
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSiderophore-based targeted antibody recruitment for promoting immune responses towards Gram-negative pathogens†
Seungwoo
Kim
a,
Ho-Sung
Park
b,
Do Young
Kim
 a,
Hyunhi
Joh
a,
Jiseok
Oh
a,
Dong Ho
Kim
b,
Min Ju
Kang
b,
Chul Hee
Choi
*b and
Hak Joong
Kim
*a
a,
Hyunhi
Joh
a,
Jiseok
Oh
a,
Dong Ho
Kim
b,
Min Ju
Kang
b,
Chul Hee
Choi
*b and
Hak Joong
Kim
*a
aDepartment of Chemistry and Center for Proteogenome Research Korea University Seoul, 02841, Republic of Korea. E-mail: hakkim@korea.ac.kr
bDepartment of Microbiology and Medical Science Chungnam National University School of Medicine Daejeon, 35015, Republic of Korea. E-mail: choich@cnu.ac.kr
First published on 16th January 2025
Abstract
Antibody-recruiting molecules (ARMs) have emerged as a promising strategy for enhancing immune responses against pathogens and cancer cells. In this study, we developed a novel class of antibacterial ARMs utilizing siderophores, small iron-chelating compounds, as targeting motifs. Siderophores naturally exhibit high specificity for bacterial pathogens due to their role in iron acquisition, making them ideal candidates for selective targeting. We identified a potent ARM, GNP3, comprising MECAM, a siderophore mimetic, and 2,4-dinitrophenyl (DNP), a motif recognized by endogenous antibodies, connected via a flexible linker. GNP3 binds simultaneously to both anti-DNP antibody and the siderophore receptor, FepA, facilitating the targeted deposition of antibodies on the surface of FepA-expressing bacterial cells, such as Escherichia coli and Pseudomonas aeruginosa. This GNP3-induced opsonization promoted robust immune responses, including complement-dependent cytotoxicity (CDC) in the presence of serum and macrophage-mediated phagocytosis. Moreover, GNP3 effectively triggered CDC activity against serum-resistant uropathogenic E. coli. The results suggest that siderophore-based ARMs, by harnessing the immune defense system, represent a promising complementary approach to traditional antibiotics for overcoming recalcitrant bacterial infections.
Introduction
The widespread antibiotic resistance poses a serious challenge to treating bacterial infections by severely limiting available therapeutic options.1,2 Additionally, many pathogens found in clinical settings show high resistance to human serum.3,4 The humoral immunity provided by serum is a crucial first-line defense mechanism against invading pathogens, primarily through complement-dependent cell killing and phagocytic clearance. Such immune responses often begin with the opsonization of bacterial cells, where their surface is labeled with antibodies. Although serum resistance develops in various ways, a common mechanism involves altering cell surface architecture, allowing pathogens to evade antibody surveillance.4,5 As such, the immunotherapy addressing these immune evasive behaviors in pathogens could provide an effective alternative and/or complement to antibiotics.Recently, several research groups have explored a new antibacterial immunotherapy approach based on the use of bifunctional molecules, called “antibody-recruiting molecules” (ARMs).6,7 These chemical modalities are designated to direct endogenous antibodies present in serum to bacterial surfaces, thereby stimulating immune responses. Structurally, they are composed of a bacterial surface-targeting ligand and a hapten recognizable by endogenous antibodies. The dinitrophenyl (DNP) moiety is the most frequently employed hapten for this purpose, because 1–2% of endogenous antibodies in human body recognize this motif.8,9 Since the pioneering work by Schultz in 1991,10 this passive vaccination approach has been applied primarily to developing anti-cancer agents. Recently, promising results have also been demonstrated in antibacterial applications.11–21
The judicious selection of a surface epitope is the crucial first step in generating a successful ARM. The epitopes exploited for bacterial killing thus far include mannose receptor,11 lectin,17 fibril protein,18 and cell wall components12–16,19–21 such as peptidoglycan and mycomembrane. Effective epitopes for bacterial killing should have high expression levels on the target pathogen, significant exposure to the extracellular environment, and cognate ligands with strong affinity.18 In this context, we reasoned that siderophore transporters would be suitable epitopes for developing antibacterial ARMs (Fig. 1).
