
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTumor microenvironment modulation innovates combinative cancer therapy via a versatile graphene oxide nanosystem
Chuxin
Cai†
a,
Qingming
Zhang†
b,
Junqiu
Ye†
a,
Sijia
Yao†
a,
Qian
Li
a,
Zhechen
Fan
a,
Sulei
Ge
a,
Yukun
Wang
a,
Dingyi
Xu
a,
Jianping
Zhou
*a,
Hao
Cheng
*a and
Yang
Ding
 *a
*a
aState Key Laboratory of Natural Medicines, Department of Pharmaceutics, China Pharmaceutical University, Nanjing 210009, China. E-mail: dydszyzf@163.com; chenghao@cpu.edu.cn; zhoujianping@cpu.edu.cn
bDepartment of Pharmacy, Jinling Hospital, No. 305 East Zhongshan Road, Nanjing 210002, Jiangsu, People's Republic of China
First published on 17th April 2025
Abstract
The tumor microenvironment (TME) emerges as a unique challenge to oncotherapy due to its intricate ecosystem containing diverse cell types, extracellular matrix, secreted factors, and neovascularization, which furnish tumor growth, progression, invasion, and metastasis. Graphene oxide (GO)-based materials have garnered increasing attention in cancer therapy owing to their vast specific surface area, flexible lamellar structure, and electronic–photonic properties. Recently, interactions of GO with the TME have been broadly investigated, including trapping biomolecules, catalysis, cancer stem cell targeting, immunoreactions, etc., which inspires combinative therapeutic strategies to overcome TME obstacles. Herein, we summarize TME features, GO modulating various dimensions of the TME, and a TME-triggerable drug delivery system and highlight innovation and merits in combinative cancer therapy based on TME modulation. This review aims to offer researchers deeper insights into the interactions between versatile GO nanomaterials and the TME, facilitating the development of rational and reliable GO-based nanomedicines for advanced oncotherapy.
1. Introduction
The tumor microenvironment (TME) is a complex ecosystem consisting of a heterogeneous mixture of cell types, extracellular matrix, secreted factors, and neovascularization, creating a unique ecological niche with characteristics of hypoxia, acidic pH, high concentrations of glutathione etc. The ecosystem dynamically supports tumor growth, progression, invasion, and metastasis,1 which poses significant challenges to therapeutic outcomes. Thus, in recent years, various TME-targeting therapies have been developed to disrupt the supportive interactions for enhanced anti-tumor efficacy.2 Targeting the TME is pivotal in cancer therapy due to its central role in driving malignant progression, including proliferation, metastasis, and therapy resistance. Current strategies to modulate the TME can be classified into four categories: (1) immune modulation (e.g., PD-1/PD-L1 inhibitors, CAR-T cell therapy, cytokine regulation); (2) vascular targeting (anti-angiogenic agents like bevacizumab); (3) stromal intervention (targeting cancer-associated fibroblasts or degrading the dense stromal matrix to enhance drug penetration); and (4) metabolic reprogramming (inhibiting lactate metabolism or modulating nutrient competition).3,4 Due to the unique properties of high loading capacity, surface modifiability, and environmental responsiveness, GO nanomaterials could effectively achieve comprehensive modulation of the TME, including hypoxia, immunosuppression, stromal barriers, and metabolic dysregulation.5 Moreover, GO nanomaterials offer a platform for integrating TME modulation and tumor cell killing for advanced therapeutic benefits.6Interactions of the TME with GO and its derived nanosystem have been extensively studied among various nanomaterials.7 GO's vast specific surface area, flexible lamellar structure, and unique electronic–photonic properties are directly linked to GO's intrinsic TME-modulation activities. For instance, GO's super-large specific surface area could efficiently trap biomolecules like vascular endothelial growth factor (VEGF) for anti-angiogenesis8 and its flexible lamellar structure makes it preferable for internalization by cancer stem cells (CSCs) with lower cellular stiffness to promote CSC differentiation or elimination.9 The electronic–photonic properties endow GO with a capacity for catalysis and photothermal conversion,10 which could generate hydroxyl radicals to aggravate hypoxia and cause hyperthermia for blood perfusion improvement and immunological effects.11 The unique TME could also be utilized for controllable drug delivery system design by introducing TME-responsive moieties on GO nanomaterials.12 A TME-responsive drug delivery strategy is supposed to improve drug selectivity on tumor sites for enhanced efficacy and reduced toxicity and meanwhile regulate TME features, including pH increase,13 hypoxia alleviation, competitive enzyme inhibition, and glutathione depletion for tumor suppression.14 Moreover, abundant oxygen-containing groups and unsaturated bonds of GO make it fit for various modifications.15 The functionalized GO nanomaterials could offer a preferable platform for co-loading multiple drugs in combinative therapies based on TME modulation.
In this review, we summarize the distinctive features of the TME and explore various intrinsic TME-modulation activities of GO nanomaterials. We further discuss GO-derived TME-sensitive drug delivery systems, which perform tumor-specific drug release and combine TME regulation after responsiveness. Taking advantage of GO's amphipathicity and wealthy chemical active groups, we give an overview of multidrug-delivery GO nanomaterials for TME-modulation-based combinative therapies. By summarizing the latest advancements and highlighting the versatile applications of GO nanomaterials in TME modulation and the corresponding combinative therapies, this review aims to provide researchers with deeper insights into the dynamic interactions between GO nanomaterials and the TME, facilitating the development of innovative and reliable GO-based nanosystems for advanced oncotherapy.
2. TME modulation based on GO's features
A comprehensive understanding of the tumor microenvironment (TME) and its relationship with cancer progression is essential for developing TME-targeting therapies.16 The TME is considered as “soil” for cancer development.17 Current concepts of the TME suggest that the ecosystem of the TME consists of unique cellular and noncellular components.18 Cell types in the TME primarily include immune cells, fibroblasts, endothelial cells, and pericytes. Immune cells within the TME play a dual role in cancer progression.19 On the one hand, they can initiate an anti-tumor immune response to eliminate cancer cells via mechanisms such as phagocytosis by macrophages and cytotoxic activity by natural killer (NK) cells and T lymphocytes.20 On the other hand, specific macrophages known as tumor-associated macrophages (TAMs) can promote tumor growth by suppressing the immune system, stimulating angiogenesis, and facilitating metastasis. Endothelial cells in the TME contribute to neovascularization, which is a critical process for supplying oxygen and nutrients,21 and dysregulation or aberrations of vessels can promote tumor growth and metastasis. Cancer-associated fibroblasts (CAFs) are also an important cellular component of the TME22 that could secrete growth factors and cytokines to support the proliferation, survival, and metastasis of tumor cells (Fig. 1).23 | ||
| Fig. 1 Schematic illustration of the main cell types, extracellular components in the TME, and hallmarks of the TME. | ||
Various noncellular components construct the extracellular matrix (ECM) of the TME with a three-dimensional network of proteins and carbohydrates that provides structural support and influences cellular behavior. In the ECM network, cytokines, chemokines, growth factors, and exosomes are among the critical communicators in this network. Cytokines and chemokines modulate immune cell recruitment and immune responses to create an immunosuppressive environment for tumor protection and promotion.24 Growth factors stimulate angiogenesis and cellular proliferation, thereby facilitating the expansion of tumors.25 These signaling molecules create a complex communication network that regulates cell–cell interactions, ECM remodeling, and overall tumor behavior (Fig. 1).
Furthermore, the unique biostructure of the TME contributes to multiple hallmarks that significantly differ from normal tissues.10 One of the critical characteristics of the TME is hypoxia, which results from the rapid proliferation of tumor cells outstripping the supply of oxygen due to poor vascularization.26 This hypoxic condition not only promotes tumor aggressiveness and resistance to therapy but also induces the expression of hypoxia-inducible factors (HIFs) that drive angiogenesis and metabolic reprogramming.27 The hypoxia drives the elevation of the glutathione (GSH) level, which protects tumor cells from oxidative stress and can impair the effectiveness of reactive oxygen species (ROS)-based therapies. Due to the Warburg effect of tumor metabolism, tumor cells rely on glycolysis followed by lactic acid fermentation for energy production; even in the presence of adequate oxygen, an acidic pH often characterizes the TME. This acidification leads to a hostile environment for immune cells and reduces the efficacy of chemotherapeutics.28 Furthermore, the TME exhibits overexpression of specific enzymes such as matrix metalloproteinases (MMPs) and cathepsins, which are involved in extracellular matrix remodeling and facilitating tumor invasion and metastasis. These unique characteristics of the TME provide insight into tumor biology and present significant opportunities for developing targeted and combinative therapeutic strategies.29
2.1 Vast specific surface area of GO for trapping biomolecules
 | ||
| Fig. 2 (A) GO nanomaterials modulate the immunosuppressive microenvironment by macrophage modulation and vaccine adjuvant function. (B) Graphene nanosheets stimulate the secretion of cytokines and chemokines in macrophages. Primary macrophages and macrophage-like RAW264.7 cells were cultured with 20 mg ml−1 graphene for 24 h.40 Reproduced from ref. 40 with permission from Elsevier, copyright (2012). (C) FACS analysis of CD4+ and CD8+ T cell populations after immunization with normal saline, 50 μg OVA, and 25 μg GO-Au@OVA. The lymphocytes were purified from immunized mouse spleens and then incubated with 10 μg mL−1 OVA for 60 h at 37 °C. The cells were then stained with anti-CD3e and anti-CD4 or anti-CD8a antibodies.41 Reproduced from ref. 41 with permission from Ivyspring International, copyright (2020). (D) Schematics showing the design and preparation of GO-APP3/28. (1) A scanning electron microscopy image showing the as-prepared GO. (2) A TEM image negatively stained with uranyl acetate showing the GO-linked protein on the flat region of the GOs. (3–5) False-coloured low-vacuum scanning electron microscopy images showing a fixed T cell (blue) interacting with a piece of GO-APP3/28 (yellow; 3), a dendritic cell (green; 4) and Bead3/28 (red; 5). Scale bars: 30 m (1), 100 nm (2), and 2 μm (3–5).39 Reproduced from ref. 39 with permission from Springer Nature, copyright (2024). | ||
Nano-GO was proved to adsorb model antigen OVA with super high efficiency at a OVA to GO ratio of over 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w). The high aspect ratio of GO allows it to be directly inserted into the cell membrane when in contact with DCs, increasing OVA uptake by about 2.5 times. Besides the lysosomal pathway (the only pathway of OVA), GO-OVA tends to traffic via the cytoplasmic pathway, resulting in the protection of antigen from enzymatic digestion and the cross-presentation of antigen to CD8+T cells, which may be related to the non-lysosomal trafficking pathway of GO into cells.37 Compared with the OVA group, GO-OVA promotes DCs to produce more cytokines and chemokines. Moreover, it significantly increases the expression of major histocompatibility complex 1 (MHC1) molecules, which offers recognition information for CD8+ T in vivo and in vitro.38 Highly efficient and compact adsorption enables GO to change the flat structure to the folded structure, thereby allowing OVA to release slowly, rendering the programmatic activation of specific CD8+ T cells that result in effectively cracking thymoma cells in vivo (the tumor volume decreased by 80%). Based on the potential of antigen absorption, a flexible GO antigen-presenting platform (GO-APP) has been developed to anchor antibodies onto the GO surface. By decorating anti-CD3 (αCD3) and anti-CD28 (αCD28) on graphene oxide (GO-APP3/28), remarkable T cell proliferation has been achieved. Of note, interactions between GO-APP3/28 and T cells closely mimic the in vivo immunological synapses between antigen-presenting cells and T cells. This immunological synapse mimicry shows a high capacity for stimulating T cell proliferation while preserving their multifunctionality and high potency. Meanwhile, GO-APP3/28 enhances CAR gene-engineering efficiency, yielding a more than fivefold increase in CAR T cell production compared with the standard protocol (Fig. 2D).39
:1 (w/w). The high aspect ratio of GO allows it to be directly inserted into the cell membrane when in contact with DCs, increasing OVA uptake by about 2.5 times. Besides the lysosomal pathway (the only pathway of OVA), GO-OVA tends to traffic via the cytoplasmic pathway, resulting in the protection of antigen from enzymatic digestion and the cross-presentation of antigen to CD8+T cells, which may be related to the non-lysosomal trafficking pathway of GO into cells.37 Compared with the OVA group, GO-OVA promotes DCs to produce more cytokines and chemokines. Moreover, it significantly increases the expression of major histocompatibility complex 1 (MHC1) molecules, which offers recognition information for CD8+ T in vivo and in vitro.38 Highly efficient and compact adsorption enables GO to change the flat structure to the folded structure, thereby allowing OVA to release slowly, rendering the programmatic activation of specific CD8+ T cells that result in effectively cracking thymoma cells in vivo (the tumor volume decreased by 80%). Based on the potential of antigen absorption, a flexible GO antigen-presenting platform (GO-APP) has been developed to anchor antibodies onto the GO surface. By decorating anti-CD3 (αCD3) and anti-CD28 (αCD28) on graphene oxide (GO-APP3/28), remarkable T cell proliferation has been achieved. Of note, interactions between GO-APP3/28 and T cells closely mimic the in vivo immunological synapses between antigen-presenting cells and T cells. This immunological synapse mimicry shows a high capacity for stimulating T cell proliferation while preserving their multifunctionality and high potency. Meanwhile, GO-APP3/28 enhances CAR gene-engineering efficiency, yielding a more than fivefold increase in CAR T cell production compared with the standard protocol (Fig. 2D).39
2.2 Carbon defect and electron conduction of GO for catalysis
Hypoxia in tumors is a critical barrier to effective cancer treatment. It promotes resistance to conventional therapies like chemotherapy and radiotherapy and is linked to more aggressive tumor phenotypes.42 The ability of GO nanomaterials to modulate ROS levels has opened new avenues for improving cancer therapy outcomes. The latest research concerning GO catalyzing ROS production was highlighted for enhanced therapeutic efficacy.43In recent years, nano-catalysts for tumor therapy have attracted much interest. Owing to their low cost, high catalytic efficiency, and biocompatibility, carbon-based metal-free catalysts have attracted intense interest for various applications, ranging from energy through environmental to biomedical technologies. Recent studies have validated that functionalized GO nanomaterials with intrinsic catalytic activity could generate ROS in the presence of H2O2.44,45 This ROS production can cause multi-level damage to subcellular components, contributing to cancer cell death. A novel graphene oxide nanoparticle (N-GO) was constructed that could mimic peroxidase-like catalytic activity, specifically in acidic and high hydrogen peroxide (H2O2) environments characteristic of tumors.44 Under such conditions, N-GOs can efficiently convert H2O2 into highly reactive and toxic hydroxyl radicals (HO˙), leading to the necrosis of tumor cells. This targeted effect is advantageous as it avoids damage to normal cells, which reside in a neutral pH environment with lower H2O2 concentrations, where N-GOs instead exhibit catalase-like activity scavenging reactive oxygen species (ROS). In mice models, N-GOs effectively inhibited tumor growth, as evidenced by extensive destruction of tumor cell membranes and nuclei, while showing minimal toxicity to other organs. Blood component analysis revealed no significant differences between N-GO-treated and control groups, confirming the low systemic toxicity of N-GOs. N-GOs offer promising prospects for cancer therapy and diagnostics, combining the benefits of tumor-selective treatment, reduced side effects, and improved tumor cell recognition capabilities. Their ability to selectively target and destroy tumor cells while sparing normal tissue makes them a potentially powerful tool in the fight against cancer.
