
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe molecular features of non-peptidic nucleophilic substrates and acceptor proteins determine the efficiency of sortagging†
Tetiana
Bondarchuk
 ab,
Elena
Zhuravel
a,
Oleh
Shyshlyk
ac,
Mykhaylo O.
Debelyy
a,
Oleksandr
Pokholenko
ad,
Diana
Vaskiv
a,
Alla
Pogribna
ae,
Mariana
Kuznietsova
a,
Yevhenii
Hrynyshyn
a,
Oleksandr
Nedialko
af,
Volodymyr
Brovarets
c and
Sergey A.
Zozulya
*a
ab,
Elena
Zhuravel
a,
Oleh
Shyshlyk
ac,
Mykhaylo O.
Debelyy
a,
Oleksandr
Pokholenko
ad,
Diana
Vaskiv
a,
Alla
Pogribna
ae,
Mariana
Kuznietsova
a,
Yevhenii
Hrynyshyn
a,
Oleksandr
Nedialko
af,
Volodymyr
Brovarets
c and
Sergey A.
Zozulya
*a
aEnamine Ltd, 78 Winston Churchill Street, Kyiv 02094, Ukraine. E-mail: s.zozulya@enamine.net; Tel: +380 67 656-4026 Web: https://www.enamine.net
bDepartment of Structural and Functional Proteomics, Institute of Molecular Biology and Genetics, 150 Zabolotnogo Street, Kyiv 03680, Ukraine
cV. P. Kukhar Institute of Bioorganic Chemistry and Petrochemistry, 1 Academician Kukhar Street, Kyiv 02094, Ukraine
dTaras Shevchenko National University of Kyiv, Department of Chemistry, 64 Volodymyrska Street, Kyiv, 01033, Ukraine
eDepartment of Cell Signal Systems, Institute of Molecular Biology and Genetics, 150 Zabolotnogo Street, Kyiv 03680, Ukraine
fV. N. Karazin Kharkiv National University, 4 Svobody Square, Kharkiv 61022, Ukraine
First published on 26th December 2024
Abstract
Sortase A-mediated ligation (SML) or “sortagging” has become a popular technology to selectively introduce structurally diverse protein modifications. Despite the great progress in the optimization of the reaction conditions and design of miscellaneous C- or N-terminal protein modification strategies, the reported yields of conjugates are highly variable. In this study, we have systematically investigated C-terminal protein sortagging efficiency using a combination of several rationally selected and modified acceptor proteins and a panel of incoming surrogate non-peptidic amine nucleophile substrates varying in the structural features of their amino linker parts and cargo molecules. Our data suggest that the sortagging efficiency is modulated by the combination of molecular features of the incoming nucleophilic substrate, including the ionization properties of the reactive amino group, structural recognition of the nucleophilic amino linker by the enzyme, as well as the molecular nature of the attached payload moiety. Previous reports have confirmed that the steric accessibility of the C-terminal SrtA recognition site in the acceptor protein is also the critical determinant of sortase reaction efficiency. We suggest a computational procedure for simplifying a priori predictions of sortagging outcomes through the structural assessment of the acceptor protein and introduction of a peptide linker, if deemed necessary.
Introduction
Targeted modification of proteins has always played a pivotal role in both the academic efforts to elucidate their biological function and many practical applications. For this purpose, in addition to engineering proteins by traditional molecular biological means, a large variety of biophysical and biochemical probes represented by small molecules of non-peptidic origin, such as biotin, fluorescent dyes, isotope labels, drugs, sugars, lipids, and click-chemistry reagents, were introduced into a multitude of protein entities by miscellaneous bioconjugation methods. Quickly evolving enzymatic techniques more and more frequently displace the conventional chemical bioconjugation methods due to their advantageous lack of stochasticity and imprecision, as well as milder reaction conditions. One of the most popular enzymatic tools for N- or C-terminal protein modifications is Sortase A (SrtA) from Staphylococcus aureus, a representative of the family of transpeptidases from Gram-positive bacteria catalyzing the covalent anchoring of certain surface proteins to the bacterial cell envelope peptidoglycan. The main structural requirements for this sortase-mediated ligation are the presence of five amino acid long SrtA recognition motif LPXTG, where X is any amino acid, at the C-terminus of the N-terminal “acceptor” protein, and at least one N-terminal glycine residue at the C-terminal incoming nucleophilic component of the reaction. During the catalytic transpeptidation, the amide bond between the threonine and glycine residues in the recognition (“sortagging”) site is cleaved and a labile thioester product is formed with participation of the threonine carboxyl group and cysteine 184 in the enzyme active site. The thioester intermediate is resolved via a nucleophilic attack by the amino group of the N-terminal Gly in the incoming substrate resulting in the formation of a new amide bond in a covalent fusion.1,2 Since the discovery of this enzyme in 19993 and its first experimental use for in vitro protein ligations,4 a multitude of methods for N- and C-terminal or internal protein and peptide ligations, including non-natural C–C and N–N linkages, cyclization, multimerization, as well as targeted attachment of proteins to a wide variety of non-peptidic cargo molecules, supramolecular structures or surfaces, were developed, as discussed in detail in several recent reviews.1,5–10 Limitations of the native enzyme such as its calcium-dependence, modest catalytic rates and lack of robustness, as well as stringent recognition of the canonical sortagging motif, were successfully eliminated by protein engineering/directed evolution. This led to the advent of multiply mutated, functionally superior versions of SrtA from S. aureus, supplemented by the gradual introduction of divergent sortases from other genera of Gram-positive bacteria, into sortagging practice.6 A number of inventive ways to suppress formation of the hydrolytic side-products of SML to increase the target conjugate yields and to simplify purification were reported.6,8 Another exciting area of innovation is the expanding use of (oligo)glycine surrogates of non-peptide origin as nucleophilic SML substrates,11–15 which opens up access to a much greater diversity of targeted chemical modifications for proteins.Despite the substantial progress in methodology, the relationships between the molecular properties of acceptor proteins and incoming nucleophiles and the efficiency of sortagging, particularly in the case of non-canonical, synthetic surrogate substrates decorated with amino groups, are poorly understood. The main goal of this study was to shed more light on this understudied practically important aspect of SML technology.
Results
The studied panel of sortagging acceptors consisted of proteins substantially different in their sizes, tertiary structures and functions (Table 1 and Fig. 1). Human epidermal growth factor (EGF) is a small monomeric polypeptide hormone functioning through binding its cognate cell surface receptor and inducing its dimerization. EGF is just 53 amino acids long (MW 6045 Da), and it has three intramolecular disulfide bonds. The soluble form of human tumour necrosis factor alpha (TNFα) is a much larger pro-inflammatory cytokine forming stable homotrimers (MW 51 kDa) in solution. Human carbonic anhydrase, type II (CAHII) is a monomeric and relatively small (MW 29.2 kDa) intracellular enzyme. In our initial attempts to utilize Sortase A for labelling these three recombinantly produced proteins, equipped with a SrtA recognition site (LPETG) immediately following their natural C-terminal sequences, we have observed widely differing results, ranging from zero conjugate yields for CAHII to 90+% yields for EGF, with TNFα showing intermediate and rather poor yields. This was consistently observed with various incoming nucleophilic substrates, suggesting that some features of the protein substrates themselves are the main culprits. Another class of small, stable proteins known to be generally amenable for highly efficient C-terminal sortagging when equipped with an SrtA recognition site is camelid VHHs (nanobodies). While no experiments with VHHs are reported in this paper, we have included a VHH example (nanobody 7D12 against human EGF receptor16) in our structural comparison (Table 1 and Fig. 1). Both our laboratory15 and others17 have recently reported sortagging data for 6 different VHHs, including 7D12, using several biotin-containing nucleophilic substrates similar or identical to the substrates used in the present work. These sets of data rank nanobodies as an example of a very efficient protein acceptor type for our comparison purposes.