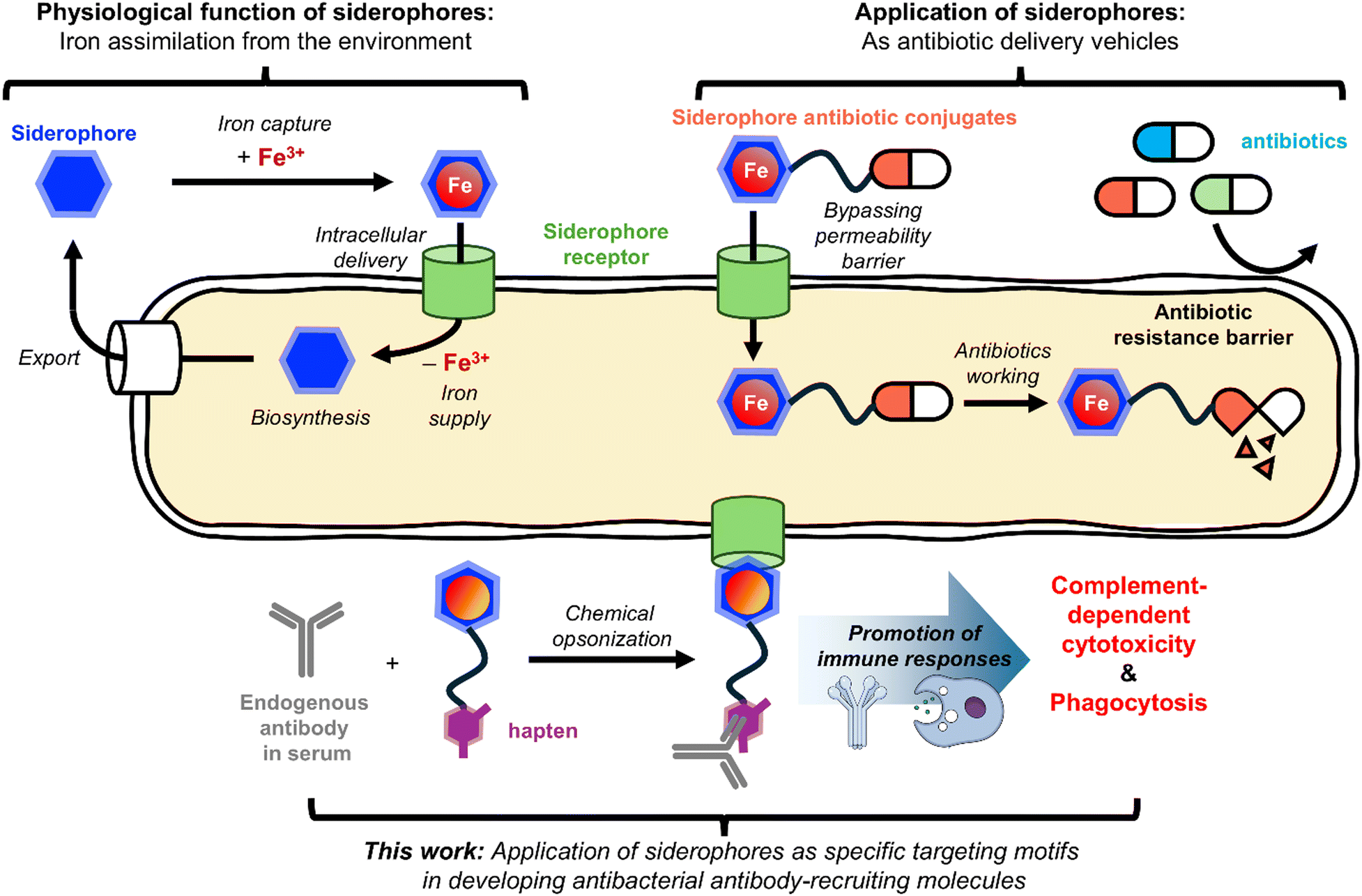 | ||
| Fig. 1 Schematic overview of this work. | ||
Siderophores are natural chelators produced by most pathogens to assimilate iron, an essential nutrient, from the environment.22–25 The intracellular iron supply via siderophores involves active transport through their cognate transporters. Siderophore transporters are displayed on membrane surfaces and, importantly, overexpressed in pathogens for their survival at infection sites. Additionally, siderophores bind tightly to their transporters,26,27 thus making the siderophore-transporter pair an ideal target for ARM development.