In the human acute monocytic leukemia cell line (THP-1), GO and vanillin-functionalized GO (V-rGO) exhibited toxic effects on THP-1 cells in a dose-dependent manner. The toxicity manifested as decreased cell viability, proliferation, and increased lactate dehydrogenase (LDH) levels, indicative of cell membrane damage. Furthermore, there was a loss of mitochondrial membrane potential (MMP), decreased ATP content, and cell death.46 The most striking observation was the elevated levels of ROS and lipid peroxidation, causing a redox imbalance within the cells. Oxidative damage led to an increase in malondialdehyde (MDA) levels and a depletion of antioxidants like glutathione (GSH), glutathione peroxidase (GPX), superoxide dismutase (SOD), and catalase (CAT). Notably, V-rGO induced a significantly higher level of ROS in THP-1 cells than GO. The increased ROS production was concentration-dependent, with a 1.5-fold and 3-fold increase in ROS levels observed after 24 hours of exposure to 100 μg mL−1 of GO and V-rGO, respectively. The results suggested that the mechanism of toxicity of V-rGO was more potent than that of GO.44 In addition to the ROS production, lipid peroxidation was measured by quantifying MDA content. Both GO and V-rGO induced lipid peroxidation in THP-1 cells in a dose-dependent manner, further supporting the hypothesis that these graphene materials induced oxidative stress.
Moreover, the defect on GO could enhance the H2O2 catalytic activity. Defect-rich graphene quantum dots (GQDs) have been shown to exhibit high catalytic efficiency in ROS generation, specifically in the H2O2-rich tumor microenvironment.42 The catalytic mechanism of GQDs in generating ROS within the TME was thoroughly investigated, revealing that GQDs possess peroxidase-like activity capable of decomposing H2O2 into highly reactive ˙OH radicals. The generation of ˙OH radicals was confirmed through electron paramagnetic resonance (EPR) spectroscopy using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin-trap, which showed a characteristic signal pattern indicative of ˙OH formation when H2O2 and GQDs were combined. The biological effects of the ROS generated by GQDs were studied in vitro using human breast cancer MCF-7 cells. It was observed that MCF-7 cells continuously consumed ˙OH radicals produced from the GQD-catalyzed H2O2 decomposition.47 Further investigation into the subcellular localization of GQDs revealed their presence in the cytoplasm and nuclei of the cancer cells, indicating successful penetration and aggregation within the cells. These findings suggest that defect-rich GQDs can effectively damage subcellular components, including nuclear structures, due to the ROS they catalytically generate. GO-derived materials open a promising avenue for developing safe and efficient catalytic nanomedicine for cancer treatment (Fig. 3).
 | ||
| Fig. 3 (A) GO nanomaterials modulate hypoxia in cancer cells and generate oxygen in normal cells. (B) Effects of GO and V-rGO on anti-oxidant markers: glutathione (GSH), glutathione peroxidase (GPx), super oxide dismutase (SOD), and catalase (CAT). THP-1 cells were treated with GO (50 μg mL−1), V-rGO (50 μg mL−1), and AgNPs (10 μg mL−1) for 24 h. (C) GO (20–100 μg mL−1) induces reactive oxygen species (ROS) generation. (D) V-rGO (20–100 μg mL−1) induces lipid peroxidation.48 Reproduced from ref. 48 with permission from MDPI, copyright (2019). (E) Catalytic performance of GQDs analysed using the Michaelis–Menten kinetics profile.49 Reproduced from ref. 49 with permission from Springer, copyright (2021). | ||
2.3 Flexible lamellar structure of GO for cancer stem cell targeting
GO and GO-derived graphene quantum dots (GQDs) have been extensively studied for their potential in targeting and manipulating cancer stem cells (CSCs), a subset of tumor cells with self-renewal capabilities that are responsible for tumor initiation, recurrence, and metastasis.50 These materials offer unique advantages due to their properties and have been shown to impact CSCs in various ways, including selective targeting and inhibition of CSCs and promotion of CSC differentiation.51 | ||
| Fig. 4 (A) GO nanomaterials can target soft CSCs selectively and inhibit CSCs. (B) GO nanomaterials kill CSCs and remodel the tumor microenvironment. (C) CSCs are resistant to free Dox but sensitive to N-GQD@Dox. Bulk tumor cells and CSCs were treated with DMSO (control), N-GQDs, free Dox, and N-GQD@Dox for 48 h, respectively. Cell viability was measured using the MTS assay. n = 3. N-GQDs@Dox is accumulated in the tumor tissues rather than the major organs. (D) The major organs (heart, liver, spleen, lungs, and kidneys) and the generated tumors.54 Reproduced from ref. 54 with permission from Elsevier, copyright (2021). (E) GDYO exhibits anti-lymphoma effects in vivo and the morphology of tumors after GDYO treatment. (F) Flow cytometric analysis of inflammatory cytokines affecting the frequency of Tregs and CSCs in vitro.55 Reproduced from ref. 55 with permission from Elsevier, copyright (2022). | ||
A recent study further demonstrates that the GO-like nanostructure of graphdiyne oxide (GDYO) nanosheets could also show CSC targeting and elimination activities.51 GDYO has been shown to effectively combat lymphoma by targeting and eliminating cancer stem cells (CSCs), which are often referred to as the “seeds” of cancer, while concurrently remodeling the TME, likened to the “soil” in which cancer grows. The mechanism of action involves GDYO significantly reducing Mif-Ackr3 signaling from the tumor to cancer-associated fibroblasts (CAFs), decreasing the levels of inflammatory cytokines within the CAF cells, and lowering the frequency of Treg cells and CSCs.56 As a result, the growth of lymphoma is inhibited. The long-term safety profile of GDYO has also been assessed, demonstrating that it does not adversely affect mouse survival even when administered for extended periods, up to 20 months. This underscores the biosafety and biocompatibility of GDYO, suggesting its potential application in clinical medicine to treat lymphoma.
Carbon quantum dots (CQDs) can serve as a physiological niche for stem cells, influencing their formation and differentiation.57 While much work has been done on stem cell imaging with CQDs, less attention has been paid to their interaction with stem cells and their potential to selectively guide differentiation towards a specific lineage.54 CQDs interact with stem cells in various ways, influencing their differentiation potential. They can modulate the expression of critical genes and proteins involved in the differentiation process, acting as signaling molecules that steer stem cells toward specific lineages. For instance, CQDs have been shown to upregulate osteogenic markers such as alkaline phosphatase (ALP), RUNX2, OCN, and BSP in bone marrow mesenchymal stem cells (MSCs), indicating the promotion of osteogenic differentiation. Specific microRNAs like miR-2861 can further enhance this effect, which promotes osteoblast differentiation (Fig. 5).53
 | ||
| Fig. 5 (A) GO nanomaterials promote the differentiation of CSCs.50 Reproduced from ref. 50 with permission from American Chemical Society, copyright (2022). (B) Immunostaining of MSCs seeded for 15 days in osteogenic differentiation medium on different substrates. MSCs growing on Si/SiO2, PDMS, and PET without graphene show OCN negative staining. Once these substrates are coated with graphene, cells are positive for OCN, indicating osteogenic differentiation. Scale bars are 100 μm.59 Reproduced from ref. 59 with permission from BioMed Central, copyright (2014). | ||
The novel mechanism by which the GO nanomaterial promotes CSC differentiation is also investigated. This research article discusses bioactive graphene quantum dots (GQDs) to target and eradicate drug-resistant cancer stem cells (CSCs), critical in cancer metastasis and recurrence.54 Conventional chemotherapy fails to eliminate these CSCs effectively due to inadequate interfacial inhibition effects. However, GQDs can self-insert into the DNA major groove (MAG) sites within cancer cells, enhancing interfacial inhibition significantly.58 Transcription factors, which regulate gene expression at MAG sites, are affected by this targeted action of GQDs, resulting in the downregulation of cancer stem genes like ALDH1, Notch1, and Bmi1. The nano-scale interface inhibition mechanism of GQDs reverses multidrug resistance (MDR) in cancer cells by inhibiting the MDR1 gene expression. When GQDs are applied at non-toxic concentrations (1/4 of the half-maximal inhibitory concentration, IC50), they act as MDR reversers. The efficacy of this approach allows for substantial blockage of CSC-mediated migration, invasion, and metastasis of cancer cells. GQDs can also sensitize clinical cytotoxic agents, improving their effectiveness in combination chemotherapy.57
2.4 Photothermal effect of GO for immunoreactions
With unique optical–thermal properties, GO offers significant advantages in modulating the tumor microenvironment for therapeutic purposes. Graphene-based materials can efficiently convert light into heat when exposed to near-infrared (NIR) light.60 This ability allows targeted hyperthermia treatment to selectively destroy cancer cells while minimizing damage to surrounding healthy tissues.61 The localized heat generation not only directly kills tumor cells but also enhances the permeability of the tumor vasculature, facilitating the delivery of therapeutic agents. Moreover, the photothermal effect of GO can stimulate an immune response against the tumor, potentially leading to a synergistic effect when combined with immunotherapy.62 The biocompatibility and tunable surface chemistry of graphene further enable its functionalization with targeting ligands or drugs, making it a versatile platform for precise and effective cancer therapy (Fig. 6). | ||
| Fig. 6 Schematic illustration of GO-derived PTT for activating anti-tumor responses via immunogenic cell death (ICD) and M2 tumor-associated macrophage elimination. | ||
GO-based PTT directly kills tumor cells and induces immunogenic cell death (ICD). During ICD, damage-associated molecular patterns (DAMPs) such as calreticulin (CRT) translocation, high-mobility group box 1 (HMGB1) release, and heat shock protein (HSP) efflux are observed. These DAMPs collectively activate antigen-presenting cells (APCs), triggering subsequent immune processes.68 Hydrophilic Fe3O4 nanoparticles were combined with rGO through electrostatic adsorption, significantly promoting DC maturation up to 30.2% under 808 nm laser irradiation.69 The GO-arginine-soy lecithin nanogenerator (GO-Arg-SL) for PTT and gas therapy (GT) effectively releases l-Arg in the cytoplasm of tumor cells, which was then catalyzed by inducible nitric oxide synthase (iNOS) to produce a large amount of nitric oxide (NO). The released NO promoted the maturation of antigen-presenting cells (DCs) and caused a significant increase in CD8+CD4−CD3+ in the spleen. Mature DCs can activate CTLs, stimulating the innate immune system and enhancing anti-tumor immunity. Tumors were significantly eliminated due to the enhanced anti-tumor effect of PTT and GT.70 Nanoparticles coated with granulocyte-macrophage colony-stimulating factor (GM-CSF@CS/GO) were synthesized, which could cause the photothermal destruction of tumors, release a large number of tumor-associated antigens and significantly promote the recruitment of DC cells and the production of anti-tumor cytokines.71 Transdermal delivery of paclitaxel (DTX) and 1-methyl-D-tryptophan (1MT) (D-1/GH) based on heparin-modified graphene oxide (GH) enhances the therapeutic effect of chemotherapy and PTT while inducing immunogenic cell death (ICD), thereby recruiting immune cells, reversing the immunosuppressive tumor microenvironment, and enhancing immune response (Fig. 7).72
 | ||
| Fig. 7 (A) Schematic showing the synthesis of GGIC and its immunostimulatory effect. (B) PBS, CpG ODNs, GGI, and GGIC were injected intratumorally in a single dose, followed by 5 min NIR irradiation or without NIR irradiation. (C) In vivo photothermal tumor heating. IR thermal images of tumor-bearing mice exposed to the NIR laser after injection with PBS, GGI, or GGIC. (D) The volumetric changes in tumor size and changes with time in body weight were achieved from mice injected with PBS, CpG ODNs, GGI, and GGIC. Data are presented as mean ± SD of five mice per group. (E–J) Histopathological studies of tissue organs of (E) PBS + NIR, (F) GGI, (G) CpG ODNs, (H) GGIC, (I) GGI + NIR, and (J) GGIC + NIR in the CT26 tumor-bearing mouse model.73 Reproduced from ref. 73 with permission from Elsevier, copyright (2014). | ||
The anti-tumor immunity triggered by PTT is severely hindered by the dynamic amplification of programmed cell death protein ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase 1 (IDO-1) in tumors. To fully unleash the efficiency of PTT, blocking the PD-L1 and IDO-1 pathways is essential to enhance effective anti-tumor immunity.74,75 Folic acid-coupled rGO loaded with IDO inhibitors (PEG-rGO-FA-IDOi) effectively blocks tryptophan catabolism and enhances T cell proliferation while simultaneously blocking PD-L1, and combining PTT, IDO inhibition, and anti-PD-L1 blockade counters immune suppression effectively. This approach enhances the presence of tumor-infiltrating lymphocytes, including CD45+ leukocytes, CD4+ and CD8+ T cells, and NK cells. It also inhibits the immunosuppressive activity of regulatory T cells (Tregs) and boosts the production of interferon-gamma (INF-γ) (Fig. 8).76 Based on rGO, the chemotherapeutic drug mitoxantrone (MTX) and the transforming growth factor β (TGF-β) inhibitor SB-431542 (SB) were loaded onto rGO-PEG through π–π stacking interactions. rGO-based PTT provides an immunogenic antigen source, forming an in situ vaccine with rGO acting as an adjuvant. The use of SB changed the tumor microenvironment, enhancing the therapeutic effect of MTX-induced chemotherapy and rGO-based PTT. The infiltration of tumor-specific cytotoxic CD8+ lymphocytes increases while the infiltration of distal Tregs decreases, providing a synergistic chemo–immuno–photothermal effect.77 GO-loaded nanoparticles with SNX-2112 and folic acid (GFS) guide LTPTT by initially inhibiting the AKT pathway, subsequently inducing the over-activation of autophagy, thereby reducing the expression of PD-L1. The results indicate that GFS-guided LTPTT can diminish tumor cell immune escape and immunosuppression. It enables T cells to regain their natural immune function, leading to the destruction of tumor cells. This process is facilitated by the combined effect of PTT and drug treatment, which activates cell autophagy and enhances immune function.78 Moreover, GO loaded with immune-related substances can synergize with PTT to activate the immune response at the tumor site. GO modified with polyethylene glycol (PEG) and polyethyleneimine (PEI) could compress CpG oligonucleotide sequences effectively to generate a GGIC nanosystem. The cellular uptake of GGIC is significantly enhanced after GO-mediated photothermal heating, effectively enhancing intracellular delivery of CpG. Subsequently, Toll-like receptor 9 (TLR9) recognizes CpG, releasing various pro-inflammatory cytokines such as tumor necrosis factor TNF-α and interleukin IL-6 for immune stimulation enhancement.79 Collectively, the interaction between heat shock and thermal immunity is significant at the cellular biology level and provides new strategies and mechanisms for the immunotherapy of tumors. In this way, the increase in heat shock proteins helps cells cope with thermal stress and effectively activates the immune system, enhancing the immune response to tumors and providing an essential target for tumor therapy.
