were in vitro transcribed overnight at 37 °C in the reaction mixture [40 mM Tris–HCl (pH 8.0), 22.5 mM MgCl2, 1 mM dithiothreitol, 1 mM spermidine, 0.01% Triton X-100, 0.12 μM T7 polymerase, 0.04 U μL−1 RNasin RNase inhibitor (Promega), 3.75 mM NTPs (pH 8.0)]. The DNA in the transcription mixture was degraded by RQ1 DNase (Promega) treatment for 1 h at 37 °C. The resulting RNAs were purified by 12% (for flexizymes) or 8% (for tRNAs) polyacrylamide gel containing 6 M urea.Preparation of aminoacyl-tRNAs
(1S,2S)-2-ACPC and AcTyr were preactivated as their 3,5-dinitrobenzyl ester (DBE) and cyanomethyl ester (CME), respectively, according to reported methods.17,25 These amino acids were then used for the tRNA aminoacylation reaction (tRNAPro1E2CAU for (1S,2S)-2-ACPC, tRNAiniCAU for AcTyr) using the corresponding flexizymes (dFx for DBE, eFx for CME) Reaction was carried out at 4 °C for 2 h (for AcTyr) and 22 h [for (1S,2S)-2-ACPC] in 600 mM MgCl2, 50 mM each buffer [Bicine pH 8.7 for (1S,2S)-2-ACPC, HEPES-KOH pH 7.5 for AcTyr]. The reaction products were purified by ethanol precipitation, then washed twice with 70% ethanol in 0.3 M AcONa and once with 70% ethanol. The resulting aminoacyl-tRNA pellets were air-dried for 5 minutes and dissolved in 1 mM AcONa immediately before use.Preparation of mRNA library encoding α/β-helical peptides
mRNA libraries encoding α/β-helical peptides were prepared by in vitro transcription using the T7 RNA polymerase. Template DNAs with an upstream T7 promoter sequence were prepared by the extension of forward and reverse extension primers followed by PCR amplification using forward and reverser PCR primers (RNA and primer sequences are shown in Table S1(a), ESI†). PCR products were purified by phenol-chloroform extraction and ethanol precipitation. The purified DNAs were in vitro transcribed overnight at 37 °C in the reaction mixture (40 mM Tris–HCl (pH 8.0), 22.5 mM MgCl2, 1 mM dithiothreitol, 1 mM spermidine, 0.01% Triton X-100, 0.12 μM T7 polymerase, 0.04 U μL−1 RNasin RNase inhibitor (Promega), 3.75 mM NTPs (pH 8.0)). DNA in the transcription mixture was degraded by RQ1 DNase (Promega) treatment for 1 h at 37 °C. The resulting RNAs were purified by 8% polyacrylamide gel containing 6 M urea, extracted in 0.3 M NaCl, and collected by ethanol precipitation. The purified RNAs were dissolved in ultrapure water to a final concentration of 10 μM.Translation of α/β-helical peptides
DNA and mRNA libraries (DNA, RNA and primer sequences are shown in Table S1(b), ESI†) were translated using FIT system for 30 min at 37 °C using the following conditions: 50 mM HEPES-KOH (pH 7.6), 100 mM KOAc, 2 mM GTP, 2 mM ATP, 1 mM CTP, 1 mM UTP, 20 mM creatine phosphate, 2 mM spermidine, 2 mM DTT, 12.3 mM Mg(OAc)2, 1.5 mg mL−1E. coli total tRNA, 1.2 μM ribosome, 0.6 μM MTF, 4 μg mL−1 creatine kinase, 3 μg mL−1 Myokinase, 0.1 μM pyrophosphatase, 0.1 μM nucleotide-diphosphatase kinase, 0.1 μM T7 RNA polymerase, 20 μM EF-Tu/EF-Ts, 0.73 μM AlaRS, 0.03 μM ArgRS, 0.38 μM AsnRS, 0.13 μM AspRS, 0.02 μM CysRS, 0.09 μM GlyRS, 0.4 μM IleRS, 0.04 μM LeuRS, 0.11 μM LysRS, 0.68 μM PheRS, 0.16 μM ProRS, 0.04 μM SerRS, 0.09 μM ThrRS, 0.02 μM TyrRS, 0.02 μM ValRS, 0.25 μM RF2, 0.17 μM RF3, 0.5 μM RRF, 2.7 μM IF1, 3 μM IF2, 1.5 μM IF3, 0.1 μM EF-G, 5 mM each α-amino acid (A, R, N, D, C, G, I, L, K, F, P, S, T, Y, and V), 50 μM each pre-charged aminoacyl-tRNA, and 1 μM mRNA library. 50 μM AcTyr-tRNAiniCAU for the initiator AUG codon and 50 μM (1S,2S)-2-ACPC-tRNAPro1E2CAU for elongator AUG codons were added in the translation mixture.Expression and purification of recombinant SARS-CoV-2 Mpro
SARS-CoV-2 Mpro was prepared as reported,30 and assays were performed exclusively using freshly purified recombinant Mpro solution.RaPID selection of peptides against SARS-CoV-2 Mpro