|
 | ||
| Fig. 1 PyMOL visualizations of the three-dimensional structures of EGF, TNFα, CAHII and VHH 7D12 modelled with the recombinant C-terminal appendages. (A) Side view of the I-TASSER model of the anti-epidermal growth factor receptor nanobody (VHH_7D12-SRT-6H). (B) Side view of the I-TASSER model of human epidermal growth factor (EGF-SRT). (C) Side and front views of the I-TASSER model of human tumor necrosis factor-α (TNFα-SRT). (D) Side and front views of the I-TASSER model of human carbonic anhydrase II (CAHII-SRT-6H). Positions of the SrtA recognition sites are highlighted in red, and 6xHis tags are shown in yellow. In all cases, the initial recombinant structures with the shortest C-terminal additions (VHH_7D12-SRT-6H, EGF-SRT, TNFα-SRT and CAHII-SRT-6H in Table 1, correspondingly) are shown. | ||
The most probable adverse SML efficiency factor selected for further investigations at this point in our study was the poor steric availability of a protein substrate C-terminus for sortase. Indeed, the beneficial effect of distancing SrtA recognition sites from the protein globule by introducing amino acid spacers was already reported as early as a decade ago in a number of research papers,18–22 as well as in general methodology publications,23,24 both for the C-terminal and N-terminal SML reactions. While this concept per se is not novel, we were curious to assess this structural factor in a comparison study using several protein substrates in combination with a large set of synthetic amine nucleophiles with varying physicochemical properties and attached payloads. In addition, the use of appropriate molecular modelling for a priori protein structure evaluation with regard to the necessity of spacer introductions might make the strategic planning of SML projects less empirical.
It is known that both the N- and C-termini of folded proteins have a strong tendency to be located on the solvent exposed protein surface.25,26 This does not, in itself, ensure their unrestricted steric availability to a polypeptide-modifying enzyme like sortase. Visual analysis of the available three-dimensional structures for EGF, TNFα, CAHII and VHH 7D12 (Protein Data Base files 2KV4, 1TNF, 1CA3, and 4KR correspondingly) provided an immediate indication that the steric availability of the C-terminal SrtA recognition site LPETG to sortase is likely to be the reason for their differential reactivity. Indeed, the crystal structures of the best SrtA substrates, EGF and VHH 7D12, show long, unstructured and solvent-exposed C-terminal tails protruding out from the tightly folded globular cores of these small proteins. In contrast, the structure of TNFα, a poor SrtA substrate, reveals that the protein C-termini are essentially hidden by the surrounding funnel-shaped depression formed by TNFα subunits forming a soluble homotrimer. In the case of CAHII, its C-terminus, while not substantially distanced from the protein globule, does not obviously appear to be sterically restrained (Fig. 1). However, our in silico modelling of the initial LPETG-containing CAHII protein construct indicated that molecular docking of the SrtA recognition site into the active site of the enzyme is energetically unfavourable. On the other hand, the introduction of the 10 amino acids long 2xG4S spacer between the native C-terminus of the CAHII and LPETG motifs dramatically improved the free energy minimization in this binding event (Fig. 2).
 | ||
| Fig. 2 PyMOL visualization of docking of the C-terminus of the CAHII-GS-SRT-6H construct in the active site of the SrtA enzyme. (A) Side view of the ClusPro Protein–Protein docking model between SrtA and the I-TASSER model of human carbonic anhydrase II (Sortase-hCAHII-2GS-SRT-6H). (B) Front view of the ClusPro Protein–Protein docking model between Sortase and the C-terminus of the I-TASSER model of hCAHII-2GS-SRT-6H. The C-terminus includes the 2xG4S linker (green), the Sortase recognition site LPETG (red), and a His-tag (yellow). Sulphur atom of the thioester intermediate forming Cys184 in the SrtA active site is coloured yellow. | ||
Encouraged by these observations, we have designed the modified protein expression constructs for both TNFα and CAHII with peptide spacer extensions between the proteins and the SrtA site. For both proteins, we have utilized a flexible unstructured linker GGGGSGGGGS (2xG4S)27 that is commonly used in recombinant protein designs. This extension linker was successfully used by other groups to increase sortagging efficiency for various proteins.19,20,22,24,25 For the case of CAHII, an additional construct with a much longer rigid PAS type linker28 was also used for comparison. Amino acid sequences of the C-terminal variants for all 4 proteins are shown in Table 1.
In order to investigate the interplay between the properties of the acceptor proteins and incoming nucleophiles in SrtA-mediated reactions, we have decided to test the 6 resulting model proteins in combination with a set of nucleophilic substrates containing the same useful payload (biotin) attached to the varying connector amino linkers. In our recent publication,15 we have screened a set of 452 unidirectionally protected diamines to identify the optimal amino linker prototypes for sortagging reactions. Here, we have selected 10 custom-synthesized biotin derivatives incorporating some of the best amino linkers discovered by us,15 as well as 5 commercially available biotin derivatives used for bioconjugations via an amino group (Fig. 3), for comparison-testing in sortagging reactions. The amino linker chains in this compound set comprise several chemotypes – aminomethyl pyridine, aminomethyl phenyl, aminomethyl cyclobutane, aminomethyl azetidine, linear aliphatic and polyethylene glycol amines and, based on our prior data, were expected to exhibit a broad range of conjugation efficiencies. One of the derivatives, compound 15 (Biocytin) containing the totally unreactive α-carbon branched primary amino group13,15 was included as a negative control in the sortagging experiments. To explore the effect of the distal part of the nucleophilic substrate on the sortagging efficiency, two derivatives of the macrocyclic chelator DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) commonly used in bioconjugations were also included (Fig. 3, compounds 16 and 17). These compounds contained the same or similar amino linkers but a bulkier and strongly negatively charged payload moiety as compared to their closest biotin-derived counterparts (Fig. 3, compounds 8, 11 correspondingly). The resulting matrix of acceptor–nucleophile pairs was tested in Sortase A reactions performed under the conditions favoring incomplete ligations for suboptimal pairs (0.5 mM nucleophile concentrations, 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 nucleophile substrate to protein ratio), to allow a more distinct relative efficiency ranking. We have also performed the same set of experiments at a two-fold higher (1 mM) nucleophile concentration and a nucleophile-to-protein ratio (10:1). This data set (see Table S1, ESI†) was not used for the comparative efficiency analysis, with the exception for pairwise comparisons of DOTA derivatives (16 and 17) and their biotin counterparts (8 and 11) as shown in Fig. 5B, since the yields for many reactions were in the 90–100% range. Outcomes of the reactions were quantitatively compared after the mass-spectrometric analysis of the reaction mixtures and identification of the reaction products and by-products, as well as unreacted acceptor proteins, following the previously described methodology.15 As expected, introduction of spacers between the natural C-termini of both proteins and the SrtA recognition site led to dramatic increases in their ability to be modified by SrtA using all tested substrates (Fig. 4 and Table S1, ESI†). Insertion of the 10 amino acid residues long flexible 2xG4S linker was sufficient to impart this property to the acceptor proteins while the 10-fold longer proline-rich PAS linker did not provide any advantage over 2xG4S, if not the opposite (Fig. 4).