 | ||
| Fig. 8 (A) A multifunctional IDO inhibitor (IDOi)-loaded reduced graphene oxide (rGO)-based nanosheets (IDOi/rGO nanosheets) with the properties of directly killing tumor cells under laser irradiation and triggering anti-tumor immune responses in situ. (B) PEG-rGO-FA-IDOi can induce DC activation in vivo after irradiation with NIR; mature DCs induced increased expressions of CD86 and CD80. (C) Individual tumor growth for a primary tumor (1#) and a distant tumor (2#). (D) Treatment with the combined PEG-rGO-FA-IDOi-mediated PTT plus PD-L1 antibody led to a remarkably increased proportion of CD45 leukocytes, CD4 T cells, and CD8 T cells in the distant tumors and approximately 1.8- to 4-fold enhancement compared to treatment with the single PEG-rGO-FA-IDOi-mediated PTT.76 Reproduced from ref. 76 with permission from American Chemical Society, copyright (2019). | ||
The results indicated that the uptake of FITC-PEG-GO did not induce polarization of macrophages towards the M1 pro-inflammatory phenotype. Instead, it promoted a controlled balance between M1 and M2 phenotypes and slightly shifted towards the M2 reparative phenotype involved in tissue repair, ensuring an appropriate immune response to these nanosheets.86 However, macrophages treated with IL-4 would show M2 polarization, while post GO-mediated PTT, the RAW264.7 cell line exhibited more polarization towards M1 and less towards M2. These findings suggest that GO itself does not possess the capability to influence TAMs in the tumor microenvironment, and PTT is a crucial factor for mediating TAM's immune function. Through thermal stress, GO-mediated PTT affects the function of M2 macrophages, potentially inducing their transformation to the M1 type, thereby inhibiting tumor progression (Fig. 9).87 The effects of GO and rGO on primary bone marrow-derived macrophages (BMDM) and the immortalized macrophage cell line (J774A.1) were also evaluated in previous studies. They found that rGO, compared to GO, induced higher levels of TNF-α and IL-6 production, indicating a more robust pro-inflammatory response in macrophages. These studies demonstrate the potential application of GO and its derivatives in promoting macrophage polarization toward the M1 phenotype.88 Additionally, leveraging the activities of M2 macrophages can synergize with PTT. A multifunctional, biologically active scaffold comprising hydrated CePO4 nanorods and chitosan (CS) forms a CePO4/CS/GO structure. During PTT, the released Ce3+ ions promote the polarization of macrophages towards the M2 phenotype. Utilizing M2 macrophages to secrete vascular endothelial growth factor (VEGF) and arginase-1 (Arg-1) fosters angiogenesis and induces bone regeneration. This approach optimizes adjuvant therapy following tumor surgery and improves prognosis.89
 | ||
| Fig. 9 (A) GP-mediated PTT affects the function of M2 macrophages through heat stress and may induce their conversion to M1, thereby inhibiting tumor progression. It involves the polarization of macrophages by heating and regulates their anti-tumor ability. (B) The IL-4 group showed significantly increased levels of the surface protein CD206, and the NIR + IL-4 group showed considerably decreased expressions of CD206. (C) The expression of M2-related genes in the IL-4 group was significantly increased, whereas the expression of these M2-related genes was sharply inhibited after PTT. (D) The NIR group could not repair the scar after 24 h of culture, and its ability to repair scratches was the worst. (E) The macrophage supernatants treated with NIR could inhibit the invasion and migration of tumor cells. (F) PTT showed an excellent anti-tumor effect in vivo via macrophage polarization.87 Reproduced from ref. 87 with permission from Elsevier, copyright (2020). | ||
3. TME-responsive GO nanomaterials control drug shuttling and TME regulation
Recent advancements in nanotechnology have contributed to the exploration of GO as a versatile drug carrier, offering innovative approaches for cancer diagnosis and therapy. Extensive studies have witnessed that GO can effectively load and deliver chemotherapeutic agents and biomolecules to enhance therapeutic efficacy and minimize systemic toxicity.90 However, GO intrinsically lacks active targeting capabilities and demonstrates limited therapeutic efficacy against cancer. Therefore, recent research has shifted towards enhancing GO's tumor specificity and therapeutic potential through strategic surface modifications.91–93 The diverse functional groups on GO's surface provide a foundational platform for such modifications, enabling the incorporation of targeted therapeutic strategies. The tumor microenvironment, with its distinct characteristics like acidic pH, overexpressed enzymes, and specific biomarkers, offers many targeting opportunities.94 By leveraging these unique environmental features, GO can be tailored to selectively accumulate in tumor tissues and share controllable release profiles, thus enhancing therapeutic agents’ delivery efficiency while reducing systemic toxicity (Fig. 10). | ||
| Fig. 10 TME-responsive GO nanosystem for selective and efficient drug delivery. | ||
3.1 Reduction/oxidation-sensitive GO nanomaterials
The TME presents unique redox characteristics that can be strategically exploited for cancer therapy. The tumor microenvironment's redox environment is pivotal in tumor biology and treatment.95 Due to their rapid proliferation and aberrant metabolic activities, tumor cells generate a substantial amount of reactive oxygen species (ROS), such as superoxide anions, hydrogen peroxide, and hydroxyl radicals.96 These ROS can promote tumor growth and spread yet simultaneously render tumor cells more susceptible to oxidative stress damage. To counteract this enhanced oxidative stress, tumor cells typically bolster their antioxidant systems, such as elevating glutathione (GSH).97 This augmented antioxidant capacity aids tumor cells in resisting the toxic effects of ROS.Researchers have modified and altered GO to enhance its therapeutic efficacy against tumors in response to this unique environment. These modifications aim to leverage the distinctive redox characteristics of the tumor microenvironment, creating more effective cancer treatment strategies. In the TME, the levels of reduced GSH, a crucial antioxidant, are typically elevated. GSH neutralizes ROS and other free radicals, protecting cancer cells from oxidative stress damage.98,99 Therefore, the responsive design targeting GSH is considered a feasible strategy. Disulfide bonds are essential in stabilizing protein structures in normal human tissues. They break in the presence of a high level of GSH. Disulfide bonds are, therefore, a classical means to target high GSH levels in the TME.100 A novel approach utilizing a hyaluronic acid (HA)-modified, oxidation–reduction-sensitive graphene oxide nanosheet (HSG) was developed for targeted and rapid delivery to tumor cells. HA was conjugated to GO via disulfide bonds, conferring active targeting capabilities to the graphene oxide-based system. Within the intracellular environment, the disulfide bonds of HA were cleaved by the overexpressed GSH, detaining conjugated HA fragments from the GO surface and accelerating the release of DOX (Fig. 11).101 The arginine–glycine–aspartic acid (RGD) peptide and amine-functionalized polyethylene glycol (6ARM-PEG-NH2, PEG-NH2) were employed to functionalize graphene oxide. A disulfide bond was introduced at one end of PEG, and DOX was covalently attached, constructing a pH and GSH dual-responsive nano-drug delivery system (RGD–GO–PEG–S–S–DOX). DOX exhibited rapid release in a PBS solution with pH 5.50 and a GSH concentration of 10 mM.102 Moreover, magnetic GO nanosheets were constructed by coupling cystamine-functionalized GO nanosheets with Fe3O4 nanoparticles.103 The magnetic GO nanosheets were loaded with DOX at a high drug/carrier ratio of 1 to create a redox-responsive delivery system for controllable chemotherapy. In drug release evaluation, the maximum drug release after 10 days was only 17.9% without GSH addition, while the maximum drug release reached 51.7% in the presence of GSH. The magnetic GO delivery system achieves highly selective drug release at tumor sites and high drug-loading efficiency, offering a novel strategy for precision chemotherapy.
 | ||
| Fig. 11 Scheme illustration of DOX released from HSG-DOX, which contained disulfide. (A) Glutathione triggers HA detachment and rapid release of DOX in the cytoplasm. (B) Based on the redox sensitivity of HSG-DOX, it can significantly improve the killing ability of DOX against MDA-MB-231 and HELF cells. The system has significant anti-tumor activity in vivo. (C) UV–vis spectra of the GO, GO-COOH, HSG, and HCG. (D) Measurement of the temperature of water, GO, GO-COOH, HSG, and HCG under NIR irradiation (808 nm, 5 W cm−2). (E) CLSM images of the PCI effect of HSG and HCG based on their photothermal ability. (F) CLSM images of MDA-MB-231 cells incubated with free DOX, HSG-DOX, HCG-DOX, and HA + HSG-DOX over time.101 Reproduced from ref. 101 with permission from Wiley, copyright (2017 ). | ||
The basis of carrier response mechanisms based on redox reactions is ROS levels in tumor tissues. H2O2 is the most typical ROS. H2O2 has a variety of catalytic activities and has been widely explored for the activation conditions of drug release.104 The ability of nanoscale graphene oxide particles (N-GO) to mimic peroxidase-like catalytic activity was investigated in the tumor microenvironment characterized by weak acidity and high levels of ROS. This dual-responsive behavior significantly and effectively inhibited tumor growth. In the neutral microenvironment of normal cells, N-GO exhibited peroxidase-like activity similar to catalase, converting H2O2 into oxygen and water, thereby protecting normal cells from harm. These biofunctions hold crucial implications for the diagnosis and prognosis of cancer.105 Similarly, a unique H2O2-triggered nanoenzyme-catalyzed photoacoustic imaging (PAI) contrast agent was developed utilizing a nanoscale graphene quantum dot fragment (GQDzyme) as the core, coupled with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS). Using nasopharyngeal carcinoma as a model, in the presence of elevated H2O2 expression at the tumor site, GQDzyme converted ABTS into its oxidized form. The oxidized ABTS exhibited strong NIR absorbance, making it an ideal PAI contrast agent for precise imaging at the tumor site and assisting in diagnostics.106
3.2 Acidic pH-responsive GO nanomaterials
The aberrant metabolic activities of tumor cells primarily drive the unique weakly acidic feature of the TME. The acidic environment is closely linked to the Warburg effect, where cancer cells preferentially produce energy through glycolysis followed by lactic acid fermentation, even in the presence of ample oxygen, leading to an excessive accumulation of lactic acid and consequent acidification.107 Additionally, the poor vascular structure of tumors results in hypoxic regions within, further promoting glycolysis.108 The increased expression of carbonic anhydrases in tumor cells also contributes to carbonic acid production, exacerbating the microenvironment's acidity.109 This acidic milieu profoundly impacts tumor invasiveness, metastatic potential, and response to therapies. Under acidic conditions, the efficacy of certain chemotherapeutic drugs is reduced, and the acidic environment may also suppress the function of immune cells, thereby weakening the anti-tumor immune response.110 GO has emerged as a promising material for targeting the acidic TME. Modifying the surface of GO endows it with the capability to modulate acidic environments. This modification facilitates interactions with the acidic milieu, enabling the development of targeted drug delivery systems and therapeutic strategies that leverage the unique pH values characteristic of cancerous tissues.111GO-based nanocarriers exhibit a unique pH-responsive drug release mechanism in the TME, primarily attributed to the π–π stacking interactions between the drug and the GO surface. Research indicates that these interactions remain relatively stable under neutral or mildly alkaline conditions.112 However, the release of drugs like DOX, which are conjugated to GO through π–π stacking, is controlled by the degree of carboxyl group protonation on the nanocarrier, which is influenced by the pH of different environments. In acidic media, most carboxyl groups on GO are protonated, altering the charge state and weakening the electrostatic interaction between the nanocarrier and DOX, leading to drug release.113 Additionally, under acidic conditions, the structure of GO may partially disintegrate or expand, further facilitating drug release from its surface. This characteristic renders GO-based nanocarriers particularly suitable for tumor microenvironment-specific drug delivery, enhancing therapeutic efficacy while reducing toxicity to normal tissues (Fig. 12).