The mRNA library encoding α/β-helical peptides was ligated with puromycin-linker at the 3′-end. The puromycin-ligated mRNA library was then translated for 30 min at 37 °C in a final volume of 150 μL (for the first round of selection) and in a final volume of 5 μL (for the second to sixth round of selection) scale. Puromycin ligated to 3′-end of mRNA was conjugated at C-terminus of corresponding translated peptides by incubation for 12 min at 25 °C. 1 μL of 100 mM EDTA (pH 8.0) was added to quench the translation reaction. The resulting mRNA–peptide conjugates were then reverse transcribed at 42 °C for 15 min with M-MLV reverse transcriptase lacking RNase H activity (Promega) and TGVTNG·R34 primer (5′-TTTCCGCCCCCCGTCCTAACCGTTAGTCACACCCGT-3′). Before the affinity-based selection, Avi-tagged Mpro was immobilized on the Dynabeads M-280 Streptavidin (Thermo Fisher). The mRNA/cDNA–peptide conjugates were applied to Mpro-immobilized Dynabeads and incubated at 4 °C for 15 min (positive selection). For the 2nd to 6th round of selection, beads binding peptides were removed from mRNA/cDNA–peptide libraries (negative selection) by mixing the same volume of negative beads and collecting the supernatant for three times before the positive selection. The beads used for positive selection and 3rd negative selection were then collected and washed with 100 μL of TBS-T buffer (50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.05%v/v Tween20) three times. cDNA was eluted from the beads in 300 μL (for 1st round) or 100 μL (for second to sixth round) of PCR buffer (10 mM Tris–HCl (pH 9.0), 50 mM KCl, 0.1%v/v Triton X-100, 0.25 mM dNTP, 2.5 mM MgCl2, 0.25 μM T7·F52 primer (5′-GGCGTAATACGACTCACTATAGGGTTGAACTTTAAGTAGGAGATATATCCAT-3′) and 0.25 μM TGVTNG·R34 primer) at 95 °C for 5 min. 1 μL of the eluate was taken and mixed with 19 μL of 1× PCR buffer that contained 0.002%v/v SYBR green I in DMSO and 1.5%v/vTaq DNA polymerase for the quantification of cDNA by real-time PCR. The PCR amplification of cDNA in the rest eluate was conducted by adding 1.5%v/vTaq DNA polymerase, followed by phenol/chloroform extraction and ethanol precipitation. The resulting DNA was dissolved in 50 mM KCl and used for in vitro transcription in reaction mixture (40 mM Tris–HCl (pH 8.0), 22.5 mM MgCl2, 1 mM dithiothreitol, 1 mM spermidine, 0.01% Triton X-100, 0.12 μM T7 polymerase, 0.04 U μL−1 RNasin RNase inhibitor (Promega), 3.75 mM NTPs (pH 8.0)) overnight at 37 °C and purified for the subsequent round of selection.Synthesis of α/β-helical peptides
NovaPEG Fmoc Rink Amide resin was added to a 5 mL tube and swelled by DMF. An automatic peptide synthesizer, Syro I, was used for the elongation of peptide sequences. The peptide N-terminus was acetylated using 250 μL of 0.5 M acetic anhydride/0.25 M DIPEA in NMP. The resins were washed with CH2Cl2 and air-dried. Peptides were then cleaved and deprotected by adding 3 mL of a universal cleavage mixture consisting of the ratio of TFA:TIS:DODT:H2O = 92.5:2.5:2.5:2.5. Crude peptide solutions were separated from the resins by filtration and collected in 15 mL tubes. The solutions were applied to centrifugal evaporator to remove TFA. 15 mL of diethyl ether was added to the residues and precipitations were washed with 5 mL of diethyl ether five times and air-dried. The precipitations were dissolved in 5 mL of DMSO. Crude peptide solutions were filtered with 0.45 μm filter and purified by HPLC. Acetonitrile was removed from the fractions using centrifugal evaporator and subsequently the fractions were lyophilized. Peptides were dissolved in DMSO and their concentrations were measured by UV-spectroscopy.