Herein, we report that siderophore-DNP conjugates can direct anti-DNP antibodies to the surface of Escherichia coli and other Gram-negative pathogens, triggering their complement-dependent cytotoxicity (CDC) and phagocytosis. Moreover, this siderophore-based ARM was able to induce antibody-mediated cytotoxicity against serum-resistant uropathogenic E. coli (UPEC) strains, showcasing the potential of the ARM strategy exploiting siderophore receptors in overcoming serum resistance.
Results
The study to validate the siderophore-based ARM concept commenced with synthesis of a series of MECAM-DNP conjugates with systematically varied linker lengths. MECAM was selected as the targeting siderophore, because it is a well-established enterobactin (ENT) mimetic28 and the uptake system for ENT is present in many Gram-negative pathogens. The synthesis process, inspired by a recent study on MECAM-antibiotic conjugates,29 began with the nitration of 1,3,5-tris(bromomethyl)benzene (1) (Scheme 1). Subsequent substitution of the bromides in compound 2 with primary amines was achieved using ammonia. Next, the O-acetyl protected catechol moiety was then introduced to all amine groups using acyl chloride 3 (Scheme S1, ESI†), resulting in compound 4. For effective incorporation of both the linker and DNP, a series of DNP-linked acyl chlorides (5a–5d) were prepared as delineated in Scheme S2 (ESI†). After reducing the nitro group of compound 4, the resulting amine intermediate was treated with 5a–5d in the presence of a base, followed by global deacetylation to yield the MECAM-DNP conjugates, 6a–6d. Finally, to prevent any perturbation in the mobile iron level during biological characterization, these apo-compounds were complexed with Ga(III) ions to produce the corresponding holo-forms, GNP1–4 (7a–7d, respectively). | ||
Scheme 1 Synthesis of Ga(III)-complexed MECAM-DNP conjugates, GNP1–4 (MECAM in blue, DNP in red). *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The presented yields indicate the results of the corresponding three-step processes. The presented yields indicate the results of the corresponding three-step processes. | ||
The first question to address was whether MECAM-DNP conjugates could recruit anti-DNP antibody to opsonize E. coli. For this purpose, E. coli BW25113 ΔentA, a mutant where ENT biosynthesis was impaired, was treated with GNP1–4 in the presence of AF488-labeled anti-DNP antibody. The fluorescence microscopy analysis showed that all four conjugates were able to deposit the antibody on the surface of E. coli, in contrast to Ga(III)-MECAM alone (Fig. 2A). Additionally, flow cytometry analysis showed that the linker length had significant influence on the antibody-recruiting efficiency, where GNP3 was identified as the best ARM (Fig. 2B). Functional characterization of GNP3 revealed that it exhibited neither cytotoxic effects on E. coli (Fig. S2 and S3, ESI†) nor promoted its growth under iron-deficient conditions (Fig. S4, ESI†).