:1 nucleophile substrate to protein ratio), to allow a more distinct relative efficiency ranking. We have also performed the same set of experiments at a two-fold higher (1 mM) nucleophile concentration and a nucleophile-to-protein ratio (10:1). This data set (see Table S1, ESI†) was not used for the comparative efficiency analysis, with the exception for pairwise comparisons of DOTA derivatives (16 and 17) and their biotin counterparts (8 and 11) as shown in Fig. 5B, since the yields for many reactions were in the 90–100% range. Outcomes of the reactions were quantitatively compared after the mass-spectrometric analysis of the reaction mixtures and identification of the reaction products and by-products, as well as unreacted acceptor proteins, following the previously described methodology.15 As expected, introduction of spacers between the natural C-termini of both proteins and the SrtA recognition site led to dramatic increases in their ability to be modified by SrtA using all tested substrates (Fig. 4 and Table S1, ESI†). Insertion of the 10 amino acid residues long flexible 2xG4S linker was sufficient to impart this property to the acceptor proteins while the 10-fold longer proline-rich PAS linker did not provide any advantage over 2xG4S, if not the opposite (Fig. 4).
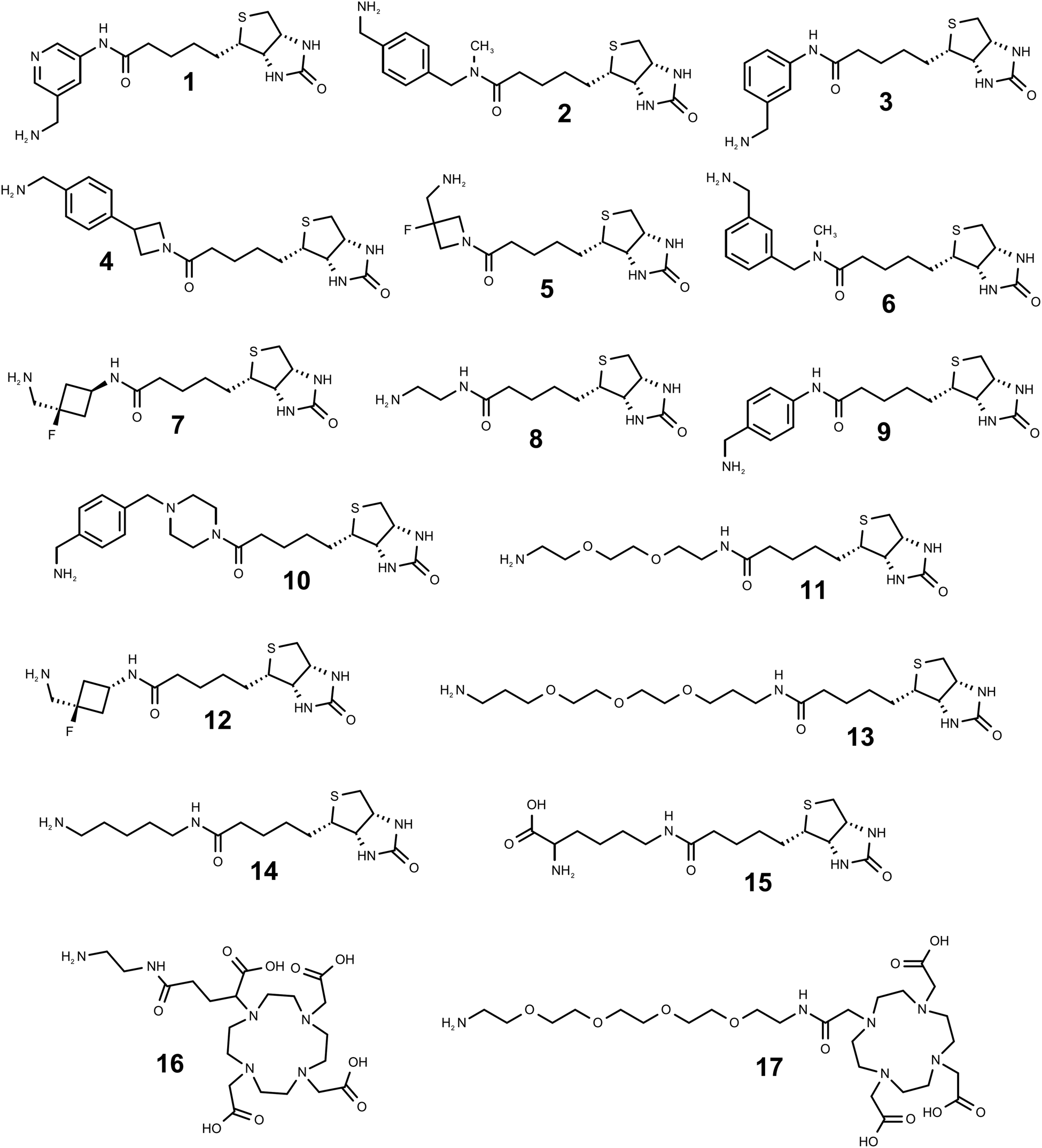 | ||
| Fig. 3 Structures of the amino linker equipped derivatives of biotin (compounds 1–15, arranged in the order of decreasing SML efficiency based on composite product yields with 4 protein acceptors) and DOTA (16, 17) used in this work. | ||
 | ||
| Fig. 4 Comparative visualization of conjugate product yields’ distribution for all the tested combinations of the 6 acceptor proteins (CAHII highlighted in red, TNFα in blue, and EGF in green) and 14 incoming biotin-containing substrates (1–14), excluding the non-reactive biocytin (15) control. | ||
The general concept of our study was to explore molecular features of the SML components with regard to their role in achieving the optimal productivity of sortagging, using a sizeable matrix of diverse protein acceptors and non-peptidic amino linker-cargo molecules.