 | ||
| Fig. 12 (A) GCE/siRNA was prepared from GO-COOH, functionalization with chitosan (GO-CS), immobilization with anti-EpCAM (GCE), and then mixed with survivin-siRNA (GCE/siRNA) in aqueous solution. (B) After targeting tumor tissues, GCE could deliver survivin-siRNA into cells successfully, and survivin-siRNA could be released from GCE/siRNA, exhibiting the anti-tumor effect. (C) Confocal images were obtained by treating the cells with PBS, naked siRNA, GCE/siRNA, and Lipo/siRNA; siRNA was labeled with the FAM fluorescent molecule (green) to show the amount of siRNA in cells and Hoechst 33342 (blue) for the cell nucleus. (D) Images were taken at different time points after the tail vein injection. (E) An image of the organs collected from each group was taken 8 h after the tail vein injection.61 Reproduced from ref. 61 with permission from Elsevier, copyright (2021). (F) Hydrolysis of the citraconic amide side chains of anionic charge-reversal polyelectrolyte (PAH-Cit) under mildly acidic conditions yields cationic PAH (poly(allylamine)). Construction of the targeted charge-reversal nanocarrier (GO-Abs/PEI/PAH-Cit/DOX). Controlled release of DOX in endosomes or lysosomes, stimulated by the pH-dependent charge-reversal of the charge-reversal polyelectrolytes on GO. (G) Cumulative DOX release from the charge-reversal polyelectrolyte coated nanocarriers GO/PEI/PAH-Cit/DOX at pH 5.0, 6.8, and 7.4. Comparison of the cumulative DOX release from GO/PEI/PAH-Cit/DOX and GO-DOX at pH 5.0. (H) Fluorescence intensities of integrin αVβ3 positive U87 MG cells (G) and integrin αVβ3 negative MCF-7 cells (H) obtained with flow cytometry after these cells were incubated with DOX, GO/PEI/PAHCit/DOX, GO-Abs/PEI/PAH-Cit/DOX and GO-Abs/DOX for 2 h, respectively. The DOX fluorescence histogram of cells was obtained from 10000 cells by flow cytometry under 488 nm excitation.114 Reproduced from ref. 114 with permission from Elsevier, copyright (2014). | ||
3.3. Enzyme-responsive GO nanomaterials
A unique spectrum of enzymes is crucial in regulating tumor development, metastasis, and therapeutic response. For instance, overexpressed proteolytic enzymes, such as matrix metalloproteinases (MMPs), hyaluronidase, and cathepsins, facilitate tumor invasion and angiogenesis by degrading the extracellular matrix.115,116 Simultaneously, the alteration in metabolic enzymes within cancer cells supports the Warburg effect, a hallmark of metabolic reprogramming.117 MMPs, zinc-containing endopeptidases, exhibit a broad substrate specificity. MMPs encompass various subtypes, including MMP-2, MMP-7, MMP-14, etc. MMPs can degrade the extracellular matrix, thereby promoting the invasion and metastasis of tumor cells, playing a pivotal role in reshaping the tumor microenvironment. MMPs are widely employed in cancer therapy as biomarkers and therapeutic targets.118 A biodegradable GO hybrid drug delivery system with triple-responsive characteristics to pH, redox, and enzymes was developed by assembling bovine serum albumin (BSA) nano units onto GO nanosheets. As natural gelatin, this system triggered doxorubicin (DOX) release under H+ action. Through the synergistic effect of MMP-2, gelatin was degraded, further promoting DOX release and leading to the rupture of small BSA-DOX particles, penetrating deep into the tumor.119 An MMP-cleavable peptide sequence (PLGVRGG) was designed for covalently binding to a polyethylene glycol-derived imaging probe and the anticancer drug bufalin IIb peptide, grafted onto the surface of GO (IPGO/BF). The cleavage of the MMP-sensitive peptide triggered imaging signals, subsequently shielding the exposure of BF on graphene oxide, used for targeted therapy in SCC7 mouse models (Fig. 13).120 Hyaluronidase, a highly active biomarker in the TME, is frequently designed to trigger controlled drug release at tumor sites. A HAase-activatable “smart” platform (HA-Ce6/GO) was developed, where chlorin e6 (Ce6) molecules were covalently coupled onto the backbone of hyaluronic acid (HA) to generate a HA-Ce6 polymer, followed by HA-Ce6 absorption onto the GO surface to construct HA-Ce6/GO.121 In the nanosystem, π–π stacking between GO and Ce6 molecules caused efficient quenching of 1O2 generation. Therefore, the HA-Ce6/GO nanosystem could remain devitalized before reaching the tumor site, while Ce6 could be released from GO nanosheets upon enzymolysis of hyaluronidase, switching on photodynamic activity. This strategy significantly improved drug delivery precision, avoiding cytotoxic effects on normal tissues during tumor treatment.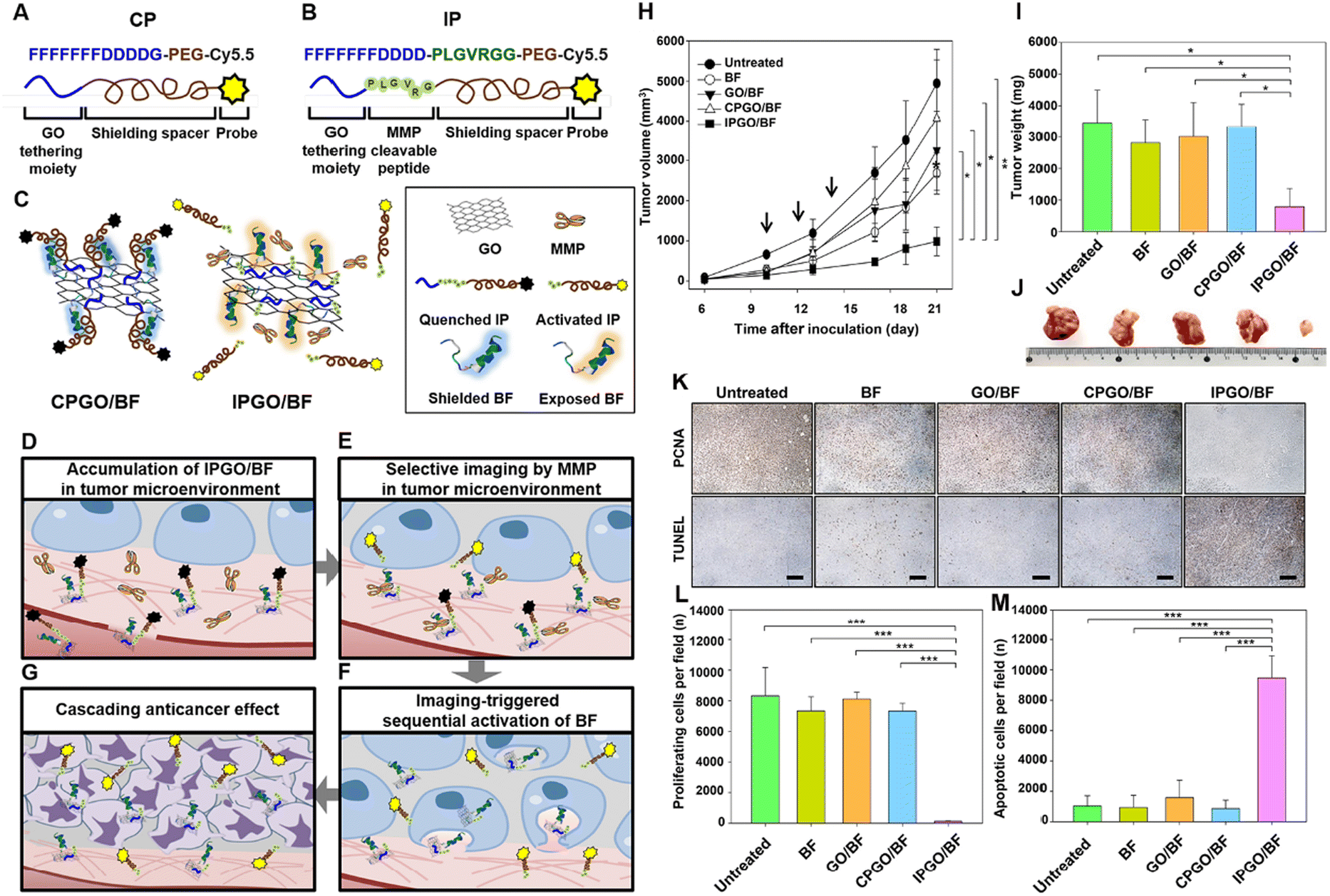 | ||
| Fig. 13 Illustration of hypothesized mechanisms involved in sequential activation of anticancer therapeutic effects triggered by selective sensitization of imaging in a tumor microenvironment. Structures of CP (A) and IP (B). Cy5.5 in IP and CP is not fluorescent upon anchoring to GO. (C) CPGO/BF does not contain MMP-cleavable peptides and remains quenched in the tumor microenvironment with the therapeutic peptide BF hidden in the PEGylated probe. IPGO/BF contains the MMP-cleavable sequence therapeutic peptide BF anchored to GO nanosheets; the PEG cloud shields the functional activity of BF. In the tumor microenvironment, MMP-responsive de-shielding of the PEGylated imaging probe activates imaging and causes cascading exposure of BF on GO. (D) In the bloodstream, the fluorescence of Cy5.5 derivatives on GO nanosheets is quenched. The presence of PEG blocks the apoptotic activity of the therapeutic peptide. GO nanosheets accumulate in tumor tissues. (E) In the tumor microenvironment, IP on GO is cleaved by MMPs, resulting in selective fluorescence activation. (F) Release of PEGylated Cy5.5 subsequently triggers the exposure of the apoptotic anticancer peptide BF into tumor cells. (G) The sequential exposure of BF on GO results in anti-tumor activity. (H) SCC7 tumor-bearing mice were intravenously injected with free BF, GO/BF, CPGO/BF, or IPGO/BF three times at 2 d intervals (injection times indicated by arrows). Tumor volume was measured periodically for 21 d after tumor inoculation. (I) Representative tumors from each group at day 21. (J) Weights of tumors for each group at day 21 (*P < 0.05; **P < 0.01). (K) For determining the immunohistochemistry of tumor tissues, SCC7 tumor-bearing mice were intravenously injected with free BF, GO/BF, CPGO/BF, or IPGO/BF three times at 2 d intervals. On day 21, after tumor inoculation, tumor tissues were sectioned for anti-PCNA antibody immunostaining (upper panels) and the TUNEL assay (lower panels). Scale bar = 100 μm. (L and M) Summary data comparing the number of proliferating (L) and apoptotic (M) cells (***P < 0.005). The results are the mean ± SD of five independent experiments.120 Reproduced from ref. 120 with permission from Elsevier, copyright (2019). | ||
4. GO nanomaterials innovate multidrug delivery for combinative therapy
GO nanomaterials have revolutionized the field of cancer therapy by enabling the precise and efficient delivery of multiple drugs, thereby enhancing the regulation of the tumor microenvironment and synergizing with other anti-tumor treatments.111 These nanocarriers can simultaneously address various aspects of tumor biology by loading GO nanoparticles with chemotherapeutic agents, immune checkpoint inhibitors, and anti-angiogenic drugs. Tumors often develop hypoxic and acidic conditions, which can promote resistance to conventional therapies and facilitate metastasis. GO nanoparticles can be designed to release oxygen-generating agents or pH-sensitive drugs that normalize the TME, thereby sensitizing the tumor to other treatments. For example, by releasing oxygen, GO can alleviate hypoxia and enhance the effectiveness of radiation and photodynamic therapy. Similarly, pH-responsive GO can release drugs specifically in the acidic tumor regions, increasing their therapeutic impact while minimizing systemic side effects. Moreover, combining GO-mediated drug delivery with other anti-tumor therapies, such as photothermal therapy (PTT) and immunotherapy, offers a synergistic approach towards cancer treatment.122 PTT, which involves the conversion of NIR light into heat by GO, can induce localized hyperthermia that directly kills cancer cells and enhances the permeability of the tumor vasculature. This increased permeability facilitates the deeper penetration of co-delivered drugs, improving their therapeutic efficacy. Additionally, the thermal ablation of tumor cells can release tumor-associated antigens, stimulating an immune response that co-administered immune checkpoint inhibitors can further potentiate.123 In summary, GO-based multidrug delivery systems provide a versatile and powerful strategy for regulating the tumor microenvironment and integrating multiple therapeutic modalities.4.1 Tumor vasculature targeting for combinative therapy
Tumor angiogenesis is a critical process in the progression and metastasis of cancer, providing a rich vascular network for the delivery of nutrients and oxygen necessary for tumor cell proliferation.124 Targeting the tumor vasculature has become a promising approach for cancer therapy, as it can disrupt the supply lines for tumor growth. The two-dimensional lamellar structure of GO, combined with its tailorable surface charge and hydrophilicity, enables robust permeability. Specifically, the lateral dimensions of GO align well with the leaky vasculature of the tumor, facilitating passive accumulation in tumor tissues. The nanoscale design of GO significantly enhances tumor targeting capability by leveraging the enhanced permeability and retention (EPR) effect in the tumor vasculature. Studies have demonstrated that when the lateral dimensions of GO are controlled within the range of 50–300 nm and its thickness is maintained at 1–2 nm, the physical characteristics of GO share optimal alignment with the interendothelial gaps in tumor blood vessels. This dimensional compatibility enables GO nanosheets to penetrate the loosely organized vascular endothelial layers efficiently and selectively accumulate in tumor tissues.125 Additionally, the negatively charged surface of GO reduces nonspecific interactions with normal endothelial cells, further enhancing tumor-specific retention. Moreover, abundant oxygen-containing functional groups (COOH, OH, and epoxy groups) on GO's surface provide versatile platforms for covalent modification of targeting ligands. For instance, covalent conjugation of anti-VEGF or anti-integrin αvβ3 antibodies to GO via amido linkage enables precise tumor vascular targeting. The incorporation of RGD peptides or vascular homing motifs (e.g., NGR peptides) could also enhance binding affinity to angiogenic endothelial cells.126 In summary, the physical properties of GO and its surface modification are significant for tumor vascular targeted therapy. By precisely regulating the physical size and chemical functionalization of GO, synergy between passive targeting and active targeting can be achieved to improve drug delivery efficiency and reduce systemic toxicity significantly.127 We summarize the recent advancements in utilizing GO nanomaterials for modulating tumor angiogenesis, highlighting their role in targeting, imaging, and therapeutic applications.Tumor vasculature-targeted nanomedicines represent an innovative and precise strategy in cancer therapy, aiming to improve efficacy while minimizing systemic toxicity. Tumors cannot survive without a constant supply of nutrients and oxygen, which they obtain through their network of blood vessels. Targeted therapies that work by inhibiting the development of these blood vessels could effectively starve the tumor and prevent its growth.128 Tumor vasculature instead of tumor cell targeting is more desirable for graphene-based nanomaterials since the targets are immediately accessible upon intravenous injection, and extravasation is not required to achieve tumor targeting.