 | ||
| Fig. 2 Analysis of the opsonization and complement-dependent cytotoxicity (CDC) of E. coli induced by MECAM-DNP conjugates combined with anti-DNP antibody. Comparison of opsonization activity among GNP1–4 using (A) fluorescence microscopy and (B) flow cytometry (n = 3). Identification of key components in the siderophore uptake machinery involved in GNP3-dependent opsonization using (C) fluorescence microscopy and (D) flow cytometry (n = 3). CDC assay results: (E) comparison of GNP1–4 in CDC induction; (F) identification of essential elements for GNP3-dependent CDC; (G) confirmation of the need for simultaneous binding of GNP3 to both the siderophore receptor and anti-DNP antibody for activity; and (H) differential responses of siderophore uptake mutants to GNP3-dependent CDC, confirming the importance of FepA in GNP3 activity. In all assays, 100 nM GNP1–4 (Fig. S1, ESI†), 0.02 mg mL−1 (ca. 130 nM) antibody, and 10% HCS (only for CDC assays) were used in pH 7.4 PBS buffer. Fluorescence images were acquired under consistent exposure settings and identically processed. Cytotoxicity was determined by colony forming unit (CFU per mL) measurements (n = 10) at 25 min time points after compound treatment (Fig. S5 and S6, ESI†). Data are presented as means ± standard error. Statistical significance was determined using two-tailed Student's t-tests (ns: not significant, *p < 0.0332, **p < 0.0021, ***p <0.0002, ****p < 0.0001). | ||
Opsonization is a cell surface event and thus the antibody-recruiting activity of GNP3 would depend on its interaction with a siderophore receptor located on the outer membrane (OM). To identify the specific OM receptor responsible for GNP3-induced opsonization, various E. coli mutant strains were treated with GNP3, along with AF488-labeled anti-DNP antibody, followed by fluorescence analysis. As illustrated in Fig. 2C and D, the labeling by GNP3 vanished near completely in the absence of FepA, the primary OM receptor for ENT uptake, indicating that FepA is likely the target receptor for GNP3. By contrast, disruption of other components involved in siderophore uptake, such as alternative OM receptors (CirA and Fiu), a periplasmic binding protein (FepB), and an ATP-binding protein of the inner membrane permease complex (FepC), had marginal effects on the labeling efficiency of GNP3. Following the confirmation of successful MECAM-DNP conjugate-dependent antibody deposition, we investigated whether this promoted opsonization could translate into enhanced CDC. To evaluate this, E. coli strains were treated with human complement serum (HCS), the MECAM-DNP conjugate, and unlabeled anti-DNP antibody. Notably, we used HCS after lysozyme depletion to focus exclusively on complement pathway activation. As shown in Fig. 2E, GNP3 exhibited the highest potency among the tested conjugates (GNP1–4), highlighting it as the most effective ARM.
The cytotoxicity induced by GNP3 was dependent on the presence of both anti-DNP antibody and active HCS, because significant activity reduction was noted in the absence of either component (Fig. 2F). Intriguingly, substantial CDC activity was observed even without the exogenously added anti-DNP antibody, supporting the presence of DNP-recognizing antibodies in the endogenous antibody pool of HCS.8,9 The failure to rescue activity by substituting anti-His antibody for anti-DNP antibody emphasized the necessity of specific antibody binding to the DNP motif. Furthermore, using heat-inactivated HCS did not induce bactericidal activity, reinforcing that GNP3-promoted cytotoxicity relies on active complement proteins. Moreover, GNP3-induced CDC appeared to require its concurrent binding to both anti-DNP antibody and FepA. This was evidenced by the observation of significant GNP3 activity attenuation when competing molecules, Ga(III)-complexed MECAM/ENT and DNP ligand (8c, Scheme S2, ESI†), were introduced in 10-fold excess (Fig. 2G). Further analysis revealed that the CDC activity of GNP3 was dependent on the presence of the FepA receptor (Fig. 2H), aligning with the opsonization test results. It is worth noting that disruption of entA enhanced the CDC activity of GNP3, likely due to the absence of competition between ENT and GNP3 for FepA binding. Collectively, these findings successfully demonstrate that a well-designed siderophore-based ARM can effectively trigger CDC against a bacterium expressing the corresponding siderophore OM receptor by recruiting complement components to the bacterial surface via antibody-mediated mechanisms, as hypothesized.