Surface plasmon resonance binding analyses
The binding affinities of the chemically synthesized α/β-helical peptides against Mpro were evaluated by surface plasmon resonance (SPR) using a Biacore T200 machine (Cytiva). Avi-tagged Mpro was immobilized on a Biotin CAPture Chip (Cytiva) to an immobilization level of 1800–2000 response units (RU) using the standard immobilization protocol. Single-cycle kinetics were measured using a five-point, two-fold serial dilution (spanning a range of 1000 nM to 62.5 nM) in running buffer consisting of 50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.05%v/v Tween 20, 2%v/v DMSO at a flow rate of 30 μL min−1. Reference flow-cell was subtracted and the resulting sensorgrams were fitted to a 1:1 binding model using a Biacore evaluation software.
Inhibition assays
Solid-phase extraction (SPE) coupled with mass spectrometry assays were performed as described.30 Inhibition of the activity of isolated recombinant SARS-CoV-2 Mpro with the final concentration of 75/50 nM was monitored by measuring the percentage hydrolysis of the 11-mer substrate TSAVLQ/SGFRKMAFPS-NH2 in the presence of variable concentrations of helical peptides.Circular dichroism analyses
Peptide samples were dried and dissolved in 50%v/v 2,2,2-trifluoroethanol in 10 mM phosphate buffer (pH 8.0) to a concentration of 75 μM, and all samples were measured using a sample cell of 2 mm path length and JASCO J-1000 spectrometer. Baseline spectra recorded with buffer were subtracted from the raw data. Dissolved samples were then measured at 25 °C with 6 times accumulation. Data were converted to ellipticity (deg cm2 dmol−1 res−1) according to the following equation:| [θ] = [θ]obs·A/(l·C) |
Serum stability assays
10 μM synthetic macrocyclic peptides (Mph3, Mph3A, Mph3B, Mph3y, Mph5, Mph5A, Mph5B) were co-incubated with 10 μM internal standard peptide (NH2-PEG5-DWDSDTDNDDDWDSDTDNDD-PEG5-CONH2) in human serum (Cosmo Bio) at 37 °C for up to 24 h. 9 μL of the mixture was sampled at each time point (0, 0.5, 1, 2, 4, 6, and 24 h), and protease reaction was quenched by methanol precipitation. 10 μL of the supernatant was mixed with 90 μL of 1%v/v TFA and centrifuged at 15310 G, 25 °C, for 3 min. The supernatant was collected for liquid chromatography/mass spectrometry analysis with a reverse-phase column (ACQUITY UPLC BEH C18 Column, 130 Å, 1.7 μm, 2.1 × 150 mm; Waters) and a Xevo G2-XS QTof system (Waters) with a linear gradient from 1% to 60% buffer B. Buffer A was H2O with 0.1%v/v formic acid and buffer B was CH3CN with 0.1%v/v formic acid.