Previous studies explored reduced graphene oxide (RGO) for tumor vasculature targeting in a breast cancer model. RGO was conjugated with the anti-CD105 antibody (anti-CD105), where CD105 is overexpressed on proliferating tumor endothelial cells. The RGO conjugate, 64Cu-RGO-anti-105, exhibited excellent stability and specificity for tumor endothelial cells in vivo. Serial PET imaging revealed rapid uptake by tumor endothelial cells, peaking at 3 h post-injection, with sustained levels over time. The results demonstrated the efficacy of RGO for tumor vasculature targeting and imaging. Apart from imaging applications, nano-GO conjugated with monoclonal antibodies against follicle-stimulating hormone receptor (FSHR) has been used for tumor vasculature targeting and inhibition in metastatic breast cancer. By focusing on angiogenic markers on the tumor vasculature, researchers can achieve more effective delivery of nanomaterials directly to the tumor site, bypassing the need for extravasation. In this work, GO nanoparticles were functionalized with a monoclonal antibody (mAb) against FSHR, a particular marker found abundantly in both primary and metastatic tumors. The resulting GO nano-conjugates, measuring approximately 120 nm in diameter, were radiolabeled with 64Cu to enable their visualization using positron emission tomography (PET) imaging. The imaging function allowed real-time monitoring of the conjugates’ distribution and tumor accumulation. The findings demonstrated that the GO conjugates accumulated significantly in metastatic tumor nodules within the lungs. The conjugates’ ability to selectively target tumor vasculature and deliver therapeutic agents efficiently makes them attractive candidates for image-guided therapeutic delivery in metastatic breast cancer treatment (Fig. 14).
 | ||
| Fig. 14 (A) GO nanomaterials modulate tumor angiogenesis by tumor vasculature targeting and VEGF adsorption. (B) Inhibition of angiogenesis by BSA-GO in the chick chorioallantoic membrane (CAM). Dotted circles represent the area of the CAM under cellulose filter paper discs. The relative branch numbers of the corresponding CAMs were quantified by counting the blood vessel branch points in each image. (C) The CAM vasculature was treated with PBS (control), NC (RGD+), NC (RGD−), SFB-loaded GCRP-NC, and free SFB on day 14 for 3 days. The total vessel network length (Tot. length), the number of branching points (Nb. branches), the number of nodes (Nb. nodes), and the number of junction points (Nb. junctions). Reproduced from ref. 32 with permission from Elsevier, copyright (2016). (D) The stereomicroscopy images of the CAM vessels. (E) Quantitative analysis using NIH ImageJ with the angiogenesis analyzer plugin. The total vessel network length (Tot. length), the number of branching points (Nb. branches), the number of nodes (Nb. nodes), and the number of junction points (Nb. junctions).130 Reproduced from ref. 130 with permission from Elsevier, copyright (2022). Data are expressed as mean ± SD (n = 6). Notes: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; NS, not significant. | ||
Another study discussed the development of a targeted drug delivery system using graphene quantum dots (GQDs) and poly(D,L-lactide-co-glycolide) (PLGA) nanocomposites to deliver sorafenib (SFB) for angiogenesis inhibition.129 SFB, a multi-tyrosine kinase inhibitor, effectively suppresses tumor progression and angiogenesis but has limitations due to its narrow therapeutic window and adverse side effects. To enhance SFB's delivery and reduce toxicity, researchers have functionalized nanocomposites with an integrin-targeting ligand, the RGD peptide. Integrin receptors, particularly the αvβ3 subtype, are overexpressed in tumor tissues and play a role in angiogenesis and metastasis. The RGD peptide binds to these receptors, facilitating selective drug transfer to cancer cells. The targeted delivery system effectively suppresses angiogenesis, making it a promising drug delivery system for poorly water-soluble therapeutic agents like SFB. The nanocomposites’ unique therapeutic and bioimaging properties make them a valuable tool for improving the curative effect of angiogenesis therapies.
4.2 Immunosuppression modulation for combinative therapy
GO-derived nanomaterials have been witnessed to facilitate tumor immunotherapy by acting as vaccine adjuvants or directly interacting with immune cells, indicating immune modulation potential.131 The large surface area and flexibility of the 2D structure enable GO to interact with cells and biological molecules, influencing the immune response in various ways. We highlight the multifaceted role of GO in modulating the immunosuppressive microenvironment, emphasizing its effects on macrophages, dendritic cells, T lymphocytes, and cytokine production.132 Moreover, GO could exhibit specificity to tumor-associated immune cells based on the biological differences between normal and tumor-associated immune cells. In both functional states and metabolic characteristics, tumor-associated and normal immune cells exhibit significant differences.133 Functionally, tumor-associated immune cells predominantly exhibit an immunosuppressive phenotype (e.g., Tregs, M2 macrophages, and exhausted T cells) and highly express inhibitory receptors such as PD-1 and CTLA-4. In contrast, normal immune cells in activated or resting states retain normal cytotoxic or antigen-presenting functions (e.g., CD8+ T cells and M1 macrophages).134 Metabolically, tumor-associated immune cells rely on glycolysis and exist in high ROS and low-pH stress microenvironments, while normal immune cells predominantly utilize oxidative phosphorylation to maintain metabolic homeostasis and more substantial antioxidant capacity.135 Leveraging these differences, GO could achieve selective effects through multiple mechanisms. On the one hand, the high ROS level in tumor-associated immune cells triggers redox-responsive reactions of GO, enabling targeted suppression of immunosuppressive cell functions. In contrast, normal immune cells efficiently counteract GO-induced oxidative stress by their inherent metabolic stability, robust antioxidant systems, and physiological pH homeostasis. On the other hand, GO's affinity for scavenger receptors enhances its enrichment in tumor-associated cells, whereas normal immune cells exhibit lower internalization efficiency due to distinct receptor distribution patterns. This targeting mechanism underpins GO's potential to disrupt tumor-promoting immunity while preserving regular immune activity selectively.GO nanomaterials have been studied for their potential to induce apoptosis in tumor-associated macrophages (TAMs), which play a critical role in the tumor microenvironment by supporting tumor growth and suppressing immune responses. Research has demonstrated that functionalized graphene and graphene oxide can induce apoptosis in macrophages through various mechanisms. Oxidative stress, disruption of the cellular cytoskeleton, and interference with cellular functions are among the pathways through which graphene-based materials affect macrophages. The interaction between graphene and macrophages can produce reactive oxygen species (ROS), which can overwhelm the cell's antioxidant defenses, leading to oxidative damage and eventual cell death.136 Moreover, GO can induce autophagy in macrophages in a concentration-dependent manner, as evidenced by the appearance of autophagic vacuoles and activation of autophagic marker proteins. This autophagy is regulated by TLR4, TLR9, and downstream adaptor proteins MyD88, TRIF, and TRAF6, highlighting the connection between autophagy and TLR signaling.137
GO nanomaterials could activate macrophages via toll-like receptor (TLR)-mediated and nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB)-related signaling pathways, leading to the secretion of cytokines such as interleukin-1 alpha (IL-1α), interleukin-6 (IL-6), interleukin-10 (IL-10), tumor necrosis factor-alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF), as well as chemokines like monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein-1 alpha (MIP-1α), macrophage inflammatory protein-1 beta (MIP-1β), and regulated upon activation, normal T-cell expressed and secreted (RANTES).138 This activation can contribute to the creation of a pro-inflammatory microenvironment. GO nanomaterials have been shown to modulate the tumor microenvironment by enhancing the infiltration of immune cells and stimulating anti-tumor immunity. GO can activate macrophages to release pro-inflammatory cytokines, which recruit and activate immune cells, contributing to tumor eradication.139 Furthermore, GO nanomaterials have been used to deliver anticancer drugs directly to tumors, improving treatment efficacy while minimizing systemic toxicity. A multifunctional nanoplatform for efficient chemo-gene combination cancer therapy is developed based on platinum-functionalized nano-GO. This system leverages GO's tumor-targeted accumulation and sustained drug release to significantly reduce nonspecific toxicity of cisplatin (CisPt). Furthermore, the platform achieves synergistic chemo-gene therapy via co-delivery of the chemotherapeutic agent CisPt and anti-miR-21. This approach simultaneously addresses drug resistance and systemic toxicity, thereby providing a novel paradigm for precision cancer therapy.140 Moreover, the nanosheets (FU/GO-PEG-GE11) made of GO were functionalized with GE11, an effective ligand for the epidermal growth factor receptor (EGFR), and loaded with the anticancer drug 5-fluorouracil. The targeting nanosystem was substantially more cytotoxic in vitro than non-targeting nanohybrids.141 According to in vivo data, targeted nanocomposites exhibited a potent anticancer effect while having almost no systemic adverse effects in mice with subcutaneous cervical cancer. Notably, the lateral size of GO nanomaterials could significantly impact macrophage activation. A series of GO samples with varying lateral sizes were constructed from the same starting material to focus specifically on the effect of size.132 They discovered that larger GO sheets exhibited stronger adsorption on the plasma membrane but were less prone to phagocytosis. The features led to more robust interactions with toll-like receptors (TLRs) and more potent activation of the NF-κB pathway. More miniature GO sheets, on the other hand, were more readily taken up by cells. Consequently, more significant GO promoted greater M1 polarization, which is associated with increased production of inflammatory cytokines and recruitment of immune cells.
4.3 Oxygen generation for combinative therapy
GO nanosheets played a dual role in the H2O2 catalysis reaction, generating oxygen. To exclusively produce oxygen, GO nanosheets are commonly doped with catalase-active materials such as MnO2 to form hybrid nanomaterials for hypoxia relief.46 This self-oxygenation mechanism has been shown to improve the effectiveness of photodynamic therapy (PDT) by providing the necessary oxygen for ROS generation. MnO2-doped GO nanosheets have enhanced oxygen levels in the tumor microenvironment, resulting in improved PDT efficacy.43To relieve hypoxia in the TME, a novel strategy was designed to enhance chemo-photodynamic therapy's efficacy through a smart nanosystem.46 This system is based on manganese dioxide (MnO2)-doped GO nanosheets, which have been engineered to simultaneously address two significant limitations in cancer therapy: tumor hypoxia and high levels of the GSH antioxidant. In this work, the MnO2 component imparts three fundamental properties. Firstly, it catalyzes the decomposition of H2O2 into oxygen, alleviating the hypoxic conditions characteristic of tumors. Secondly, it depletes the intracellular GSH levels, making tumor cells more susceptible to chemotherapeutic agents. Thirdly, MnO2 generates Mn2+ ions, which can participate in a Fenton-like reaction, further contributing to the anti-tumor activity. This work not only achieves chemo-photodynamic synergistic therapy but also significantly enhances therapeutic efficacy by modulating the TME, thereby providing a novel strategy for developing intelligent nanodrug delivery systems.
Analogously, gold nanoparticles are also hybridized with GO nanosheets for oxygen generation. A polypyrrole (PPy)-based multifunctional nanocomposite, abbreviated as PGPAI, has been developed for dual-modal imaging and enhanced synergistic phototherapy against cancer cells.142 This composite is composed of PPy nanoparticles, graphene oxide (GO) sheets, polyethylene glycol (PEG) chains, gold nanoparticles (Au NPs), and IR820 molecules. Notably, the Au NPs possess catalase-like activity, enabling them to decompose hydrogen peroxide (H2O2), which is upregulated in tumor environments, to produce oxygen. This oxygen production enhances the efficacy of oxygen-dependent PDT.
4.4 Heat shock modulation for enhanced photothermal therapy
In addition to their potential in drug delivery and natural biological activities, graphene-based materials possess a unique advantage due to their excellent NIR absorption capabilities and high photothermal conversion efficiency, rendering them suitable for PTT treatments. GO and its derivatives exhibit an approximate 50% efficiency in photothermal conversion in the near-infrared region, effectively transforming light energy into heat, thereby generating localized hyperthermia in the tumor microenvironment.143 Exogenous stimuli provide robust and efficient activation pathways in response to external physical conditions compared to poorly controlled endogenous responses. External physical stimuli are more reliable in clinical practice. PTT can provide spatial and temporal control of the activation of GO-based intelligent platforms. Therefore, toxic effects can be induced directly at the tumor site, minimizing side effects on healthy tissues and achieving additional therapeutic effects.144In current oncological research, PTT mediated by GO has garnered widespread attention as an innovative treatment strategy. Studies have demonstrated that the application of GO in PTT significantly influences the biological behavior of tumor cells.145 However, the substantial local temperature increase in the tumor microenvironment can induce thermal stress, leading to the heat shock effect. Heat shock is a biological response where cells react to high temperatures or other external stressors, serving as an adaptive mechanism to environmental stress. Closely associated with cellular stress, cells undergo heat shock in response to elevated temperatures, hypoxia, or exposure to toxins as a protective measure.146 A characteristic feature of this response is the increased expression of heat shock proteins (HSPs). HSPs, a group of proteins whose expression is upregulated in response to heat stress, assist in maintaining proper folding and stability of cellular proteins, preventing aggregation of damaged proteins. In PTT, this local thermal stress triggers the production of intracellular HSPs as a natural cellular protective response. During PTT treatment of cancer, the expression of HSPs can rapidly confer protection to cancer cells and reduce the efficacy of PTT, leading to insufficient apoptosis and tumor recurrence.147 In conventional PTT, the surrounding normal tissue of the tumor site may suffer from high-temperature burns. Therefore, conducting mild-temperature photothermal therapy (MTPTT) at lower temperatures is a promising approach for tumor treatment. However, in MTPTT, the overexpression of HSPs increases the thermal tolerance of tumor cells, thereby limiting its therapeutic effect.148
The temperature increase caused by PTT may induce more significant heat shock protein Hsp70 activity. With more intense treatment, the expression of the Hsp70 gene increases to counteract the therapeutic effect. Thus, the expression of the Hsp70 gene may be used as an indicator of treatment efficacy because it is upregulated under extreme thermal stress.149 Based on reduced graphene oxide (rGO), IR780 was covalently conjugated to rGO. The system showed an eightfold increase in HSP70 protein synthesis compared to the control group. This time-dependent upregulation of HSP70 protein expression confirms the mechanism of near-infrared light-induced PTT, making HSP70 a potential target for enhancing the efficacy of PTT.150 HSP90 is significantly believed to interact with AKT (protein kinase B). In many tumor cells, HSP90 is overexpressed and promotes tumor growth and survival by stabilizing essential signaling proteins such as AKT.20 Therefore, inhibiting the activity of HSP90 has been viewed as a potential anti-tumor treatment strategy, which can reduce the stability and activity of AKT, thereby inhibiting the growth and survival of tumor cells.