Discussion
Structural modelling of the acceptor protein substrates
The I-TASSER online service (https://zhanggroup.org/I-TASSER/)29–31 was used to model the structures of the recombinant proteins. This tool allows for protein modeling by leveraging known structures that are partially identical or homologous to the target protein, as published in the RCSB Protein Data Bank (https://www.rcsb.org/).32 A useful feature of I-TASSER is its ability to incorporate amino acid sequences with unknown structures into molecules with established structures or their structural homologs. These additional sequences are modeled based on their intrinsic physical properties, which are determined by their primary structure and influenced by the physical parameters of the known structures within the molecular complex. Using I-TASSER, we were able to append the Sortase A recognition tag LPETG to the C-termini of the known protein structures. The modeling also included the associated amino acid sequences, peptide spacers and 6xHis affinity purification tags, where applicable. Using I-TASSER, we predicted the structures of the following recombinant protein variants: camelid VHH against human EGF receptor (VHH_7D12-SRT-6H); human carbonic anhydrase type II variants CAHII-SRT-6H, CAHII-2GS-SRT-6H and CAHII-PAS-SRT-6H; human epidermal growth factor (EGF-SRT); and human tumour necrosis factor alpha variants TNFα-SRT and TNFα-GS-SRT. The modeled protein structures were visualized using the PyMOL software (DeLano Scientific LLC). For all the structures mentioned above, except for the TNFα-SRT protein, steric accessibility of the Sortase A recognition tag (LPETG) was apparent (Fig. 1), explaining the high sortagging efficiency of both EGF-SRT and VHH 7D12 proteins. However, this primary assessment contrasted with our experimental results showing no SrtA-mediated enzymatic reaction with the CAHII-SRT-6H protein. To investigate this discrepancy, we conducted molecular docking of S.aureus Sortase A (PDB structure 1T2W) with CAHII-SRT-6H using the ClusPro Protein–Protein Docking web server (https://cluspro.org/).33–36 As a positive control, we also performed docking for the Sortase A–CAHII-2GS-SRT-6H complex (Fig. 2). We found that SrtA forms a specific complex2 with CAHII-2GS-SRT-6H, whereas no such complex was formed with CAHII-SRT-6H. The docking parameters of the Sortase A–CAHII-2GS-SRT-6H and Sortase A–CAHII-SRT-6H complexes differ significantly. For the Sortase A–CAHII-2GS-SRT-6H complex, the parameters are as follows: balanced docking, Cluster 0, 115 members, with a representative center weighted score of −508.6 and a representative lowest energy weighted score of −620.5. In contrast, for the Sortase A–CAHII-SRT-6H complex, the parameters are as follows: balanced docking, Cluster 0, 66 members, with a representative center weighted score of −137.7 and a representative lowest energy weighted score of −170.5. These differences suggest that the probability of forming the Sortase A–CAHII-SRT-6H complex is low, which likely explains the lack of activity observed in the Sortase reaction for CAHII-SRT-6H. Visualization of the superposition of LPETG motifs in the sortase active site comparing our CAHII-2GS-SRT-6H model with the published data on the peptide substrate interactions with SrtA,37 which shows good matching, is included in the ESI† (Fig. S3).Thus, prior to planning sortagging experiments, it is advisable to assess the 3D structures of the proteins of interest, if available, for the possible C-terminal steric accessibility constraints. In cases where structural predictions alone might be inconclusive, we recommend complementing I-TASSER modeling with molecular docking of the predicted structures with Sortase using the ClusPro service. Alternatively, a flexible linker, such as 2xG4S, can be introduced before the SrtA recognition site as a precautionary measure, in case the relevant structural information does not exist or is not unambiguously interpretable.
pKa and nucleophilicity of incoming substrates
Acylation of amines in aqueous buffers requires a portion of the amine to be in a non-protonated state, which participates in the acylation reaction. Consequently, an increase in the fraction of non-protonated amine increases the acylation rate. The larger degree of deprotonation of the primary amino group on the incoming substrate would make it a more reactive nucleophile in sortagging reactions. A comprehensive compilation study of the experimentally measured pKa values for ionizable groups in folded proteins38 reports the average pKa (negative logarithm of the acid dissociation constant) of the protonated amino group of their N-termini as 7.7 (range of 6.8–9.1 on 16 measurements), while the more recent database39 provides the average pKa as 7.64 (range of 6.91–9.14 on 22 measurements). The same parameter measured by several laboratories for various model peptides, including oligo-glycine peptides, is in the narrow range of 7.5–8.1.40 Theoretically, exceeding the pKa of a reactive amino group of a substrate by one pH unit would lead to deprotonation of 90% of the amino group, while exceeding the pKa by two pH units would bring this number to 99%. This allows us to conclude that conducting sortagging reactions at the often-used pH 9 is a reasonable choice for the incoming nucleophiles which are either N-Gly containing proteins or N-Gly peptide-based substrates. The choice of this pH for the reaction buffer is essentially a trade-off between decreasing the positive charge on the nucleophilic primary amino group by increasing the reaction buffer pH, and at the same time not deviating too drastically from the optimal pH for the enzymatic activity of SrtA – which is around 8 and drops steeply above pH 9.41For the synthetic non-peptidic amine substrates, there is a potential possibility of designing improved amino group-containing linkers with depressed pKa values hence better reactivity, as long as the associated structural modifications do not affect the substrate recognition by the SrtA enzyme (and hopefully even improve it). Such an accomplishment could enhance reaction yields, as well as reduce the consumption of the enzyme and substrate. At a constant pH, the fraction of non-protonated amine increases if the pKa of the protonated amine decreases, which can be achieved by introducing adjacent electronegative groups. However, electronegative groups also decrease the acylation reaction rate constant by withdrawing electron density from the nucleophilic centre, making it less reactive. Thus, electronegative substituents exert two opposing effects, and the predominance of one will either facilitate or hinder amine acetylation. A well-known example of acylation being facilitated by an electronegative substituent is the pH-dependent selectivity in acylation reactions for the α-amino group at the N-terminus versus the ε-amino groups of lysine residues in proteins.42 Since the use of chemically diverse non-peptidic incoming nucleophiles efficiently mimicking the conventional (oligo)glycine substrates is a developing trend in the practice of sortagging, studies of structure–activity relationships in such molecules are of great interest. Since the experimentally determined pKa values for the biotin derivatives used in this study were not available, we have calculated the corresponding pKa for their primary amino groups using four well-validated software tools of both the corporate (Chemaxon, ACD/Labs, Schrodinger) and academic (MolGpka server43) origin. The reliable computation of ionization properties for chemical compounds is still a challenging task,44 and there is significant variability in pK values calculated using a variety of reported methods. Therefore, we have used arithmetic means of the four algorithmically different pKa calculations performed using reputable software tools for evaluating the correlation of pKa values with the observed reactivity of the compounds (Fig. 5A). Complete numerical data used in this graph can be found in the ESI† (Table S1). Despite the intrinsic imprecision of the plotted product yield versus pKa data, the expected trend of inverse dependency between the pKa values, reflecting the nucleophilicity of the compounds, and their sortagging efficiency, expressed as composite product yields with four different protein acceptors, is apparent.