Active uptake of ENT has been observed not only in E. coli, but also in other Gram-negative bacteria, suggesting that GNP3 could potentially enhance the CDC against pathogens beyond E. coli. To explore this, the ability of GNP3 to opsonize representative Gram-negative pathogens, Acinetobacter baumannii, Klebsiella pneumoniae, and Pseudomonas aeruginosa, was tested using fluorescence analysis. As shown in Fig. 3A and B, P. aeruginosa was the most efficiently opsonized organism by GNP3 among these three bacteria, consistent with a recent report by Fritsch et al. that demonstrated efficient iron delivery by MECAM in P. aeruginosa.30 Surprisingly, A. baumannii and K. pneumoniae were poorly labeled, despite both possessing close homologs to FepA, suggesting a likelihood of their interacting with MECAM.31,32 Consistent with the opsonization results, P. aeruginosa was susceptible to GNP3-promoted CDC enhancement as shown in Fig. 3C, supporting the therapeutical potential of GNP3 in controlling other Gram-negative pathogens.
 | ||
| Fig. 3 Assessment of GNP3 activity, in combination with anti-DNP antibody, against (A)–(C) various Gram-negative bacteria beyond E. coli and (D)–(F) serum-resistant uropathogenic E. coli strains. Opsonization activity assay results from (A) and (D) fluorescence microscopy and (B) and (E) flow cytometry (n = 3). (C) and (F) CDC activity assay results in the presence of HCS (n = 10) (Fig. S8 and S9, ESI†). In all assays, 100 nM GNP3, 0.02 mg mL−1 antibody, and 10% HCS (only for CDC assays) were used in pH 7.4 PBS buffer. Data are represented as means ± standard error. Statistical significance was determined by two-tailed Student's t-tests (ns: not significant, *p < 0.0332, **p < 0.0021, ***p <0.0002, ****p < 0.0001). | ||
Uropathogenic E. coli (UPEC) is the primary causative bacterium responsible for urinary tract infection and poses growing concerns due to its rising resistance to both antibiotics and humoral immunity.33–35 A recent meta-analysis found that over 50% of UPEC strains exhibit genotypic traits linked to serum resistance.36 To address this challenge, we explored a siderophore-based ARM strategy to determine whether it could increase the vulnerability of UPEC to CDC, thereby alleviating their serum resistance. We selected three UPEC strains for this investigation: CFT073, a well-established model strain, and two patient-derived strains, UPEC 26-1 and UPEC 39,37 all of which demonstrated serum resistance. As illustrated in Fig. 3D and E, the fluorescence-based opsonization test revealed that GNP3 could recruit anti-DNP antibodies to CFT073 and UPEC 26-1, but not to UPEC 39. Although the recruitment efficiency for CFT073 and UPEC 26-1 was lower than for E. coli BW25113, treatment with a combination of GNP3, HCS, and anti-DNP antibody resulted in significant bactericidal activity against these two strains (Fig. 3F). As predicted by the opsonization test results, no CDC activity was observed for UPEC 39. At this stage, the reason for the variation in opsonization efficiency across different strains is unclear. Intriguingly, all UPEC strains tested exhibited higher fepA expression levels compared to E. coli BW25113 (Fig. S10, ESI†), suggesting that the reprogrammed membrane structure, such as capsule formation and modifications in the lipopolysaccharide layer, might be responsible for limiting the accessibility of anti-DNP antibodies and hence complement components. Nevertheless, the observed CDC induction by GNP3 in CFT073 and UPEC 26-1 highlights that the antibody-recruiting strategy holds potential for counteracting serum resistance associated with UPECs.
Antibody deposition on the bacterial surface not only triggers CDC, but also enhances phagocytosis by immune cells such as macrophages and neutrophils. To investigate whether GNP3-promoted opsonization of E. coli could indeed enhance phagocytosis by macrophages, THP-1-derived macrophage cells were co-cultured with E. coli BW25113 cells, treated with either PBS or 100 nM GNP3 in the presence of anti-DNP antibody. Following harvest of the co-cultured cells, the infected cells were stained with fluorescence dyes, AF568-conjugated phalloidin for actin and DAPI for nuclei visualization. As shown in Fig. 4A, confocal microscopic analysis revealed clearly enhanced phagocytosis in the sample IV treated with both GNP3 and anti-DNP antibody, compared to the other controls (samples I–III). For more quantitative assessment, the intracellular bacterial cells engulfed by macrophages were counted by determining CFU per mL, which indicated that the enhancement of phagocytosis occurred owing to the presence of both GNP3 and anti-DNP antibody (Fig. 4B). These observations align with the CDC assay results, supporting the conclusion that siderophore-based ARMs like GNP3 can effectively promote bacterial clearance through enhancement of not only CDC, but also phagocytosis.
 | ||
| Fig. 4 Antibody-dependent phagocytosis assays. (A) Confocal microscopy images showing DAPI staining of nuclei in both macrophages and E. coli (blue), with AF568-conjugated phalloidin staining F-actin filaments (red). White arrows indicate engulfed E. coli cells. (B) Quantification of engulfed E. coli based on colony-forming unit (CFU) counts. Data are represented as means ± standard error (n = 10). Statistical significance was determined by two-tailed Student's t-tests (ns: not significant, ****p < 0.0001). | ||
Conclusion
In this study, we demonstrated that siderophores can serve as effective bacteria-targeting motifs for developing ARMs. Specifically, the MECAM-DNP conjugate, GNP3, successfully opsonized bacterial cells expressing FepA in the presence of anti-DNP antibody, thereby promoting complement-mediated killing and phagocytosis. Using siderophores as targeting motifs offers several advantages. First, siderophore-based ARMs may be less prone to resistance development, as the siderophore uptake machinery is a key virulence factor essential for the survival of many pathogens at infection sites.22–25 Second, the highly specific interactions between siderophores and their corresponding OM receptors suggest that siderophore-based ARMs would operate within a narrow spectrum, targeting only pathogens that express compatible OM receptors and thus minimizing perturbations in the human microbiota. This specificity is important from a safety perspective, because broad-spectrum ARMs carry a risk of excessive immune activation, which could be harmful to patients. Lastly, the small molecule nature of siderophores makes them more suitable for therapeutic development compared to previously reported antibacterial ARMs that use peptides, glycoconjugates, and aptamers as targeting motifs,11,14,17,18,20 or require metabolic labeling to prime the bacterial surface.12,13,15,19,21The utilization of siderophores in antibacterial development is not new. In particular, over the past few decades, they have been widely exploited as vehicles for the intracellular delivery of antibiotic drugs, with the primary goal of overcoming the membrane permeability barrier found in many antibiotic-resistant pathogens (Fig. 1).38,39 These efforts have been fruitful, culminating in the recent clinical approval of the siderophore-based antibiotic, cefiderocol.40 However, despite this progress, several challenges remain in developing effective siderophore-based antibiotic delivery systems. These include a limited molecular understanding of siderophore transport mechanisms and difficulties in delivering cytoplasmic antibiotics to target Gram-negative pathogens.41 The current study has explored another new application of siderophores in ARM development. We specifically envisioned that the siderophore-based ARM modality could help alleviate serum resistance observed in serious pathogens. While the CDC assays using our most potent ARM, GNP3, against UPEC strains have shown encouraging results, the observed selective responses present the next challenge to address. This will require an in-depth examination of the serum resistance mechanisms and further structural optimization of the ARM. Nevertheless, given the rich repertoire of siderophores and their synthetic accessibility, we anticipate that next generation siderophore-based ARMs will overcome the current limitations of GNP3 and be tailored to target a broader range of serious pathogens beyond E. coli and P. aeruginosa in near future.
Author contributions
S. W. Kim and H. J. Kim conceptualized the project. S. W. Kim, D. Y. Kim, H. H. Joh, and J. S. Oh performed synthesis and purification. S. W. Kim performed imaging of bacterial cells, flow cytometry, and CDC assay. H. S. Park, D. H. Kim, and M. J. Kang performed phagocytosis assay. H. J. Kim and C. H. Choi supervised the project and provided scientific guidance.Data availability
All supplementary data, biological characterization procedures, and characterization data of all synthesized compounds are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Bio&Medical Technology Development Program and the Mid-career Research Grant of the National Research Foundation of Korea (NRF) funded by the Korean government (MSIT) (RS-2023-00259824 and NRF-2021R1A2C1094331, respectively) and the Korea Health Technology R&D Project Grant HR22C1734 through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare.Notes and references
- A. R. Collaborators, C. J. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. R. Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. K. Hamadani, E. A. P. Kumaran, B. McManigal, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. D. Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, M. Khorana, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, N. Mturi, T. Munera-Huertas, P. Musicha, M. M. Mussi-Pinhata, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, A. Y. Peleg, C. Perrone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, K. E. Rudd, N. Russell, J. Schnall, J. A. G. Scott, M. Shivamallappa, J. Sifuentes-Osornio, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, H. Vu, T. Walsh, S. Waner, T. Wangrangsimakul, T. Wozniak, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Lancet, 2022, 399, 629 CrossRef PubMed.
- S. Walesch, J. Birkelbach, G. Jézéquel, F. P. J. Haeckl, J. D. Hegemann, T. Hesterkamp, A. K. H. Hirsch, P. Hammann and R. Müller, EMBO Rep., 2023, 24, e56033 CrossRef CAS PubMed.
- P. Lê-Bury, H. Echenique-Rivera, J. Pizarro-Cerdá and O. Dussurget, FEMS Microbiol. Rev., 2024, 48, fuae013 CrossRef PubMed.
- H. Miajlovic and S. G. Smith, FEMS Microbiol. Lett., 2014, 354, 1 CrossRef CAS PubMed.
- J. D. Lambris, D. Ricklin and B. V. Geisbrecht, Nat. Rev. Microbiol., 2008, 6, 132 CrossRef CAS PubMed.
- P. J. McEnaney, C. G. Parker, A. X. Zhang and D. A. Spiegel, ACS Chem. Biol., 2012, 7, 1139 CrossRef CAS PubMed.
- W. Z. Charles, C. R. Faries, Y. T. Street, L. S. Flowers and B. R. McNaughton, ChemBioChem, 2022, 23, e202200092 CrossRef CAS.
- F. S. Farah, Immunology, 1973, 25, 217 CAS.
- R. T. C. Sheridan, J. Hudon, J. A. Hank, P. M. Sondel and L. L. Kiessling, ChemBioChem, 2014, 15, 1393 CrossRef CAS PubMed.
- K. M. Shokat and P. G. Schultz, J. Am. Chem. Soc., 1991, 113, 1861 CrossRef CAS.
- C. R. Bertozzi and M. D. Bednarski, J. Am. Chem. Soc., 1992, 114, 5543 CrossRef CAS.
- P. Kaewsapsak, O. Esonu and D. H. Dube, ChemBioChem, 2013, 14, 721 CrossRef CAS PubMed.
- J. M. Fura, M. J. Sabulski and M. M. Pires, ACS Chem. Biol., 2014, 9, 1480 CrossRef CAS PubMed.
- S. A. Kristian, J. H. Hwang, B. Hall, E. Leire, J. Iacomini, R. Old, U. Galili, C. Roberts, K. B. Mullis, M. Westby and V. Nizet, J. Mol. Med., 2015, 93, 619 CrossRef CAS PubMed.
- M. J. Sabulski, S. E. Pidgeon and M. M. Pires, Chem. Sci., 2017, 8, 6804 RSC.
- M. S. Feigman, S. Kim, S. E. Pidgeon, Y. Yu, G. M. Ongwae, D. S. Patel, S. Regen, W. Im and M. M. Pires, Cell Chem. Biol., 2018, 25, 1185 CrossRef CAS.
- J. Y. Hyun, C.-H. Lee, H. Lee, W.-D. Jang and I. Shin, ACS Macro Lett., 2020, 9, 1429 CrossRef CAS.
- M. N. Idso, A. S. Akhade, M. L. Arrieta-Ortiz, B. T. Lai, V. Srinivas, J. P. Hopkins, A. O. Gomes, N. Subramanian, N. Baliga and J. R. Heath, Chem. Sci., 2020, 11, 3054 RSC.
- B. E. Dalesandro and M. M. Pires, ACS Infect. Dis., 2021, 7, 1116 CrossRef CAS PubMed.
- B. E. Dalesandro and M. M. Pires, J. Med. Chem., 2023, 66, 503 CrossRef CAS PubMed.
- P. Dzigba, A. K. Rylski, I. J. Angera, N. Banahene, H. W. Kavunja, M. C. Greenlee-Wacker and B. M. Swarts, ACS Chem. Biol., 2023, 18, 1548 CrossRef CAS PubMed.
- M. Miethke and M. A. Marahiel, Microbiol. Mol. Biol. Rev., 2007, 71, 413 CrossRef CAS PubMed.
- R. Golonka, B. S. Yeoh and M. Vijay-Kumar, J. Innate Immun., 2019, 11, 249 CrossRef CAS PubMed.
- J. Kramer, Ö. Özkaya and R. Kümmerli, Nat. Rev. Microbiol., 2020, 18, 152 CrossRef CAS PubMed.
- I. J. Schalk, Nat. Rev. Microbiol., 2025, 23, 24 CrossRef CAS PubMed.
- Z. Cao, Z. Qi, C. Sprencel, S. M. C. Newton and P. E. Klebba, Mol. Microbiol., 2000, 37, 1306 CrossRef CAS PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637 RSC.
- D. J. Ecker, B. F. Matzanke and K. N. Raymond, J. Bacteriol., 1986, 167, 666 CrossRef CAS.
- L. Pinkert, Y.-H. Lai, C. Peukert, S.-K. Hotop, B. Karge, L. M. Schulze, J. Grunenberg and M. Brönstrup, J. Med. Chem., 2021, 64, 15440 CrossRef CAS.
- S. Fritsch, V. Gasser, C. Peukert, L. Pinkert, L. Kuhn, Q. Perraud, V. Normant, M. Brönstrup and I. J. Schalk, ACS Infect. Dis., 2022, 8, 1134 CrossRef CAS.
- R. Koczura and A. Kaznowski, Microb. Pathog., 2003, 35, 197 CrossRef CAS PubMed.
- S. Subashchandrabose, S. Smith, V. DeOrnellas, S. Crepin, M. Kole, C. Zahdeh and H. L. T. Mobley, mSphere, 2015, 1, e00013–e00015 Search PubMed.
- M.-D. Phan, K. M. Peters, S. Sarkar, S. W. Lukowski, L. P. Allsopp, D. G. Moriel, M. E. S. Achard, M. Totsika, V. M. Marshall, M. Upton, S. A. Beatson and M. A. Schembri, PLoS Genet., 2013, 9, e1003834 CrossRef PubMed.
- M. Putrinš, K. Kogermann, E. Lukk, M. Lippus, V. Varik and T. Tenson, Infect. Immun., 2015, 83, 1056 CrossRef PubMed.
- M. E. Terlizzi, G. Gribaudo and M. E. Maffei, Front. Microbiol., 2017, 8, 1566 CrossRef PubMed.
- G. K. Bunduki, E. Heinz, V. S. Phiri, P. Noah, N. Feasey and J. Musaya, BMC Infect. Dis., 2021, 21, 753 CrossRef CAS PubMed.
- J. H. Lee, B. Subhadra, Y.-J. Son, D. H. Kim, H. S. Park, J. M. Kim, S. H. Koo, M. H. Oh, H.-J. Kim and C. H. Choi, Lett. Appl. Microbiol., 2016, 62, 84 CrossRef CAS PubMed.
- P. E. Klebba, S. M. C. Newton, D. A. Six, A. Kumar, T. Yang, B. L. Nairn, C. Munger and S. Chakravorty, Chem. Rev., 2021, 121, 5193 CrossRef CAS PubMed.
- B. Rayner, A. D. Verderosa, V. Ferro and M. A. T. Blaskovich, RSC Med. Chem., 2023, 14, 800 RSC.
- T. Aoki, H. Yoshizawa, K. Yamawaki, K. Yokoo, J. Sato, S. Hisakawa, Y. Hasegawa, H. Kusano, M. Sano, H. Sugimoto, Y. Nishitani, T. Sato, M. Tsuji, R. Nakamura, T. Nishikawa and Y. Yamano, Eur. J. Med. Chem., 2018, 155, 847 CrossRef CAS PubMed.
- Y.-M. Lin, M. Ghosh, P. A. Miller, U. Möllmann and M. J. Miller, Biometals, 2019, 32, 425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00293h |
| This journal is © The Royal Society of Chemistry 2025 |