To maximize PTT efficiency of graphene oxide (GO), a primary strategy involves suppressing heat shock protein (HSP)-mediated thermoresistance. This was achieved by non-covalent loading of the HSP90 inhibitor NVP-AUY922 onto GO nanosheets, constructing a multifunctional GO/NVP-AUY922 nanoplatform. The nanosystem synergistically combines GO's photothermal conversion capability with NVP-AUY922's HSP90 inhibition, effectively disrupting the cytoprotective heat shock response and enhancing tumor ablation under near-infrared (NIR) irradiation. This system was applied for photothermal therapy in HeLa cells, significantly inhibiting the expression of HSP90 in cancer cells, thereby helping to overcome the thermal stress resistance of tumor cells.151 SNX-2112, another Hsp90 inhibitor, primarily functions by binding to the N-terminal ATP binding site of Hsp90. HSP90 inhibitor SNX2112 was loaded onto the surface of GO. By non-covalently binding chitosan (CHI) onto the GO surface to improve biocompatibility and grafting hyaluronic acid (HA) as a targeting ligand for CD44, they achieved specific recognition of tumor cells and enhanced efficiency in anti-tumor drug delivery. Under NIR irradiation, the GO-CHI-HA/SNX-2112 nanosystem released SNX-2112 molecules at the tumor site through a photothermal effect. This photothermal therapy mechanism enhances the drug's efficacy by raising the temperature at the tumor site, simultaneously synergizing to reverse drug resistance, effectively inhibiting and killing A549 lung cancer cells while exhibiting lower toxicity to normal cells.152
Simultaneously targeting HSP as a therapeutic endpoint, combined with PTT and other treatment modalities such as chemotherapy, immunotherapy, and PDT, is anticipated to enhance PTT's efficacy synergistically. Elevated temperatures induced by PTT can augment blood flow, facilitating increased intracellular transport and accumulation of chemotherapeutic agents within tumor cells. Additionally, complementary treatments may render cancer cells more susceptible to PTT, effectively improving therapeutic outcomes and prognosis. A stimulus-responsive scaffold composed of poly(acrylic acid)-grafted polylactic acid (PAA-g-PLLA) modified GO with gambogic acid (GA) and polycaprolactone (PCL) was constructed and further pre-implanted with adipose-derived stem cells (ADSCs). GA, a chemical small molecule inhibitor of heat shock protein synthesis, enhances the therapeutic effect of MTPTT. Under mild-temperature photothermal conditions, tumor cells were directly killed, and ADSCs also survived better and differentiated into adipocytes, thus promoting the formation of new fatty tissue. This method is efficient in treating tumors and provides strong support for postoperative tissue regeneration (Fig. 15).153 Additionally, some methods can circumvent the protective mechanism of HSPs. A mitochondria-targeted low-temperature photothermal therapy (LTPTT) nanocomposite was constructed based on berberine derivatives (BD). This nanocomposite, composed of PEG-modified GO and BD combined with tumor-targeting folic acid (FA), effectively accumulates in the mitochondria of osteosarcoma cells, achieving enhanced mitochondrial-targeted LPTT effects. It directly activates the mitochondria's intrinsic apoptotic pathway, overcoming the tumor thermal resistance caused by HSPs in MTPTT.68
 | ||
| Fig. 15 (A) Schematic illustration of the therapeutic process of an ADSC-loaded GO-GA-polymer scaffold with the pH-triggered dual release of GO and glycyrrhetinic acid (GA). (B) Under NIR irradiation, the tumor site temperature of tumor-bearing mice implanted with the GO-GA-polymer scaffold significantly increased compared to those implanted with PCL and GO-polymer scaffolds. (C) The GO-GA-polymer scaffold significantly promotes the adhesion of ADSCs under NIR irradiation, enhances the expression of adipogenic genes such as PPAR-γ, C/EBP-α, adiponectin, etc., ultimately stimulating the adipogenic differentiation of ADSCs and promoting the formation of adipose tissue in vivo, making it an ideal candidate material for achieving both photothermal tumor therapy and adipose tissue regeneration.153 Reproduced from ref. 153 with permission from Wiley, copyright (2019). | ||
5. Conclusions and perspectives
In conclusion, the intricate nature of the tumor microenvironment (TME) presents significant barriers to effective cancer therapy. However, the advent of graphene oxide (GO)-derived nanomaterials has introduced novel approaches for overcoming these challenges, offering promising avenues for advanced cancer treatment. The unique properties of GO, including its high specific surface area, ultrathin structure, and electronic and photonic characteristics, have been harnessed for multifaceted applications in cancer therapeutics. Beyond its established role in drug delivery, GO has emerged as a powerful tool for TME modulation, enabling catalytic decomposition of reactive oxygen species (ROS), targeting cancer stem cells, anti-angiogenesis, and enhancement of anti-tumor immunity.Recent studies have highlighted the efficacy of GO-based nanomedicines in altering the TME to create a less hospitable environment for tumor growth. GO's ability to scavenge ROS can normalize the redox balance within the TME, mitigating inflammation and disrupting the conditions that support tumor proliferation. Its anti-angiogenic effects contribute to tumor regression by inhibiting the formation of new blood vessels necessary for tumor sustenance. Moreover, GO's interaction with immune cells can convert the immunosuppressive TME into one that promotes immune cell infiltration and activation, thereby enhancing the body's natural defenses against cancer. The utilization of GO in cancer theranostics has expanded to include photothermal therapy. GO acts as a photothermal converter to generate heat upon light exposure, effectively destroying cancer cells while minimizing damage to surrounding healthy tissues. This approach can be augmented with other modalities, such as chemotherapy and immunotherapy, to provide a more comprehensive treatment strategy. Furthermore, GO's capacity for targeted drug delivery, facilitated by its responsive surface modifications, allows for the precise shuttling of drugs directly to the tumor site, improving therapeutic efficacy and reducing side effects.
GO demonstrates innovative potential in oncotherapy due to its two-dimensional structure, high specific surface area, abundant oxygen-containing functional groups, and excellent photothermal conversion efficiency. As a nanocarrier, GO enables the loading and tumor-targeted delivery of various drug molecules including chemotherapeutics, nucleic acids, immunomodulators etc. Meanwhile, the intrinsic PTT and PDT capabilities of GO could induce effective tumor ablation upon near-infrared irradiation. The first strictly controlled human exposure clinical trial indicates that inhaling GO causes no short-term adverse effects on lung or cardiovascular function (NCT03659864). The positive outcomes of the clinical study provide essential evidence for GO biosafety validation. However, future development of GO-based nanomedicines still faces various critical challenges. Long-term biocompatibility and in vivo metabolic pathways of GO remain unclear. To address the challenge, GO biodistribution and metabolic products could be tracked using the isotope labelling method. Chemical structure modification of GO is supposed to be an effective approach for improving biocompatibility. Biodegradable chemical bonds could be introduced into the 2D structure skeleton. Optimization of surface modifications to reduce immunogenicity and toxicity becomes imperative, such as a stable protein corona. On the other hand, large-scale production processes of GO suffer from issues of batch-to-batch variability and residual impurities. These variations critically impair GO's performance in biomedical applications, especially its biosafety. Future investigations should prioritize the development of green synthesis techniques to address process-induced variations and ensure production consistency. Additionally, clinical translation of GO nanomaterials necessitates GLP/GMP-compliant validation and comprehensive pharmacodynamic, pharmacokinetic, and toxicological profiling.
Author contributions
Y. D., H. C., J. Z., C. C., Q. Z., J. Y., and S. Y. wrote the manuscript; Q. L., Z. F., and S. G. curated the data; H. C., Y. D., and Y. W. summarised the literature studies; Y. D., H. C., J. Z., and D. X. revised the manuscript. All authors have read and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the National Natural Science Foundation of China (82372113, 82404565 and 82373815), the National Ten Thousand Talents Program for Young Top-notch Talents, the Excellent Youth Foundation of the Jiangsu Scientific Committee (BK20240096), the Jiangsu 333 High-level Talent Training Project ([2022]3-16-190), the Double First-Rate Construction Plan of China Pharmaceutical University (CPU2022QZ18), the Natural Science Foundation of Jiangsu Province (BK20231018), the China Postdoctoral Science Foundation (2023M733892), the Jiangsu Province Outstanding Postdoctoral Program (2023ZB784), and the Fundamental Research Funds for the Central Universities (2632023GR18). This study was supported by the Guizhou Provincial Scientific and Technological Innovation Base (No. [2023]003).References
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201–204 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- L. Bejarano, M. J. C. Jordāo and J. A. Joyce, Cancer Discovery, 2021, 11, 933–959 CrossRef CAS PubMed.
- A. Glaviano, H. S. Lau, L. M. Carter, E. H. C. Lee, H. Y. Lam, E. Okina, D. J. J. Tan, W. Tan, H. L. Ang, D. Carbone, M. Y. Yee, M. K. Shanmugam, X. Z. Huang, G. Sethi, T. Z. Tan, L. H. K. Lim, R. Y. Huang, H. Ungefroren, E. Giovannetti, D. G. Tang, T. C. Bruno, P. Luo, M. H. Andersen, B. Z. Qian, J. Ishihara, D. C. Radisky, S. Elias, S. Yadav, M. Kim, C. Robert, P. Diana, K. A. Schalper, T. Shi, T. Merghoub, S. Krebs, A. P. Kusumbe, M. S. Davids, J. R. Brown and A. P. Kumar, J. Hematol. Oncol., 2025, 18, 6 CrossRef PubMed.
- J. Wu, H. Lin, D. J. Moss, K. P. Loh and B. Jia, Nat. Rev. Chem., 2023, 7, 162–183 CrossRef PubMed.
- J. Hu, L. Zhang, H. Xia, Y. Yan, X. Zhu, F. Sun, L. Sun, S. Li, D. Li, J. Wang, Y. Han, J. Zhang, D. Bian, H. Yu, Y. Chen, P. Fan, Q. Ma, G. Jiang, C. Wang and P. Zhang, Genome Med., 2023, 15, 14 CrossRef CAS PubMed.
- L. Huang, J. Feng, J. Zhu, J. Yang, W. Xiong, X. Lu, S. Chen, S. Yang, Y. Li, Y. Xu and Z. Shen, Adv. Healthc. Mater., 2023, 12, e2203362 CrossRef PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- Y. Xu, J. Xiong, X. Sun and H. Gao, Acta Pharm. Sin. B, 2022, 12, 4327–4347 CrossRef CAS PubMed.
- T. Bohn, S. Rapp, N. Luther, M. Klein, T. J. Bruehl, N. Kojima, P. Aranda Lopez, J. Hahlbrock, S. Muth, S. Endo, S. Pektor, A. Brand, K. Renner, V. Popp, K. Gerlach, D. Vogel, C. Lueckel, D. Arnold-Schild, J. Pouyssegur, M. Kreutz, M. Huber, J. Koenig, B. Weigmann, H. C. Probst, E. von Stebut, C. Becker, H. Schild, E. Schmitt and T. Bopp, Nat. Immunol., 2018, 19, 1319–1329 CrossRef CAS PubMed.
- J. M. Chan, L. Zhang, R. Tong, D. Ghosh, W. Gao, G. Liao, K. P. Yuet, D. Gray, J. W. Rhee, J. Cheng, G. Golomb, P. Libby, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 CrossRef CAS PubMed.
- Z. Zhao, L. Xue, L. Zheng, L. Ma, Z. Li, N. Lu, Q. Zhan and Y. Song, Signal Transduction Targeted Ther., 2023, 8, 29 CrossRef CAS PubMed.
- X. Huang and W. Zhang, Small Methods, 2023, e2301326, DOI:10.1002/smtd.202301326.
- Y. Xi, Y. Shen, L. Chen, L. Tan, W. Shen and X. Niu, Cytokine Growth Factor Rev., 2023, 73, 78–92 CrossRef CAS PubMed.
- S. S. Narlawar and S. Gandhi, Mater. Today Adv., 2021, 12, 100174 CrossRef CAS.
- M. Certo, C. H. Tsai, V. Pucino, P. C. Ho and C. Mauro, Nat. Rev. Immunol., 2021, 21, 151–161 CrossRef CAS PubMed.
- A. Casazza, G. Di Conza, M. Wenes, V. Finisguerra, S. Deschoemaeker and M. Mazzone, Oncogene, 2014, 33, 1743–1754 CrossRef CAS PubMed.
- K. E. de Visser and J. A. Joyce, Cancer Cell, 2023, 41, 374–403 CrossRef CAS PubMed.
- M. Kochetkova and M. S. Samuel, Trends Cell Biol., 2022, 32, 285–294 CrossRef CAS PubMed.
- Z. Liu, D. Wang, J. Zhang, P. Xiang, Z. Zeng, W. Xiong and L. Shi, Cancer Lett., 2023, 577, 216409 CrossRef CAS PubMed.
- T. L. Whiteside, J. Clin. Invest., 2020, 130, 5115–5117 CAS.
- J. Ma, L. Huang, D. Hu, S. Zeng, Y. Han and H. Shen, J. Exp. Clin. Cancer Res., 2021, 40, 327 CrossRef CAS PubMed.
- S. Ugel, S. Cane, F. De Sanctis and V. Bronte, Annu. Rev. Pytopathol., 2021, 16, 93–122 CrossRef CAS PubMed.
- I. Li and B. Y. Nabet, Mol. Cancer, 2019, 18, 32 CrossRef PubMed.
- X. Wang, J. Mao, T. Zhou, X. Chen, H. Tu, J. Ma, Y. Li, Y. Ding, Y. Yang, H. Wu and X. Tang, Theranostics, 2021, 11, 209–221 CrossRef CAS PubMed.