Chloroalkane penetration assay (CAPA)
Cell penetration assays using CAPA were carried out according to the protocols of Perato et al.36 HaloTag-GFP-Mito cells used for the assay were HEK293 cells. Cell lines were generated in the laboratory and cultured using DMEM (High Glucose) (Nacalai Tesque) + 10% heat-inactivated FBS + 1% Pen/Strep and kept at 37 °C with 5% CO2. Cells were seeded in Collagen-Coated Microplate 96 well with Lid Collagen Type I (IWAKI) at a density of 4 × 104 cells per well before the day of the experiment. After the attraction of DMEM buffer in each well, peptide solutions dissolved in Opti-MEM®I (1×) (Nacalai Tesque) + 5%v/v FBS containing 1% DMSOv/v were added, followed by 24 h incubation at 37 °C with 5% CO2. Cells were washed by 15 min incubation in Opti-MEM + 5% FBS after attracting peptide solutions, and then incubated with 5 μM ct-TAMRA dissolved in Opti-MEM + 5% FBS containing 0.5%v/v DMSO for 15 min. After removing the media containing ct-TAMRA, the cells were rinsed twice with Opti-MEM + 5% FBS. The cells were trypsinized and resuspended in PBS and then the fluorescence of GFP and ct-TAMRA was measured by CytoFLEX S flow cytometer (Beckman Coulter). All assays were performed in triplicate.Structural calculation using molecular dynamics simulation
The crystallographically-observed helical structures of α/β-helical peptides, including an (1S,2S)-2-ACPC-containing ααβ peptide (PDB 3C3H) and an αααβ peptide (PDB 3C3F),31 provided structural templates for the in silico modeling of the peptides in this work. Peptide models were constructed by modifying sequences to match the peptides of interest, using the mutagenesis tool in PyMOL (v. 2.3.0).43 The most favorable backbone-dependent rotamer for each residue was selected.44 The structures of the α-Ala- or β-Ala-substituted peptide analogs were prepared by removal of atoms from (1S,2S)-2-ACPC. Molecular mechanics (MM) parameters were taken from the Amber ff99SB-ILDN force field,45 except for the charges of the nonstandard β-amino acids [(1S,2S)-2-ACPC and β-Ala] which were calculated using the RESP protocol (Fig. S13 and S14, ESI†).46,47Simulations were performed using pmemd (Amber20).48 Each peptide was placed in a periodic octahedral simulation box with a 1.1 nm minimum distance from the box edges and solvated with TIP3P water molecules.49 The system was neutralized by addition of Na+/Cl− ions, then subjected to a 1000-step steepest descent MM energy minimization, resulting in a geometrically reasonable helical peptide structure free of unfavorable clashes. The system was then for 200 ps in the NVT ensemble (1 kcal mol−1 Å−2 restraint on nonhydrogen atoms), using a 2 fs time steps and with hydrogen-containing bonds constrained by SHAKE.50 The temperature of the system was maintained at 298.15 K using the Langevin thermostat with a collision frequency of (1 ps−1). This was followed by 200 ps unrestrained MD simulations in the NPT at 298.15 K and 1 bar (Berendsen barostat with a relaxation time of 2 ps)51 and further unrestrained NVT relaxation (4.6 ns). Peptide conformational sampling was performed in triplicate using the Gaussian accelerated MD protocol,52 with a total simulation length of 504.6 ns, consisting of 2 ns conventional MD (first 400 ps used as preparation), 2.6 ns boosted MD (first 400 ps as preparation), and 500 ns production phase. Dual boost on both dihedral and total potential energies (σ0 = 6 kcal mol−1) was applied with thresholds set to the lower bounds. Long-range electrostatic interactions were calculated using Particle Mesh Ewald.53 The sampled peptide coordinates were analyzed using GROMACS tools (v. 2019.2),54 including clustering (15000 structures per peptide) with a 3 Å Cα RMSD cut-off by the gromos algorithm.55 Secondary structure was assigned by DSSP (v. 2.0.4).56,57 PyMOL (v. 2.3.0)43 was used for analysis and visualization of peptide structures and polar contacts. Note that the environment for the MD simulations were performed in aqueous solution, whereas the CD measurements were performed in 50%v/v TFE due to the relatively low water solubility of peptides.
Author contributions
M. K., T. K. conceived the study. M. K., T. R. M., H. T. H. C., A. T., L. B., E. S., N. T. planned and performed the experiments. M. K., T. R., H. T. H. C., L. B., T. K., A. K., C. J. S., F. D., and H. S. wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for JSPS Fellows (21J11468) to M. K.; Grant-in-Aid for Scientific Research (A) (22H00439) and Grant-in-Aid for Challenging Research (Pioneering) (21K18233 and 24K21267) to T. K.; Grant-in-Aid for Specially Promoted Research (20H05618) to H. S.Notes and references
- T. Katsu, M. Kuroko, T. Morikawa, K. Sanchika, H. Yamanaka, S. Shinoda and Y. Fujita, Biochim. Biophys. Acta, 1990, 1027, 185–190 CrossRef CAS PubMed.
- K. K. Hou, H. Pan, G. M. Lanza and S. A. Wickline, Biomaterials, 2013, 34, 3110–3119 CrossRef CAS PubMed.
- K. T. O’Neil and W. F. DeGrado, Trends Biochem. Sci., 1990, 15, 59–64 CrossRef PubMed.
- W. N. Hait, L. Grais, C. Benz and E. C. Cadman, Cancer Chemother. Pharmacol., 1985, 14, 202–205 CrossRef CAS PubMed.
- M. Moreno and E. Giralt, Toxins, 2015, 7, 1126–1150 CrossRef CAS PubMed.
- M. J. I. Andrews and A. B. Tabor, Tetrahedron, 1999, 55, 11711–11743 CrossRef CAS.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3–33 CAS.
- C. Cabrele, T. A. Martinek, O. Reiser and Ł. Berlicki, J. Med. Chem., 2014, 57, 9718–9739 CrossRef CAS PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- B. F. Fisher, S. H. Hong and S. H. Gellman, J. Am. Chem. Soc., 2018, 140, 9396–9399 CrossRef CAS PubMed.
- L. M. Johnson and S. H. Gellman, Methods Enzymol., 2013, 523, 407–429 CAS.
- M. A. Schmitt, S. H. Choi, I. A. Guzei and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 4538–4539 CrossRef CAS PubMed.
- S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 2917–2924 CrossRef CAS PubMed.
- M. A. Schmitt, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 417–428 CrossRef CAS PubMed.
- M. D. Boersma, H. S. Haase, K. J. Peterson-Kaufman, E. F. Lee, O. B. Clarke, P. M. Colman, B. J. Smith, W. S. Horne, W. D. Fairlie and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 315–323 CrossRef CAS PubMed.
- J. W. Checco, E. F. Lee, M. Evangelista, N. J. Sleebs, K. Rogers, A. Pettikiriarachchi, N. J. Kershaw, G. A. Eddinger, D. G. Belair, J. L. Wilson, C. H. Eller, R. T. Raines, W. L. Murphy, B. J. Smith, S. H. Gellman and W. D. Fairlie, J. Am. Chem. Soc., 2015, 137, 11365–11375 CrossRef CAS PubMed.
- H. Murakami, A. Ohta, H. Ashigai and H. Suga, Nat. Methods, 2006, 3, 357–359 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Nat. Protoc., 2011, 6, 779–790 CrossRef CAS PubMed.
- T. Katoh, Y. Iwane and H. Suga, Nucleic Acids Res., 2017, 45, 12601–12610 CrossRef CAS PubMed.
- T. Katoh and H. Suga, J. Am. Chem. Soc., 2018, 140, 12159–12167 CrossRef CAS PubMed.
- T. Dale and O. C. Uhlenbeck, Trends Biochem. Sci., 2005, 30, 659–665 CrossRef CAS PubMed.
- T. Dale, L. E. Sanderson and O. C. Uhlenbeck, Biochemistry, 2004, 43, 6159–6166 CrossRef CAS PubMed.
- T. Katoh, I. Wohlgemuth, M. Nagano, M. V. Rodnina and H. Suga, Nat. Commun., 2016, 7, 11657 CrossRef CAS PubMed.
- T. Katoh and H. Suga, J. Am. Chem. Soc., 2020, 142, 16518–16522 Search PubMed.
- T. Katoh, T. Sengoku, K. Hirata, K. Ogata and H. Suga, Nat. Chem., 2020, 12, 1081–1088 CrossRef CAS PubMed.
- T. Katoh and H. Suga, BCSJ, 2021, 94, 549–557 Search PubMed.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562–1570 CrossRef CAS PubMed.
- T. Fujino, Y. Goto, H. Suga and H. Murakami, J. Am. Chem. Soc., 2016, 138, 1962–1969 CrossRef CAS PubMed.
- P. V’kovski, A. Kratzel, S. Steiner, H. Stalder and V. Thiel, Nat. Rev. Microbiol., 2021, 19, 155–170 CrossRef PubMed.
- T. R. Malla, A. Tumber, T. John, L. Brewitz, C. Strain-Damerell, C. D. Owen, P. Lukacik, H. T. H. Chan, P. Maheswaran, E. Salah, F. Duarte, H. Yang, Z. Rao, M. A. Walsh and C. J. Schofield, Chem. Commun., 2021, 57, 1430–1433 RSC.
- W. S. Horne, J. L. Price and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9151–9156 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- D. Roccatano, G. Colombo, M. Fioroni and A. E. Mark, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12179–12184 CrossRef CAS PubMed.
- K. J. Peterson-Kaufman, H. S. Haase, M. D. Boersma, E. F. Lee, W. D. Fairlie and S. H. Gellman, ACS Chem. Biol., 2015, 10, 1667–1675 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756 Search PubMed.