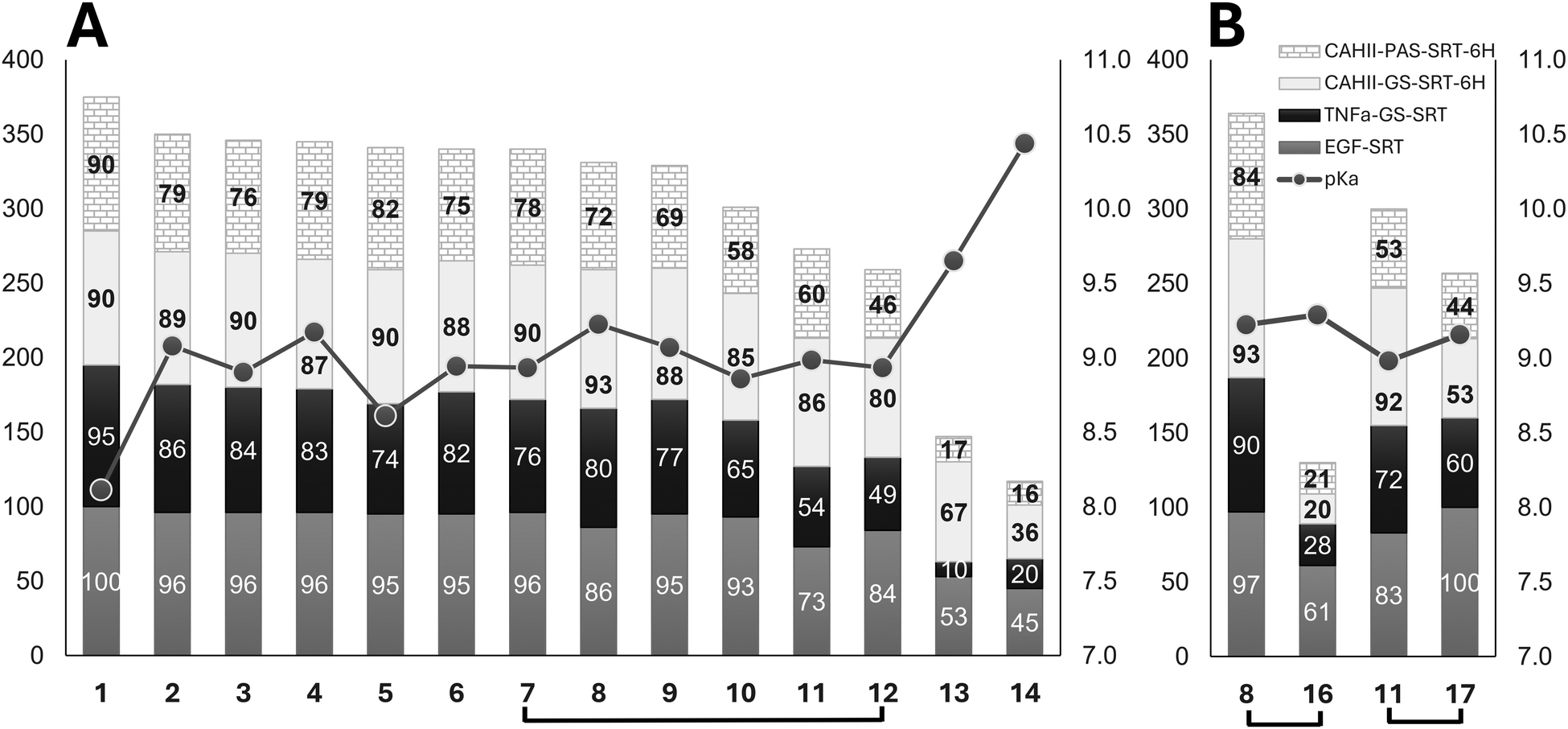 | ||
| Fig. 5 (A) Composite reaction yields (%, left scale) for 4 protein acceptors, excluding the non-reactive proteins, and 14 biotin derivatives (1–14), excluding the non-reactive biocytin (15). The stacked bars (EGF – grey, TNFα-GS-SRT – black, CAHII-GS-SRT-6H – light grey, CAHII-PAS-SRT-6H – brick pattern) are sorted left to right in the order of decreasing composite yield. The line connecting mean pKa values (right scale) for each substrate is overlayed over the bar graph. Brackets connect the compounds for pairwise comparisons. (B) Same graphical representation as in (A) for comparing two DOTA derivatives (16 and 17) with their corresponding biotin counterparts (8 and 11). Yields correspond to SML reactions done at 0.5 mM (panel A) or 1 mM (panel B) nucleophile concentrations. | ||
Interestingly, despite the fact that the 10 custom-synthesized biotin derivatives (compounds 1–7, 9, 10, and 12) containing the most efficient amino linkers were selected as a result of a comparative sortagging screen of 452 diverse model monoprotected diamines,15 their computed amino group pKa values are close to the experimentally determined pKa for N-terminal glycines in peptidic substrates.37–39 This creates a potential for further improvement in the nucleophilicity of the substrates, e.g. via introduction of electrophilic substituents in the vicinity of the reactive amino group. Since the correlation of amine basicity and nucleophilicity is not perfect and the predictability of a substituent effect on both the nucleophilicity and substrate–enzyme recognition is limited, such an endeavor would clearly require mostly a trial-and-error molecular screening effort.
Structural features of nucleophilic substrates
In addition to nucleophilicity, structural features of SrtA substrates determining their interaction with the enzyme must be also among the factors affecting their reaction efficiency. Indeed, the compounds 7 and 12, which are stereoisomers of each other and therefore have identical pKa values and nucleophilicity, show distinctly different sortagging efficiency (Fig. 5A). In this case, the conformation-modifying chiral center is located in the immediate vicinity of the active site of SrtA, and the direct substrate–enzyme recognition is apparently affected. To explore this aspect further, we have included two commercially available amino linked derivatives of DOTA, a chelator used in bioconjugations for producing targeted imaging and therapeutic radiopharmaceuticals or MRI contrast agents, in our panel of sortagging substrates. DOTA is a bulky macrocyclic compound decorated with 3 or 4 free carboxylic groups in the two selected derivatives, which imparts a strong negative charge to the DOTA payload moiety in contrast to a smaller and non-charged biotin cargo. DOTA-derived compound 16 and biotin-derived compound 8 contain the same SrtA active site-proximal ethylenediamine linker, have almost identical calculated pKa values for the nucleophilic amino group, as well as approximately the same overall linker distance from the nucleophilic amino group to the payload attachment point. However, the DOTA derivative 16 shows much lower conjugation yields than 8 with all 4 acceptor proteins (Fig. 5B). On the other side, the second DOTA derivative, compound 17, with a much longer linker, shows substantially improved conjugation yields – close to those of the biotin derivative 11 with a similar polyethylene glycol linker (Fig. 3). Again, these two compounds also have very close pKa values of their amino groups. These pairwise comparisons clearly suggest a detrimental effect of the DOTA moiety, contrary to biotin, on the substrate interaction with the enzyme, which is relieved by increasing the length of the separating spacer. The subjects of structure and nucleophilicity of noncanonical attacking nucleophiles with regard to their performance in SML were raised by others;11,12,14 the goal of this and our preceding study15 was to gain deeper insight into the issue.A potential caveat in our conclusions may stem from the fact that two out of the 17 tested substrates (compounds 13, 14) were procured as trifluoroacetic acid (TFA) salts, while the rest were supplied as either free bases or hydrochloride salts. Since trifluoroacetate was reported to interfere with biological activities in some cell-based and biochemical assays,45 we have checked whether TFA had an inhibitory effect on Sortase A, which may have potentially led to reaction efficiency underestimation for the corresponding nucleophiles. This was tested by running control sortagging reactions with TNFα-GS-SRT and CAHII-GS-SRT-6H and compound 11 (biotin ethylenediamine) in parallel in the presence or absence of 0.5 mM of sodium trifluoroacetate. No inhibition of SrtA by TFA was detected in these assays (Table S2, ESI†). Another potential issue is a solubility limit below 0.5 mM, which might compromise the reactivity data for some biotin derivatives. Biotin itself is sufficiently well soluble in both DMSO and aqueous buffers. However, decoration of the biotin moiety by amino linkers containing alkyl, aryl or polyethylene glycol chains of various lengths could potentially decrease the solubility of some derivatives. The solubility of biotin cadaverine (compound 13), likely one of the most hydrophobic compounds from the tested set also showing inferior efficiency in sortagging, is reported to be around 5 mM in aqueous buffers by some reagent vendors. Our visual microscopic examination of 0.5–1 mM solutions of compound 13 in the aqueous sortase reaction buffer, which additionally contains 5% of solubility enhancing DMSO, also did not show any signs of insolubility.
It is worth noting that for compounds 1–10 (Fig. 3), which are all biotin derivatives decorated with the most efficient amino linkers discovered by us previously in an MS-based SML screen of a model tert-butyloxycarbonyl (Boc) group monoprotected diamine library,15 most of the observed conjugate product yields fall into the 90–100% range for the effective protein acceptors, especially for the experiments run at 1:10 acceptor–substrate ratio (Table S1 and Fig. 5A, ESI†). The observed high sortagging yields could result from either the high reaction rates, reaction irreversibility, or both. In-depth mechanistic study of the many acceptor–nucleophile combinations explored here is beyond the focus and the scope of this work and must be the subject of a separate investigation. However, the selected time course experiments on TNFα-GS-SRT and compounds 1 and 7 (see Fig. S4, ESI†) indicate both the fast reaction kinetics and probable irreversibility of SML for these substrate–nucleophile pairs. The latter characteristic is not an unexpected finding for the sortagging of a synthetic non-peptidic molecule to a protein, which a priori is likely to lead to the formation of an unnatural amide bond product which can no longer be attacked by sortase. Many inventive approaches to imparting irreversibility to SML by protein acceptor and nucleophile engineering, as well as reaction setup modifications, including those employing the formation of unnatural conjugated bonds, are referenced in several comprehensive literature reviews.6,8,9 The use of optimized surrogate non-peptidic linkers may well become the preferred way of attaching a useful chemical cargo to protein C-termini due to its simplicity and consistently achievable high yields. As graphically illustrated in Fig. 4 and 5, the relative reactivity ranking of the 14 biotin derivatives was very similar for all the tested proteins, indicating the absence of any specific interactivity within the acceptor–substrate pairs, which could have modulated the efficiency of sortagging. These results imply a general suitability and effectiveness of these well performing non-peptidic linkers for different proteins and cargo molecule types.
Materials and methods
Reagents
Compounds 16 – NH2-DOTA-GA (Cat. #C116) and 17 – NH2-PEG4-DOTA (Cat. #C125) were purchased at Chematech. Five biotin derivatives – biotin ethylenediamine, hydrochloride (Cat. #90075); biotin cadaverine, trifluoroacetate (Cat. #90063); biocytin (ε-biotinyl-L-lysine) (Cat. #90055); biotin-PEO2-PPO2-amine, trifluoroacetate salt (Cat. #90078) and biotin-PEO3-amine (Cat. #90067) were all from Biotium. Ni-The NTA affinity sorbent was from G-Biosciences. All standard biochemicals, including Tris, isopropyl-β-D-galactopyranoside (IPTG), 2-mercaptoethanol (2-ME), dithiothreitol (DTT), ampicillin (Amp), dimethyl sulfoxide (DMSO), bacterial growth media components and mineral salts were purchased from either MilliporeSigma or Bio Basic. Ten additional biotin derivatives (compounds 1–7 and 9, 10, and 12, Fig. 1) were custom synthesized by enamine; the corresponding synthetic and analytical data can be found in the ESI,† except for the previously described compounds 1, 3, 4, 5.14Recombinant proteins
All recombinant proteins used in this work were produced in our laboratory according to the protocols shown below to a purity level of ≥95%, as controlled by SDS-PAAG and QTOF LC/MS (Fig. S1 and S2, ESI†). Expression and purification of SrtA7M, the heptamutant Sortase A version46,47 used in this work, was described before.15:100 to 0.7 L of LB medium with 100 µg mL−1 Amp. The culture was then grown at 37 °C on an orbital shaker (180 rpm). When the OD600 reached ∼0.8, the shaker temperature was switched to 18 °C, and the protein expression was induced by adding IPTG to 1 mM concentration. The culture was grown at 18 °C with shaking at 200 rpm for 18-20 hours, then the cells were harvested by centrifugation at 6000 g, 4 °C for 10 min. The cell pellet (10 g) was resuspended in 50 mL of buffer A (25 mM Tris HCl, 300 mM NaCl, 10 mM imidazole, 5% glycerol, 5 mM 2-mercaptoethanol (2-ME), pH 7.5) containing 1 mM PMSF (5 mL g−1 cell pellet) and sonicated for 5 min on ice in 50–50 pulse mode with 70% amplitude using a Branson SFX250 250 ultrasonic cell disintegrator with 1/2″ horn. A few crystals of DNAse I were added to the lysate before centrifuging it at 20000g at 4 °C for 20 min. The collected supernatant was mixed with the Ni-NTA affinity sorbent (9 mL) pre-equilibrated with buffer A and then incubated for 2 h at 4 °C on a rotator. The slurry was transferred to a disposable chromatography column and washed successively with buffer A supplemented with 0.1% Triton X-100 (5 bed volumes, BV), buffer A (9 BV) and then with buffer A containing increasing concentrations of imidazole – 20 mM (2 BV), 40 mM (2 BV) and 60 mM (1 BV). The bound protein was eluted from the column with buffer A supplemented with 250 mM imidazole (3 BV). Collected fractions were analysed using 12% SDS-PAGE and combined as appropriate. The elution buffer was exchanged for buffer B (50 mM Tris–HCl, 150 mM NaCl, 5% glycerol, 2-ME pH 7.5) using desalting Sephadex G25M PD-10 columns (Cytiva). In the next step, the fusion protein was digested by SUMO-specific Ulp1 protease (Ulp1:protein = 1:30 by weight) at 30 °C on a rotator for 1.5 hours. To separate His-tagged SUMO and Ulp1 from the cleaved-off EGF, the reaction mix was supplied with Ni-NTA sorbent (5 mL) and incubated on a rotator for 20 min at room temperature, then loaded onto a disposable column and washed with buffer C (50 mM Tris HCl, 150 mM NaCl, pH 7.5, 4 BV), buffer C + 20 mM imidazole (2 BV), and buffer C + 250 mM imidazole. 18% SDS-PAGE was run to analyse the fractions stored at 4 °C before the subsequent concentration (Vivaspin Turbo 4, 3 kDa MWCO, Sartorius). The final polishing chromatography was run on an FPLC Superdex 200 16/600 column at a flow rate of 0.8 mL min−1 in 25 mM Tris–HCl, pH 7.5, and 200 mM NaCl. The collected fractions were analysed on 18% SDS-PAGE, appropriately combined, concentrated and frozen in liquid nitrogen. Typical yields of the purified EGF-LPETGG were 1–2 mg L−1 of bacterial culture. Full verification of the protein sequence indicating formation of 3 internal disulfide bonds was done by LC/MS-QTOF. The biological activity of EGF-LPETGG was tested in the A431 cell line proliferation assay and found to be consistent with the literature data50,51 (data not shown).
000g for 30 min and the supernatant was filtered through a 0.45 nylon membrane. Ni-NTA affinity resin equilibrated with lysis buffer was added to the supernatant (∼1 mL of resin per 10–20 mg of protein) and incubated for 1 h at +4 °C on a rotator. The resin was loaded onto a disposable column and washed sequentially with 10 column volumes of lysis buffer and 6 column volumes each of the same buffer containing 20 mM and 35 mM imidazole. Purified protein was eluted with 4 column volumes of elution buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 250 mM imidazole, 5 mM 2-ME, 10% glycerol). After the buffer exchange on the PD-10 gel-filtration column (GE Healthcare), the SUMO-tag was cleaved off from the purified fusion protein by adding 6xHis-tagged ULP1 protease (at a ratio of 12 ug of ULP1 protease per 1 mg of 6xHis-SUMO-TNFα) in 20 mM Tris–HCl pH 8.0, 150 mM NaCl, and 2 mM DTT for 1 h at 30 °C. Upon the completion of the cleavage, Ni-NTA resin equilibrated with the same buffer was added to the proteolysis mixture (∼1 mL of resin per 10–20 mg of protein), incubated with rotation for 1 h and separated by gravity flow. The flow-through was collected, and fractions were analysed by SDS-PAAG and concentrated to 4–5 mg mL−1 protein using a centrifugal concentrator with 10000 MW cut-off (Amicon Ultra). The final yields of the TNFα proteins were ∼15 mg L−1 of the bacterial culture.
000g and 4 °C for 40 min, the supernatant was collected and filtered through 0.2 µm syringe filters prior to chromatography. Proteins were purified using nickel affinity chromatography using gravity-flow columns with a bed volume of 7 mL. The column was washed sequentially with lysis buffer containing 0, 20, 40 and 60 mM imidazole, 5 column volumes of each. Elution was done with a buffer containing 250 mM imidazole (3 column volumes). Protein fractions were analysed using SDS-PAAG and dialyzed against 25 mM Tris–HCl, pH 7.5, and 200 mM NaCl overnight at +4 °C. Dialyzed protein was concentrated to ∼5 mg mL−1 using Sartorius Turbo15 30000 MWCO centrifugal concentrators, aliquoted and snap-frozen in liquid nitrogen. The yields of the proteins were in the range of 10–15 mg L−1 of bacterial culture. N-terminal methionine residue was post-translationally removed by E. coli Met aminopeptidase in all three versions of CAHII as indicated by LC/TOF-MS.
Sortagging efficiency comparisons for the protein acceptor–nucleophile pairs
794/17680 Da) TNFα-GS-SRT (18538/18422 Da), CAH-SRT-6H (30435/29555 Da), CAH-GS-SRT-6H (31069/30186 Da), and CAH-PAS-SRT-6H (34570/33688 Da). Deconvolution deviations were all within 4 Da. Detected peaks for the SrtA7M enzyme (17722.4 Da) were excluded from further calculations. The peak height ratios for the initial protein, conjugate product and the side-product were assumed to be equal to their molar ratio. Based on this assumption, for each well the absolute peak heights (counts) for the identified molecules were normalized to their sum and the resulting values were reported as their % molar fractions. Most of the experiments were performed in singleton, based on the sufficiently high quantitative reproducibility observed in this and our previous15 large-scale mass-spectrometry based study. Examples of the experimental reproducibility are shown in the ESI,† Fig. S4 and S5.
Computational methods
Conclusions
We carried out a systematic study of the relative efficiency of C-terminal sortagging using a large panel of protein acceptors and incoming non-peptidic nucleophile substrates, with the goal of elucidating the factors, specific for the acceptor–nucleophile combinations that may affect the SML reactions. We concur with the previously published research18–24 implying the highly critical role of the C-termini of acceptor proteins bearing the SrtA recognition site on ligation efficiency. In agreement with the existing studies, introduction of a short peptide linker extending the sortagging site-bearing C-tail out from the sterically constrained protein markedly augments the SML efficiency. We have developed a structural protein assessment and modelling procedure aimed at rationalizing the planning of SML experiments in this regard. Other factors, the role of which are apparent from our results, are the basicity (nucleophilicity) of the attacking amino group of the substrate in combination with the structural recognition of the adjacent linker part of the molecule by the enzyme, including the reactive amino group-proximal stereochemistry. An interesting avenue for further improvement in sortagging technology would be the attempt to create synthetic amino linkers with enhanced nucleophilicity and optimized enzyme–substrate interactions by introducing appropriate substituents in the vicinity of the reactive amino groups. The chemical nature of the linker-attached cargo molecules appears to be able to negatively modulate the yields as well, possibly due to the size- or charge-dependent interference with the adjacent enzyme. We hope that these practical insights will help the research community to better design and implement bioconjugation experiments using sortases.Abbreviations
| SML | Sortase-mediated ligation |
| PDB | Protein Data Bank |
| DTT | Dithiothreitol |
| 2-ME | 2-Mercapthethanol |
| PMSF | Phenylmethylsulphonyl fluoride |
| DMSO | Dimethyl sulfoxide |
| Ni-NTA | Nickel(II)-nitrilotriacetic acid |
| Amp | Ampicillin |
| IPTG | Isopropyl β-D-1-thiogalactopyranoside |
| MWCO | Molecular weight cut-off |
| SrtA | Sortase A from S. aureus |
| TFA | Trifluoroacetic acid |
Author contributions
The manuscript was written through significant contributions of all authors. All authors have given approval to the final version of the manuscript. Below, roles of each author are specified according to the CRediT (https://credit.niso.org/) descriptors. Conceptualization: TB, OS, SAZ; data curation: TB, OS, SAZ; formal analysis: MOD, OP; investigation: TB, DV, AP, MK, YM, ON; methodology: MOD, YM, ON; project administration: SAZ; software: MOD, OP; resources: AP, MK; supervision: EZ, OS; visualization: TB, EZ, MOD; writing – original draft: SAZ; writing – review and editing: TB, EZ, OS, VB, SAZ.Data availability
The supporting data for this article was included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
All authors are grateful to Prof. Andrei Tolmachev for supporting this work. We would also like to thank Anton Cherednichenko for assisting with the pKa calculations and Olga Kuchuk for valuable discussions and help with illustrations.References
- J. F. Amacher and J. M. Antos, Trends Biochem. Sci., 2024, 49, 596–610 CrossRef CAS PubMed.
- U. Ilangovan, H. Ton-That, J. Iwahara, O. Schneewind and R. T. Clubb, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6056–6061 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- N. Braga Emidio and R. W. Cheloha, Curr. Opin. Chem. Biol., 2024, 80, 102443 CrossRef CAS PubMed.
- Z. Zou, Y. Ji and U. Schwaneberg, Angew. Chem., Int. Ed., 2024, 63, e202310910 CrossRef CAS PubMed.
- E. M. Obeng, A. J. Fulcher and K. M. Wagstaff, Biotechnol. Adv., 2023, 64, 108108 CrossRef CAS PubMed.
- H. E. Morgan, W. B. Turnbull and M. E. Webb, Chem. Soc. Rev., 2022, 51, 4121–4145 RSC.
- X. Dai, A. Boker and U. Glebe, RSC Adv., 2019, 9, 4700–4721 RSC.
- N. Pishesha, J. R. Ingram and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 2018, 34, 163–188 CrossRef CAS PubMed.
- S. Baer, J. Nigro, M. P. Madej, R. M. Nisbet, R. Suryadinata, G. Coia, L. P. Hong, T. E. Adams, C. C. Williams and S. D. Nuttall, Org. Biomol. Chem., 2014, 12, 2675–2685 RSC.
- J. E. Glasgow, M. L. Salit and J. R. Cochran, J. Am. Chem. Soc., 2016, 138, 7496–7499 CrossRef CAS PubMed.
- Z. Zou, M. Nöth, F. Jakob and U. Schwaneberg, Bioconjugate Chem., 2020, 31, 2476–2481 CrossRef CAS PubMed.
- E. M. Obeng, D. L. Steer, A. Fulcher and K. M. Wagstaff, Bioconjugate Chem., 2023, 34, 1667–1678 CrossRef CAS PubMed.
- T. Bondarchuk, D. Vaskiv, E. Zhuravel, O. Shyshlyk, Y. Hrynyshyn, O. Nedialko, O. Pokholenko, A. Pohribna, O. Kuchuk, V. Brovarets and S. Zozulya, Bioconjugate Chem., 2024, 35, 1172–1181 CrossRef CAS PubMed.
- K. R. Schmitz, A. Bagchi, R. C. Roovers, P. M. van Bergen en Henegouwen and K. M. Ferguson, Structure, 2013, 21, 1214–1224 CrossRef CAS PubMed.
- E. M. Obeng, D. L. Steer, A. J. Fulcher and K. M. Wagstaff, Nanoscale Adv., 2023, 5, 2251–2260 RSC.
- D. J. Williamson, M. A. Fascione, M. E. Webb and W. B. Turnbull, Angew. Chem., 2012, 51, 9377–9380 CrossRef CAS PubMed.
- T. Heck, P. Pham, F. Hammes, L. Thöny-meyer and M. Richter, Bioconjugate Chem., 2014, 25, 1492–1500 CrossRef CAS PubMed.
- K. Wagner, M. J. Kwakkenbos, Y. B. Claassen, K. Maijoor, M. Böhne, K. F. van der Sluijs, M. D. Witte, D. van Zoelen, L. A. Cornelissen, T. Beaumont, A. Q. Bakker, H. L. Ploegh and H. Spits, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16820–16825 CrossRef CAS PubMed.
- Q. Chen, Q. Sun, N. M. Molino, S. Wang, E. T. Boder and W. Chen, Chem. Commun., 2015, 51, 12107–12110 RSC.
- S. Reed, D. Brzovic, S. Takasaki, K. V. Boyko and J. M. Antos, Bioconjugate Chem., 2020, 31, 1463–1473 CrossRef CAS PubMed.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 CrossRef PubMed.
- J. M. Antos, J. Ingram, T. Fang, N. Pishesha, M. C. Truttmann and H. L. Ploegh, Curr. Protoc. Protein Sci., 2017, 89, 15.3.1–15.3.19 Search PubMed.
- E. Jacob and R. Unger, Bioinformatics, 2007, 23, e225–e230 CrossRef CAS PubMed.
- S. Sharma and M. R. Schiller, Crit. Rev. Biochem. Mol. Biol., 2019, 54, 85–102 CrossRef CAS PubMed.
- X. Chen, J. L. Zaro and W. C. Shen, Adv. Drug Delivery Rev., 2013, 65, 1357–1369 CrossRef CAS PubMed.
- J. Breibeck and A. Skerra, Biopolymers, 2018, 109, e23069 CrossRef PubMed.
- J. Yang and Y. Zhang, Nucleic Acids Res., 2015, 43, W174–W181 CrossRef CAS PubMed.
- W. Zheng, C. Zhang, Y. Li, R. Pearce, E. W. Bell and Y. Zhang, Cells Rep. Methods, 2021, 1, 100014 CrossRef CAS PubMed.
- X. Zhou, W. Zheng, Y. Li, R. Pearce, C. Zhang, E. W. Bell, G. Zhang and Y. Zhang, Nat. Protoc., 2022, 17, 2326–2353 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- D. Kozakov, D. Beglov, T. Bohnuud, S. E. Mottarella, B. Xia, D. R. Hall and S. Vajda, Proteins, 2013, 81, 2159–2166 CrossRef CAS PubMed.
- D. Kozakov, D. R. Hall, B. Xia, K. A. Porter, D. Padhorny, C. Yueh, D. Beglov and S. Vajda, Nat. Protoc., 2017, 12, 255–278 CrossRef CAS PubMed.
- I. T. Desta, K. A. Porter, B. Xia, D. Kozakov and S. Vajda, Structure, 2020, 28, 1071–1081 CrossRef CAS PubMed.
- S. Vajda, C. Yueh, D. Beglov, T. Bohnuud, S. E. Mottarella, B. Xia, D. R. Hall and D. Kozakov, Proteins, 2017, 85, 435–444 CrossRef CAS PubMed.
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 CrossRef CAS PubMed.
- G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2009, 18, 247–251 CrossRef CAS PubMed.
- N. Ancona, A. Bastola and E. Alexov, J. Comput. Biophys. Chem., 2023, 22, 515–524 CrossRef CAS PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15, 1214–1218 CrossRef CAS PubMed.
- Z. Wu, H. Hong, X. Zhao and X. Wang, Bioresour. Bioprocess, 2017, 4, 13 CrossRef PubMed.
- A. Tantipanjaporn and M.-K. Wong, Molecules, 2023, 28, 1083 CrossRef CAS PubMed.
- X. Pan, H. Wang, C. Li, J. Z. H. Zhang and C. Ji, J. Chem. Inf. Model., 2021, 61, 3159–3165 CrossRef CAS PubMed.
- J. Wu, Y. Kang, P. Pan and T. Hou, Drug Discovery Today, 2022, 27, 103372 CrossRef CAS PubMed.
- C. Ardino, F. Sannio, C. Pasero, L. Botta, E. Dreassi, J. D. Docquier and I. D'Agostino, Mol. Diversity, 2023, 27, 1489–1499 CrossRef CAS PubMed.
- H. Hirakawa, S. Ishikawa and T. Nagamune, Biotechnol. J., 2015, 10, 1487 CrossRef CAS PubMed.
- L. Chen, J. D. Cohen, X. Song, A. Zhao, Z. Ye, C. J. Feulner, P. J. Doonan, W. S. Somers, L. Lin and P. R. Chen, Sci. Rep., 2016, 6, 31899 CrossRef CAS PubMed.
- Z. Su, Y. Huang, Q. Zhou, Z. Wu, X. Wu, Q. Zheng, C. Ding and X. Li, Protein Pept. Lett., 2006, 13, 785–792 CrossRef CAS PubMed.
- Y. Ma, J. Yu, J. Lin, S. Wu, S. Li and J. Wang, BioMed Res. Int., 2016, 3758941 Search PubMed.
- D. W. Barnes, J. Cell Biol., 1982, 93, 1–4 CrossRef CAS PubMed.
- J. Y. Song, S. W. Lee, J. P. Hong, S. E. Chang, H. Choe and J. Choi, Cancer Lett., 2009, 283, 135–142 CrossRef CAS PubMed.
- R. C. Johnston, K. Yao, Z. Kaplan, M. Chelliah, K. Leswing, S. Seekins, S. Watts, D. Calkins, J. Chief Elk, S. V. Jerome, M. P. Repasky and J. C. Shelley, J. Chem. Theory Comput., 2023, 19, 2380–2388 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables, and supplementary methods. See DOI: https://doi.org/10.1039/d4cb00246f |
| This journal is © The Royal Society of Chemistry 2025 |