- Q. Wu, L. You, E. Nepovimova, Z. Heger, W. Wu, K. Kuca and V. Adam, J. Hematol. Oncol., 2022, 15, 77 CrossRef CAS PubMed.
- H. R. Wang, X. Han, Z. L. Dong, J. Xu, J. Wang and Z. Liu, Adv. Funct. Mater., 2019, 29, 1902440 CrossRef.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- H. Wahyudi, A. A. Reynolds, Y. Li, S. C. Owen and S. M. Yu, J. Controlled Release, 2016, 240, 323–331 CrossRef CAS PubMed.
- M. Yamakuchi, M. Okawa, K. Takenouchi, A. Bibek, S. Yamada, K. Inoue, K. Higurashi, A. Tabaru, K. Tanoue, Y. Oyama, S. Higashi, C. Fujisaki, H. Kanda, H. Terasaki, T. Sakamoto, Y. Soga and T. Hashiguchi, PLoS One, 2023, 18, e0284131 CrossRef CAS PubMed.
- S. S. Acharya and C. N. Kundu, Cancer Treat. Rev., 2024, 127, 102749 CrossRef CAS PubMed.
- P. X. Lai, C. W. Chen, S. C. Wei, T. Y. Lin, H. J. Jian, I. P. Lai, J. Y. Mao, P. H. Hsu, H. J. Lin, W. S. Tzou, S. Y. Chen, S. G. Harroun, J. Y. Lai and C. C. Huang, Biomaterials, 2016, 109, 12–22 CrossRef CAS PubMed.
- W. Cao, L. He, W. Cao, X. Huang, K. Jia and J. Dai, Acta Biomater., 2020, 112, 14–28 CrossRef CAS PubMed.
- S. Huang, Y. Li, S. Zhang, Y. Chen, W. Su, D. J. Sanchez, J. D. H. Mai, X. Zhi, H. Chen and X. Ding, J. Controlled Release, 2024, 365, 716–728 CrossRef CAS PubMed.
- S. Zhu, C. Yu, N. Liu, M. Zhao, Z. Chen, J. Liu, G. Li, H. Huang, H. Guo, T. Sun, J. Chen, J. Zhuang and P. Zhu, Bioact. Mater., 2022, 13, 119–134 CAS.
- A. Sinha, B. G. Cha, Y. J. Choi, T. L. Nguyen, P. J. Yoo, J. H. Jeong and J. Kim, Chem. Mater., 2017, 29, 6883–6892 CrossRef CAS.
- M. J. Feito, M. Cicuéndez, L. Casarrubios, R. Diez-Orejas, S. Fateixa, D. Silva, N. Barroca, P. A. A. P. Marques and M. T. Portolés, Int. J. Mol. Sci., 2022, 23, 10625 CrossRef CAS PubMed.
- G.-Y. Chen, H.-J. Yang, C.-H. Lu, Y.-C. Chao, S.-M. Hwang, C.-L. Chen, K.-W. Lo, L.-Y. Sung, W.-Y. Luo, H.-Y. Tuan and Y.-C. Hu, Biomaterials, 2012, 33, 6559–6569 CrossRef CAS PubMed.
- E. Zhu, J. Yu, Y. R. Li, F. Ma, Y. C. Wang, Y. Liu, M. Li, Y. J. Kim, Y. Zhu, Z. Hahn, Y. Zhou, J. Brown, Y. Zhang, M. Pelegrini, T. Hsiai, L. Yang and Y. Huang, Nat. Nanotechnol., 2024, 19, 1914–1922 CrossRef CAS PubMed.
- H. Zhou, K. Zhao, W. Li, N. Yang, Y. Liu, C. Chen and T. Wei, Biomaterials, 2012, 33, 6933–6942 CrossRef CAS PubMed.
- M. Fan, Y. Han, S. Gao, H. Yan, L. Cao, Z. Li, X. J. Liang and J. Zhang, Theranostics, 2020, 10, 4944–4957 CrossRef CAS PubMed.
- L. Xu, J. Wang, S.-Y. Lu, X. Wang, Y. Cao, M. Wang, F. Liu, Y. Kang and H. Liu, Langmuir, 2019, 35, 9246–9254 CrossRef CAS PubMed.
- H. Lu, W. Li, P. Qiu, X. Zhang, J. Qin, Y. Cai and X. Lu, Nanoscale Adv., 2022, 4, 4304–4313 RSC.
- B. Lin, H. Chen, D. Liang, W. Lin, X. Qi, H. Liu and X. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 11157–11166 CrossRef CAS PubMed.
- Y. Tao, L. Zhu, Y. Zhao, X. Yi, L. Zhu, F. Ge, X. Mou, L. Chen, L. Sun and K. Yang, Nanoscale, 2018, 10, 5114–5123 RSC.
- P. Liu, X. Xie, M. Liu, S. Hu, J. Ding and W. Zhou, Acta Pharm. Sin. B, 2021, 11, 823–834 CrossRef CAS PubMed.
- X. Wang, C. Hu, Z. Gu and L. Dai, J. Nanobiotechnol., 2021, 19, 340 CrossRef CAS PubMed.
- S. Gurunathan, M. H. Kang, M. Jeyaraj and J. H. Kim, Int. J. Mol. Sci., 2019, 20, 247 CrossRef PubMed.
- X. Wang, C. Hu, Z. Gu and L. Dai, J. Nanobiotechnol., 2021, 19, 340 CrossRef CAS PubMed.
- M. Majood, P. Garg, R. Chaurasia, A. Agarwal, S. Mohanty and M. Mukherjee, ACS Omega, 2022, 7, 28685–28693 CrossRef CAS PubMed.
- Y. Su, K. Ye, J. Y. Hu, Z. L. Zhang, Y. Wang, B. J. Geng, D. Y. Pan and L. X. Shen, Adv. Healthcare Mater., 2024, 13, e2304648 CrossRef PubMed.
- X. Chen, Y. Fan, J. Sun, Z. Zhang, Y. Xin, K. Li, K. Tang, P. Du, Y. Liu, G. Wang, M. Yang and Y. Tan, Acta Biomater., 2021, 135, 493–505 CrossRef CAS PubMed.
- J. X. Li, Q. W. Wang, H. Lu, Y. L. Han, L. L. Jiang, W. C. Qian, M. Zhu, B. N. Wang, J. S. Min, Y. Hou, S. N. Xu, Z. C. Xiong, H. B. Liu, Y. L. Li, C. Y. Chen, Y. Liu and P. X. Qian, Nano Today, 2022, 46, 101622 CrossRef CAS.
- X. Chen, Y. Fan, J. Sun, Z. Zhang, Y. Xin, K. Li, K. Tang, P. Du, Y. Liu, G. Wang, M. Yang and Y. Tan, Acta Biomater., 2021, 135, 493–505 CrossRef CAS PubMed.
- J. Li, Q. Wang, H. Lu, Y. Han, L. Jiang, W. Qian, M. Zhu, B. Wang, J. Min, Y. Hou, S. Xu, Z. Xiong, H. Liu, Y. Li, C. Chen, Y. Liu and P. Qian, Nano Today, 2022, 46, 101622 CrossRef CAS.
- D. Toomeh, S. M. Gadoue, S. Yasmin-Karim, M. Singh, R. Shanker, S. P. Singh, R. Kumar, E. Sajo and W. Ngwa, Phys. Med., 2018, 55, 8–14 CrossRef PubMed.
- Y. Su, K. Ye, J. Hu, Z. Zhang, Y. Wang, B. Geng, D. Pan and L. Shen, Adv. Healthc. Mater., 2024, 13, e2304648 CrossRef PubMed.
- N. An, X. Yan, Q. Qiu, Z. Zhang, X. Zhang, B. Zheng, Z. Zhao, J. Guo and Y. Liu, J. Nanobiotechnol., 2024, 22, 133 CrossRef CAS PubMed.
- E. Bressan, L. Ferroni, C. Gardin, L. Sbricoli, L. Gobbato, F. S. Ludovichetti, I. Tocco, A. Carraro, A. Piattelli and B. Zavan, J. Transl. Med., 2014, 12, 296 CrossRef PubMed.
- W. Guo, Z. Chen, X. Feng, G. Shen, H. Huang, Y. Liang, B. Zhao, G. Li and Y. Hu, J. Nanobiotechnol., 2021, 19, 146 CrossRef CAS PubMed.
- S. Chen, S. Zhang, Y. Wang, X. Yang, H. Yang and C. Cui, Asian J. Pharm. Sci., 2021, 16, 598–611 Search PubMed.
- Y. Luo, B. Li, X. Liu, Y. Zheng, E. Wang, Z. Li, Z. Cui, Y. Liang, S. Zhu and S. Wu, Bioact. Mater., 2022, 18, 421–432 CAS.
- G. Covarrubias, M. E. Lorkowski, H. M. Sims, G. Loutrianakis, A. Rahmy, A. Cha, E. Abenojar, S. Wickramasinghe, T. J. Moon, A. C. S. Samia and E. Karathanasis, Nanoscale Adv., 2021, 3, 5890–5899 RSC.
- H. L. Lei, Q. G. Li, Z. F. Pei, L. Liu, N. L. Yang and L. Cheng, Small, 2023, 19, e2303438 CrossRef PubMed.
- B. H. Kang and H. K. Lee, Int. J. Mol. Sci., 2022, 23, 7325 CrossRef CAS PubMed.
- M. J. Bou-Dargham, L. L. Sha, D. B. Sarker, M. Z. Krakora-Compagno, Z. Chen, J. F. Zhang and Q. X. A. Sang, Int. J. Mol. Sci., 2023, 24, 9355 CrossRef CAS PubMed.
- P. Bonaventura, T. Shekarian, V. Alcazer, J. Valladeau-Guilemond, S. Valsesia-Wittmann, S. Amigorena, C. Caux and S. Depil, Front. Immunol., 2019, 10, 168 CrossRef CAS PubMed.
- H. Z. Hu, Q. C. Song, W. B. Yang, Q. W. Zeng, Z. H. Liang, W. J. Liu, Z. W. Shao, Y. R. Zhang, C. Chen and B. C. Wang, Front. Chem., 2023, 11, 1114434 CrossRef CAS PubMed.
- L. Wang, M. Wang, B. Q. Zhou, F. F. Zhou, C. Murray, R. A. Towner, N. Smith, D. Saunders, G. Xie and W. R. Chen, J. Mater. Chem. B, 2019, 7, 7406–7414 RSC.
- H. A. Tian, J. Y. Lin, F. K. Zhu, J. Q. Li, S. H. Jiang, L. Y. Xie, Y. Li, P. Y. Wang, Z. Q. Hou and J. X. Mi, Chin. Chem. Lett., 2023, 34, 107577 CrossRef CAS.
- W. F. Ren, X. M. Cai, J. Chen, L. F. Ruan, H. R. Lu, J. Y. Zhang, Y. Hu and J. M. Gao, Oncologie, 2021, 23, 359–371 Search PubMed.
- X. Y. Du, X. Y. Yang, Y. Zhang, S. Gao, S. G. Liu, J. B. Ji and G. X. Zhai, Nano Today, 2022, 46, 101565 CrossRef CAS.
- Y. Tao, E. Ju, J. Ren and X. Qu, Biomaterials, 2014, 35, 9963–9971 CrossRef CAS PubMed.
- L. Zhou, B. Feng, H. Wang, D. G. Wang and Y. P. Li, Nano Today, 2022, 44, 101466 CrossRef CAS.
- L. P. Huang, Y. N. Li, Y. N. Du, Y. Y. Zhang, X. X. Wang, Y. Ding, X. L. Yang, F. L. Meng, J. S. Tu, L. Luo and C. M. Sun, Nat. Commun., 2019, 10, 4871 CrossRef CAS PubMed.
- M. M. Yan, Y. J. Liu, X. H. Zhu, X. L. Wang, L. X. Liu, H. F. Sun, C. Wang, D. L. Kong and G. L. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 1876–1885 CrossRef CAS PubMed.
- F. F. Zhou, M. Wang, T. Luo, J. L. Qu and W. R. Chen, Biomaterials, 2021, 265, 120421 CrossRef CAS PubMed.
- X. Y. Deng, W. Guan, X. C. Qing, W. B. Yang, Y. M. Que, L. Tan, H. Liang, Z. C. Zhang, B. C. Wang, X. M. Liu, Y. L. Zhao and Z. W. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 4265–4275 CrossRef CAS PubMed.
- Y. Tao, E. G. Ju, J. S. Ren and X. G. Qu, Biomaterials, 2014, 35, 9963–9971 CrossRef CAS PubMed.
- Y. Y. Pan, Y. D. Yu, X. J. Wang and T. Zhang, Front. Immunol., 2020, 11, 583084 CrossRef CAS PubMed.
- L. He, T. Nie, X. Xia, T. Liu, Y. Huang, X. Wang and T. Chen, Adv. Funct. Mater., 2019, 29, 1901240 CrossRef.
- A. J. Boutilier and S. F. Elsawa, Int. J. Mol. Sci., 2021, 22, 6995 CrossRef CAS PubMed.
- N. Kumari and S. H. Choi, J. Exp. Clin. Cancer Res., 2022, 41, 68 CrossRef CAS PubMed.
- J. Gao, Y. Z. Liang and L. Wang, Front. Immunol., 2022, 13, 888713 CrossRef CAS PubMed.
- Q. Li, Z. Huang, Q. Wang, J. Gao, J. Chen, H. Tan, S. Li, Z. Wang, X. Weng, H. Yang, Z. Pang, Y. Song, J. Qian and J. Ge, Biomaterials, 2022, 284, 121529 CrossRef CAS PubMed.
- M. J. Feito, R. Diez-Orejas, M. Cicuéndez, L. Casarrubios, J. M. Rojo and M. T. Portolés, Colloids Surf., B, 2019, 176, 96–105 CrossRef CAS PubMed.
- X. Y. Deng, H. Liang, W. B. Yang and Z. W. Shao, J. Photochem. Photobiol., B, 2020, 208, 111913 CrossRef CAS PubMed.
- Y. K. Wu, F. F. Wang, S. H. Wang, J. Ma, M. Xu, M. Gao, R. Liu, W. Chen and S. J. Liu, Nanoscale, 2018, 10, 14637–14650 RSC.