- L. Peraro, K. L. Deprey, M. K. Moser, Z. Zou, H. L. Ball, B. Levine and J. A. Kritzer, J. Am. Chem. Soc., 2018, 140, 11360–11369 CrossRef CAS PubMed.
- T. Miura, T. R. Malla, C. D. Owen, A. Tumber, L. Brewitz, M. A. McDonough, E. Salah, N. Terasaka, T. Katoh, P. Lukacik, C. Strain-Damerell, H. Mikolajek, M. A. Walsh, A. Kawamura, C. J. Schofield and H. Suga, Nat. Chem., 2023, 15, 998–1005 CrossRef CAS PubMed.
- J. Johansen-Leete, S. Ullrich, S. E. Fry, R. Frkic, M. J. Bedding, A. Aggarwal, A. S. Ashhurst, K. B. Ekanayake, M. C. Mahawaththa, V. M. Sasi, S. Luedtke, D. J. Ford, A. J. O’Donoghue, T. Passioura, M. Larance, G. Otting, S. Turville, C. J. Jackson, C. Nitsche and R. J. Payne, Chem. Sci., 2022, 13, 3826–3836 RSC.
- K. Harrison, P. W. Carlos, S. Ullrich, A. Aggarwal, J. Johansen-Leete, V. M. Sasi, I. Barter, J. W. C. Maxwell, M. J. Bedding, M. Larance, S. Turville, A. Norman, C. J. Jackson, C. Nitsche and R. J. Payne, Chemistry, 2024, e202401606 CrossRef CAS PubMed.
- Y. Tan, J. Yang, M. Wang, Q. Peng, Y. Li, L. Fu, M. Zhang, J. Wu, G. Yang, C. J. Hipolito, Y. Zhang, J. Qi, Y. Shi and Y. Yin, J. Med. Chem., 2024, 67, 20258–20274 CrossRef CAS PubMed.
- Y. Mesrouze, H. Gubler, F. Villard, R. Boesch, J. Ottl, J. Kallen, P. C. Reid, C. Scheufler, A. L. Marzinzik and P. Chène, ACS Chem. Biol., 2023, 18, 643–651 CrossRef CAS PubMed.
- F. Hink, J. Aduriz-Arrizabalaga, X. Lopez, H. Suga, D. De Sancho and J. M. Rogers, J. Am. Chem. Soc., 2024, 146, 24348–24357 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 2.3.0, Schrödinger LLC Search PubMed.
- R. L. Dunbrack Jr and F. E. Cohen, Protein Sci., 1997, 6, 1661–1681 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, K. Palmo, P. Maragakis, J. L. Klepeis, R. O. Dror and D. E. Shaw, Proteins, 2010, 78, 1950–1958 CrossRef CAS PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly and P. A. Kollman, J. Am. Chem. Soc., 1993, 115, 9620–9631 CrossRef CAS.
- D. A. Case, H. M. Aktulga, K. Belfon, D. S. Cerutti, G. A. Cisneros, V. W. D. Cruzeiro, N. Forouzesh, T. J. Giese, A. W. Götz, H. Gohlke, S. Izadi, K. Kasavajhala, M. C. Kaymak, E. King, T. Kurtzman, T.-S. Lee, P. Li, J. Liu, T. Luchko, R. Luo, M. Manathunga, M. R. Machado, H. M. Nguyen, K. A. O’Hearn, A. V. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, A. Risheh, S. Schott-Verdugo, A. Shajan, J. Swails, J. Wang, H. Wei, X. Wu, Y. Wu, S. Zhang, S. Zhao, Q. Zhu, T. E. Cheatham 3rd, D. R. Roe, A. Roitberg, C. Simmerling, D. M. York, M. C. Nagan and K. M. Merz Jr, J. Chem. Inf. Model., 2023, 63, 6183–6191 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- Y. Miao, V. A. Feher and J. A. McCammon, J. Chem. Theory Comput., 2015, 11, 3584–3595 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 10089–10092 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- X. Daura, K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren and A. E. Mark, Angew. Chem., Int. Ed., 1999, 38, 236–240 CrossRef CAS.
- W. G. Touw, C. Baakman, J. Black, T. A. H. te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS PubMed.
- R. P. Joosten, T. A. H. te Beek, E. Krieger, M. L. Hekkelman, R. W. W. Hooft, R. Schneider, C. Sander and G. Vriend, Nucleic Acids Res., 2011, 39, D411–D419 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00021a |
| This journal is © The Royal Society of Chemistry 2025 |