- Y. W. Ge, X. L. Liu, D. G. Yu, Z. A. Zhu, Q. F. Ke, Y. Q. Mao, Y. P. Guo and J. W. Zhang, J. Nanobiotechnol., 2021, 19, 91 CrossRef PubMed.
- H. Sharma and S. Mondal, Int. J. Mol. Sci., 2020, 21, 6280 CrossRef CAS PubMed.
- H. S. Liu, X. Y. Liu, F. B. Zhao, Y. Liu, L. J. Liu, L. H. Wang, C. B. Geng and P. Huang, J. Colloid Interface Sci., 2020, 562, 182–192 CrossRef CAS PubMed.
- A. M. Itoo, S. L. Vemula, M. T. Gupta, M. V. Giram, S. A. Kumar, B. Ghosh and S. Biswas, J. Controlled Release, 2022, 350, 26–59 CrossRef CAS PubMed.
- S. Gurunathan and J. H. Kim, Int. J. Nanomed., 2022, 17, 5697–5731 CrossRef PubMed.
- X. H. Wang, H. Zhang, X. H. Chen, C. R. Wu, K. Ding, G. Y. Sun, Y. Luo and D. B. Xiang, Acta Biomater., 2023, 166, 42–68 CrossRef CAS PubMed.
- M. M. Chen, D. P. Liu, F. S. Liu, Y. N. Wu, X. J. Peng and F. L. Song, J. Controlled Release, 2021, 332, 269–284 CrossRef CAS PubMed.
- Y. C. Yang, Y. Zhu, S. J. Sun, C. J. Zhao, Y. Bai, J. Wang and L. T. Ma, Front. Immunol., 2023, 14, 1259797 CrossRef CAS PubMed.
- S. H. Jia, S. K. Ke, L. Tu, S. Q. Chen, B. K. Luo, Y. Q. Xiong, Y. Li, P. Y. Wang and S. F. Ye, J. Colloid Interface Sci., 2023, 652, 329–340 CrossRef CAS PubMed.
- S. P. Feng, J. H. Wang, X. Y. Mu, G. L. Gu, Y. F. Wang, J. Y. Lu, S. L. Wang and Q. F. Zhao, Colloids Surf., B, 2023, 222, 113095 CrossRef CAS PubMed.
- B. Y. Niu, K. X. Liao, Y. X. Zhou, T. Wen, G. L. Quan, X. Pan and C. B. Wu, Biomaterials, 2021, 277, 121110 CrossRef CAS PubMed.
- B. J. Sun, C. Luo, H. Yu, X. B. Zhang, Q. Chen, W. Q. Yang, M. L. Wang, Q. M. Kan, H. T. Zhang, Y. J. Wang, Z. G. He and J. Sun, Nano Lett., 2018, 18, 3643–3650 CrossRef CAS PubMed.
- T. Yin, J. Liu, Z. Zhao, Y. Zhao, L. Dong, M. Yang, J. Zhou and M. Huo, Adv. Funct. Mater., 2017, 27, 1604620 CrossRef.
- M. Wei, T. C. Lu, Z. Z. Nong, G. Li, X. Pan, Y. M. Wei, Y. F. Yang, N. N. Wu, J. Huang, M. S. Pan, X. H. Li and F. Y. Meng, J. Drug Delivery Sci. Technol., 2019, 53, 101202 CrossRef CAS.
- A. R. Sarikhani, M. Abedi, S. S. Abolmaali, S. Borandeh and A. M. Tamaddon, BMC Chem., 2024, 18, 189 CrossRef CAS PubMed.
- D. Kalyane, D. Choudhary, S. Polaka, H. Goykar, T. Karanwad, K. Rajpoot and R. Kumar Tekade, Prog. Mater. Sci., 2022, 130, 100974 CrossRef CAS.
- B. P. Ling, H. T. Chen, D. Y. Liang, W. Lin, X. Y. Qi, H. P. Liu and X. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 11157–11166 CrossRef PubMed.
- H. Ding, Y. J. Cai, L. Z. Gao, M. M. Liang, B. P. Miao, H. W. Wu, Y. Liu, N. Xie, A. F. Tang, K. L. Fan, X. Y. Yan and G. H. Nie, Nano Lett., 2019, 19, 203–209 CrossRef CAS PubMed.
- R. Zhang, C. Wang, X. H. Zheng, S. L. Li, W. C. B. Zhang, Z. Kang, S. Yin, J. Y. Chen, F. Chen and W. B. Li, Cancer Med., 2023, 12, 20639–20654 CrossRef CAS PubMed.
- J. L. Bi, F. F. Bi, X. Pan and Q. Yang, J. Transl. Med., 2021, 19, 382 CrossRef CAS PubMed.
- S. J. Lee, N. J. Toft, T. V. Axelsen, M. S. Espejo, T. M. Pedersen, M. Mele, H. L. Pedersen, E. Balling, T. Johansen, M. Burton, M. Thomassen, P. Vahl, P. Christiansen and E. Boedtkjer, Breast Cancer Res., 2023, 25, 46 CrossRef CAS PubMed.
- E. Boedtkjer and S. F. Pedersen, in Annual Review Of Physiology, ed. M. T. Nelson and K. Walsh, 2020, vol. 82, pp. 103–126 Search PubMed.
- N. Bellier, P. Baipaywad, N. Ryu, J. Y. Lee and H. Park, Biomater. Res., 2022, 26, 65 CrossRef CAS PubMed.
- H. R. Zhu, L. Gao, X. L. Jiang, R. Liu, Y. T. Wei, Y. L. Wang, Y. L. Zhao, Z. F. Chai and X. Y. Gao, Chem. Commun., 2014, 50, 3695–3698 RSC.
- A. Mihanfar, N. Targhazeh, S. Sadighparvar, S. G. Darband, M. Majidinia and B. Yousefi, Biomol. Concepts, 2021, 12, 8–15 CrossRef CAS PubMed.
- T. Zhou, X. Zhou and D. Xing, Biomaterials, 2014, 35, 4185–4194 CrossRef CAS PubMed.
- G. Gonzalez-Avila, B. Sommer, E. Flores-Soto and A. Aquino-Galvez, Int. J. Mol. Sci., 2023, 24, 16887 CrossRef CAS PubMed.
- A. Mustafa, F. Elkhamisy, N. Arghiani and M. Z. I. Pranjol, Biomed. Pharmacother., 2023, 164, 114932 CrossRef CAS PubMed.
- A. M. Itoo, S. L. Vemula, M. T. Gupta, M. V. Giram, S. A. Kumar, B. Ghosh and S. Biswas, J. Controlled Release, 2022, 350, 26–59 CrossRef CAS PubMed.
- Q. Yao, L. F. Kou, Y. Tu and L. Zhu, Trends Pharmacol. Sci., 2018, 39, 766–781 CrossRef CAS PubMed.
- B. Z. Wu, M. P. Li, L. D. Wang, Z. Iqbal, K. Q. Zhu, Y. H. Yang and Y. L. Li, J. Mater. Chem. B, 2021, 9, 4319–4328 RSC.
- G. Shim, Q. V. Le, J. Suh, S. Choi, G. Kim, H. G. Choi, Y. B. Kim, R. B. Macgregor and Y. K. Oh, J. Controlled Release, 2019, 298, 110–119 CrossRef CAS PubMed.
- F. Li, S. J. Park, D. Ling, W. Park, J. Y. Han, K. Na and K. Char, J. Mater. Chem. B, 2013, 1, 1678–1686 RSC.
- Y. Chen, Y. Yang, Y. Xian, P. Singh, J. Feng, S. Cui, A. Carrier, K. Oakes, T. Luan and X. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 352–360 CrossRef CAS PubMed.
- J. Pi, D. Chen, J. Wang, E. Yang, J. Yang, Y. Liu, J. Yu, J. Xia, X. Huang, L. Chen, Y. Ruan, J. F. Xu, F. Yang and L. Shen, Pharmacol. Res., 2024, 208, 107379 CrossRef CAS PubMed.
- S. Shi, K. Yang, H. Hong, H. F. Valdovinos, T. R. Nayak, Y. Zhang, C. P. Theuer, T. E. Barnhart, Z. Liu and W. Cai, Biomaterials, 2013, 34, 3002–3009 CrossRef CAS PubMed.
- K. Yang, J. Wan, S. Zhang, B. Tian, Y. Zhang and Z. Liu, Biomaterials, 2012, 33, 2206–2214 CrossRef CAS PubMed.
- L. Lu, H. Qi, J. Zhu, W. X. Sun, B. Zhang, C. Y. Tang and Q. Cheng, Biomed. Pharmacother., 2017, 92, 187–195 CrossRef CAS PubMed.
- D. Yang, L. Feng, C. A. Dougherty, K. E. Luker, D. Chen, M. A. Cauble, M. M. Banaszak Holl, G. D. Luker, B. D. Ross, Z. Liu and H. Hong, Biomaterials, 2016, 104, 361–371 CrossRef CAS PubMed.
- Z. Rahiminezhad, A. Tamaddon, A. Dehshahri, S. Borandeh, S. S. Abolmaali, H. Najafi and N. Azarpira, Biomater. Adv., 2022, 137, 212851 CrossRef CAS PubMed.
- P.-X. Lai, C.-W. Chen, S.-C. Wei, T.-Y. Lin, H.-J. Jian, I. P.-J. Lai, J.-Y. Mao, P.-H. Hsu, H.-J. Lin, W.-S. Tzou, S.-Y. Chen, S. G. Harroun, J.-Y. Lai and C.-C. Huang, Biomaterials, 2016, 109, 12–22 CrossRef CAS PubMed.
- Z. Rahiminezhad, A. Tamaddon, A. Dehshahri, S. Borandeh, S. S. Abolmaali, H. Najafi and N. Azarpira, Biomater. Adv., 2022, 137, 212851 CrossRef CAS PubMed.
- B. Vakili, M. Karami-Darehnaranji, E. Mirzaei, F. Hosseini and N. Nezafat, Int. Immunopharmacol., 2023, 125, 111062 CrossRef CAS PubMed.
- J. Ma, R. Liu, X. Wang, Q. Liu, Y. Chen, R. P. Valle, Y. Y. Zuo, T. Xia and S. Liu, ACS Nano, 2015, 9, 10498–10515 CrossRef CAS PubMed.
- K. J. Hiam-Galvez, B. M. Allen and M. H. Spitzer, Nat. Rev. Cancer, 2021, 21, 345–359 CrossRef CAS PubMed.
- R. Saleh and E. Elkord, Semin. Cancer Biol., 2020, 65, 13–27 CrossRef CAS PubMed.
- E. N. Arner and J. C. Rathmell, Cancer Cell, 2023, 41, 421–433 CrossRef CAS PubMed.
- H. Zhou, K. Zhao, W. Li, N. Yang, Y. Liu, C. Chen and T. Wei, Biomaterials, 2012, 33, 6933–6942 CrossRef CAS PubMed.
- X. Zhi, H. Fang, C. Bao, G. Shen, J. Zhang, K. Wang, S. Guo, T. Wan and D. Cui, Biomaterials, 2013, 34, 5254–5261 CrossRef CAS PubMed.
- I. M. Martinez Paino, F. Santos and V. Zucolotto, J. Biomed. Mater. Res., Part A, 2016, 105, 728–736 CrossRef PubMed.
- W. Cao, L. He, W. Cao, X. Huang, K. Jia and J. Dai, Acta Biomater., 2020, 112, 14–28 CrossRef CAS PubMed.
- P. Liu, S. Wang, X. Liu, J. Ding and W. Zhou, Eur. J. Pharm. Sci., 2018, 121, 319–329 CrossRef CAS PubMed.
- Z. Qiu, J. Hu, Z. Li, X. Yang, J. Hu, Q. You, S. Bai, Y. Mao, D. Hua and J. Yin, Colloids Surf., A, 2020, 593, 124585 CrossRef CAS.
- S. Gurunathan, M.-H. Kang, M. Jeyaraj and J.-H. Kim, Int. J. Mol. Sci., 2019, 20, 247 CrossRef PubMed.
- B. Han, Y. L. Zhang, Q. D. Chen and H. B. Sun, Adv. Funct. Mater., 2018, 28, 1802235 CrossRef.
- Y. W. Chen, Y. L. Su, S. H. Hu and S. Y. Chen, Adv. Drug Delivery Rev., 2016, 105, 190–204 CrossRef CAS PubMed.
- L. González-Rodríguez, S. Pérez-Davila, M. López-Alvarez, S. Chiussi, J. Serra and P. González, J. Nanobiotechnol., 2023, 21, 196 CrossRef PubMed.
- X. L. Ding, M. D. Liu, Q. Cheng, W. H. Guo, M. T. Niu, Q. X. Huang, X. Zeng and X. Z. Zhang, Biomaterials, 2022, 281, 121369 CrossRef CAS PubMed.
- Z. F. Wen, F. Y. Liu, G. X. Liu, Q. Y. Sun, Y. H. Zhang, M. Muhammad, Y. Q. Xu, H. J. Li and S. G. Sun, J. Colloid Interface Sci., 2021, 590, 290–300 CrossRef CAS PubMed.
- P. Wang, B. Q. Chen, Y. Y. Zhan, L. G. Wang, J. Luo, J. Xu, L. L. Zhan, Z. H. Li, Y. G. Liu and J. C. Wei, Pharmaceutics, 2022, 14, 2279 CrossRef CAS PubMed.
- C. C. Barrera, H. Groot, W. L. Vargas and D. M. Narváez, Int. J. Nanomed., 2020, 15, 6421–6432 CrossRef PubMed.
- B. S. Dash, Y. J. Lu, P. Pejrprim, Y. H. Lan and J. P. Chen, Biomater. Adv., 2022, 136, 212764 CrossRef CAS PubMed.
- X. Chang, M. Q. Zhang, C. Wang, J. P. Zhang, H. X. Wu and S. P. Yang, Carbon, 2020, 158, 372–385 CrossRef CAS.
- X. Liu, X. Z. Cheng, F. Z. Wang, L. B. Feng, Y. Wang, Y. F. Zheng and R. Guo, Carbohydr. Polym., 2018, 185, 85–95 CrossRef CAS PubMed.
- G. Bai, P. Yuan, B. Cai, X. Qiu, R. Jin, S. Liu, Y. Li and X. Chen, Adv. Funct. Mater., 2019, 29, 1904401 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |