
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a nano-vaccine for high-grade serous ovarian cancer†
Chayanika
Saha
a,
Ahmed
Elkashif
a,
Elaine J.
Gilmore
a,
Binyumeng
Jiang
a,
Ying
Sun
a,
Raj Kumar
Duary
 b,
Niamh
Buckley
a,
Nicholas J.
Dunne
acdefghij and
Helen O.
McCarthy
*a
b,
Niamh
Buckley
a,
Nicholas J.
Dunne
acdefghij and
Helen O.
McCarthy
*a
aSchool of Pharmacy, Queen's University of Belfast, Belfast, UK. E-mail: h.mccarthy@qub.ac.uk
bDepartment of Food Engineering and Technology, Tezpur University, Tezpur, India
cSchool of Mechanical and Manufacturing Engineering, Dublin City University, Dublin, Ireland
dCentre for Medical Engineering Research, School of Mechanical and Manufacturing Engineering, Dublin City University, Dublin, Ireland
eBiodesign Europe, Dublin City University, Dublin, Ireland
fDepartment of Mechanical and Manufacturing Engineering, School of Engineering, Trinity College Dublin, Dublin, Ireland
gAdvanced Manufacturing Research Centre (I-Form), Dublin City University, Dublin, Ireland
hAdvanced Materials and Bioengineering Research Centre (AMBER), Dublin City University, Dublin, Ireland
iAdvanced Processing Technology Research Centre, Dublin City University, Dublin, Ireland
jTrinity Centre for Biomedical Engineering, Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin, Ireland
First published on 8th May 2025
Abstract
High-Grade Serous Carcinoma (HGSC) is characterised by aggressive malignant tumours and poor prognosis accounting for 75% of ovarian cancer. Conventional treatments often result in relapse, with a need for innovative therapeutic approaches. This study aimed to develop and evaluate a DNA vaccine targeting the preferentially expressed antigen of melanoma, PRAME, a cancer tumour antigen (CTA) overexpressed in HGSC. PRAME demonstrated the highest differential gene expression between normal fallopian tubes and HGSC tumour tissues in a range of patient datasets. The PRAME DNA was condensed by the cationic cell-penetrating peptide RALA to form nanoparticles (NPs). These self-assembling NPs exhibited a mean hydrodynamic size <150 nm and zeta potential >10 mV at N![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratios ≥4 with ≤3% free DNA. The NPs successfully transfected NCTC-929 and DC 2.4 cells with PRAME overexpression, with negligible cytotoxicity. Vaccination with the NPs in vivo elevated CD4+ and CD8+ T-cell activation with increased expression of INF-γ and IL-2 cytokines. Vaccination also significantly improved survival rates in a PRAME-expressing tumour model in vivo. This study demonstrated the utility of a PRAME-targeted DNA vaccine for HGSC treatment which warrants further investigation.
:P ratios ≥4 with ≤3% free DNA. The NPs successfully transfected NCTC-929 and DC 2.4 cells with PRAME overexpression, with negligible cytotoxicity. Vaccination with the NPs in vivo elevated CD4+ and CD8+ T-cell activation with increased expression of INF-γ and IL-2 cytokines. Vaccination also significantly improved survival rates in a PRAME-expressing tumour model in vivo. This study demonstrated the utility of a PRAME-targeted DNA vaccine for HGSC treatment which warrants further investigation.
1. Introduction
Ovarian cancer (OC) is a silent gynaecological cancer, ranked eighth for incidence and mortality (GLOBOCAN Statistics, 2020) with a five-year survival rate of ∼30% worldwide (2023) for patients with advanced disease.1 OC has subtypes: type I and type II (Fig. 1) with High-Grade Serous Carcinoma (HGSC) the most prevalent (70%) of type II tumours. The pre-cursor lesion, serous tubal intraepithelial carcinoma (STIC) of HGSC originates from the distal, fimbrial end of the fallopian tubes (Fig. 1A and B).2 | ||
| Fig. 1 (A) The origin of HGSC OC. Precursor lesions known as serous tubal intraepithelial carcinoma (STIC) in the fallopian tube are considered as the site of origin of the malignant cells and involve the ovaries only secondarily. (B) Ovarian cancer types. All tumours associated with OC have been broadly classified into two subtypes: (i) type I and (ii) type II. Malignant type II comprises >75% of the total OC. (C) DNA vaccine targeting DC cells to induce an immune response. Immature APC, macrophages and other immune cells are recruited. The cargo enters the immature APC, trafficks to the nucleus where the RNA transcription occurs followed by protein translation in the cytoplasm and is then represented on the cellular surface as MHC I. These cells then trigger the CD8+ CTL response. However, if the plasmid is taken up by the non-APC cells, they either present them in MHC I and subsequently release the antigenic peptide on apoptosis or directly release the peptide by proteolysis by proteasome. These peptides are then taken up by the APC and are either cross-presented in MHC I or presented on MHC II which stimulates the CD4+ T cell response which further generates CD8+ CTL and memory cells. (D) Barriers to gene delivery. The epithelial layer, high interstitial fluids and the dense extracellular matrix are some of the barriers which can rapidly degrade the gene before it reaches the nucleus. Once it reaches the cell, the cell membrane acts as a natural barrier that restricts its entry. The negatively charged phospholipid bilayer of the cell membrane makes it difficult for negatively charged nucleic acids, such as DNA to pass through efficiently. Intracellular trafficking, endosomal escape and the nuclear membrane are all key considerations for DNA delivery systems. | ||
The most common biomarker used to detect OC is cancer antigen 125 (CA125), but its specificity and accuracy have always been sub-optimal. Elevated levels >35 U mL−1 can be indicative of OC but are also observed in non-cancerous conditions such as endometriosis, uterine fibroids, and benign ovarian cysts and in healthy premenopausal women.3 The current main therapy for OC includes cytoreductive surgery and platinum/taxane-based chemotherapy (e.g., paclitaxel, cisplatin, carboplatin). Even though these therapies have been effective in getting some response from patients, the recurrence rate is 70%–80% which is typically fatal.4
Targeted therapies and antibodies that augment the immune response such as Bevacizumab (monoclonal antibody targeting VEGF), Pembrolizumab (monoclonal antibody targeting PD1/PDL1 checkpoint), Olaparib (Poly ADP-ribose polymerase-PARP inhibitor) or Niraparib (PARP 1 and 2 inhibitor) have contributed to the improvement of the 10 year survival responses from 18% in 1970s to ∼35% in 2017–2019 in the UK (Cancer Research UK). Immunotherapies include: (i) immune checkpoint inhibitors with specific antibodies,5 (ii) monoclonal antibodies against specific tumour antigens,6 (iii) adoptive T-cell therapy involving reinfusion of modified autologous T-cells,7 (iv) naturally occurring or genetically modified oncolytic virus therapies,8 (v) cancer vaccines (Fig. 1C).9–13
Unlike prophylactic vaccines, therapeutic vaccines are used to treat OC via (i) autologous dendritic cell-based (ii) peptide or protein-based, (iii) nucleic acid-based. Gray et al. conducted a Phase IIb clinical trial of Cvac, a MUC1 autologous dendritic cell (DC) vaccine, in patients with Stage III/IV ovarian cancer. Immunoassays showed a stronger CD8+ CTL response (0.8% CD8+/IFN-γ+) than CD4+ T helper cell response (0.6% CD4+/IFN-γ+), with a higher MUC1-specific T-cell response in patients receiving 10 doses of the vaccine over 56 weeks. Progression-free survival improved (13 vs. 9 months) and overall survival was longer in patients in second remission (13 vs. 5 months). However, DC-based vaccines face limitations, such as low success rates, high costs, labour-intensive production, and challenges in DC maturation and longevity, prompting the exploration of alternative vaccination approaches. In a Phase I/IIa clinical trial conducted by Brown et al., a peptide vaccine targeting folate-binding proteins (E39 + GM-CSF) demonstrated a 77.9% disease-free survival (DFS) rate at 24 months in ovarian cancer patients receiving the 1000 μg dose, compared to 31.2% with the 100 μg/500 μg doses and 40% in the control group.14 The most advanced clinical trials involving nucleic acid vaccines for ovarian cancer (Stage III/IV) have only progressed to Phase I (NCT01322802, completed in 2020). In this trial, a multi-epitope plasmid DNA therapeutic vaccine, AST-201 (pUMVC3-hIGFBP-2), developed by Disis et al. at the University of Washington, was administered intradermally at 100 μg doses monthly for three months to patients with advanced or recurrent ovarian cancer.15 The vaccine was well tolerated and elicited a Th1-cell immune response targeting IGFBP-2 (Insulin-Like Growth Factor Binding Protein 2).16 A phase 2 trial with AST-201 + GM-CSF adjuvant (3 doses with 3 weeks interval) in combination with 6 doses of standard chemotherapy in 3 weeks interval (paclitaxel/carboplatin) post debulking surgery has already been initiated in 2023 (CornerStone-004 study, NCT05794659) and will complete in November 2025.17 Despite all of these trials, to date no HGSC vaccines have gained clinical approval.18
Naked nucleic acid vaccines require delivery to the nucleus of antigen-presenting cells to evoke the necessary T-cell response. The delivery system must overcome extracellular barriers such as the skin epithelium, bypass nuclease degradation and the negatively charged phospholipid bilayer of the cell membrane. Intracellular barriers include the endosome, cytoplasm and nuclear membrane (Fig. 1D).
One of the less explored non-viral methods for nucleic acid vaccination is to use cell-penetrating peptides (CPP).19–22 Cell penetrating peptides (CPPs) are a type of non-viral delivery system characterised by positively charged, amphipathic small peptides (5–30 amino acids) that can penetrate through the extracellular and intracellular barrier of the cells to release the nucleic acid cargo at the destination site.19,20 RALA is one such synthetic cationic CPP which can form self-assembling NPs with a negatively charged nucleic acid cargo which can enter the cells via clathrin-mediated endocytosis.23 While Lipid-based nanoparticles (LNPs) are widely recognised as effective non-viral delivery systems, RALA offers several unique advantages that make it particularly suitable for vaccine application. Unlike LNPs, which may trigger immune responses or cytotoxicity due to the lipid composition, RALA has been shown to maintain low toxicity profiles while achieving comparable transfection efficiency with pDNA, siRNA, and mRNA forming stable complexes through electrostatic interactions, resulting in nanoparticles with a net positive charge.19,20 The preparation of RALA–pDNA nanoparticles is relatively straightforward compared to LNPs, which involve precise optimisation of multiple lipid components (e.g., ionisable lipids, PEGylated lipids) to balance encapsulation efficiency, stability, and toxicity. This simplicity reduces manufacturing complexity and costs. RALA nanoparticles are highly biocompatible and versatile, supporting the delivery of various nucleic acids without requiring extensive chemical modifications.
The adjuvant is derived from the word “adjuvare” meaning help in Latin. One such adjuvant is Granulocyte Macrophage-Colony stimulating factor (GM-CSF) which is a hematopoietic growth factor that stimulates the production of granulocytes and monocytes. GM-CSF is a proinflammatory cytokine that stimulates the proliferation of dendritic cells. It has been investigated as a vaccine adjuvant and can promote a stronger antigen-specific immune response. For example, an autologous melanoma tumour cell vaccine (107 cells injected ID and subcutaneously at 7, 14 and 28 day intervals) that was engineered to secrete GM-CSF enhanced DC antigen presentation. This resulted in both CD4+ and CD8+ T-cell response and antibody-mediated immunity in metastatic non-small-cell lung carcinoma. The autologous vaccine eradicated almost 80% of the tumour in all patients (n = 16).24 GM-CSF has been evaluated as an adjuvant to cancer vaccines in clinical trials for prostate cancer, melanoma, skin cancer, breast cancer, lung cancer, ovarian cancer etc. as a mono or combinational therapy.25,26 GM-CSF–DNA has been demonstrated as a potent adjuvant for melanoma DNA vaccines in both preclinical and clinical studies. In a Phase I/II trial, administration of pGM-CSF at doses of 100, 400, and 800 μg subcutaneously, combined with a peptide vaccine (gp100 and tyrosinase at 500 μg each), elicited CD8+ T-cell responses in 42% of Stage III/IV melanoma patients (n = 8). However, no significant differences in response were observed between dose levels, and dose-limiting toxicity was noted.27
This study aimed to identify and develop an OC-specific DNA vaccine to be delivered using a 30 amino acid CPP in combination with pGM-CSF DNA as a vaccine adjuvant.
2. Materials and methods
2.1. Antigen selection
To identify an antigenic target, the gene expression of several tumour antigens (TA) was analysed using publicly available microarray gene expression patient datasets from the studies conducted at Queen's University Belfast, UK (QUB cohort),28 University of Houston, USA (GSE69429 and GSE69428 cohort)29 and University Health Network, Canada (GSE10971 cohort).30 These datasets were used to analyse the expression of TA in HGSC throughout different stages of cancer using the Kruskal–Wallis non-parametric test and unpaired t-tests at alpha level 0.05.2.2. Cloning of plasmid encoding selected antigenic target
A 1 μg sample of universal human reference RNA (Thermo, UK) was transcribed to cDNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche, UK) The PRAME gene was amplified from the resultant cDNA (Thermo, UK) using KOD Xtreme Hot Start Polymerase (Merck, UK). The amplified fragments, flanked by restriction sites were isolated using a Monarch Nucleic Acid Purification Kit (NEB, UK) and cloned into the pEF-GFP vector (Addgene, UK) at the XmaI and NotI sites. The resulting plasmid was transformed by heat shock into competent DH5α cells (Thermo, UK) and propagated using a HiPure Plasmid Maxiprep Kit (Thermo, UK) according to the manufacturer's instructions. Sequences were verified by Sanger sequencing (Eurofins, Germany).2.3. Nanoparticle formulation
The lyophilised RALA (Biomatik, USA) was stored at −20 °C and reconstituted with DNase/RNase-free UltraPure™ distilled water (Life Technologies, Invitrogen, UK). RALA/pDNA complexes were formed at N:P ratios ranging from 0–12 by adding an appropriate amount of RALA peptide to the specific amount of pDNA cargo. N:P ratios are indicative of the molar ratio of positively charged nitrogen atoms in the peptide to negatively charged phosphates in the DNA backbone.
2.4. Nanoparticle characterisation
The NPs were analysed for mean hydrodynamic size (z-average) and Zeta potential (charge) by dynamic light scattering and by laser Doppler velocimetry on a Nano ZS zetasizer with DTS software (Malvern Instruments, UK). The encapsulation efficiency of the NPs was measured using PicoGreen Reagent assay in a 96-well black plate in triplicate with a volume of 50 μL per well. 50 μL of the prepared reagent (1:200 dilution in 1× TAE buffer) was added to each well and the plate was then read in a FLUOstar Omega microplate reader (BMG Labtech, Germany) with absorbance measured at 520/480 nm. Free pDNA cargo was measured using Ion exchange chromatography (IEC). pDNA or RALA/pDNA NP was loaded into a frit column with anionic SP-Sephadex (Sigma-Aldrich, SPC25120, GER) resin, eluted with ultrapure (DNase/RNase free) H2O and analysed using a Nanodrop UV spectrophotometer (Thermo Scientific, MA, USA) at 260 nm. To analyse the morphology of the NPs TEM analysis was performed by loading NPs onto carbon-reinforced 400 mesh copper grids (TAAB laboratories, UK) and staining with Uranyless EM stain (EMS, USA). The samples were imaged using a JEOL-JEM 1400 plus TEM (JEOL, USA) at 80 kV accelerating voltage.
2.5. Gel retardation analysis of the nanoparticles
NPs were loaded in 1.5% Agarose gel prepared with SYBR-safe DNA gel stain (Thermo Fisher Scientific, UK) and 1× TAE buffer. Nucleic acid sample buffer was added to the samples at 10% concentration before adding to a well of the gel. The gel was run in the buffer tank filled with 1× TAE buffer at 100 V for 60 min and imaged using a UV analyser (UVITEC) with the NineAlliance Mini HD9 Auto software.2.6. In vitro cellular transfection assay
NCTC-929 murine fibroblast cells and DC2.4 murine dendritic cells were obtained from ATCC (USA), grown as monolayers, incubated in an ESCO CelCulture CO2 Incubator (Davidson and Hardy Ltd, UK) at 37 °C, 5% CO2 and 1% O2 using MEM 1× and RPMI 1640 1× + L Glutamine medium (Gibco-Thermo Fisher Scientific, UK) respectively with 10% FBS. A routine Mycoplasma test was performed on all the cell lines used once every 1–2 months to ensure they were contamination-free using a Mycoplasma test kit (Analab, UK). Cells were seeded at a density of 20000 cells per well in 96-well tissue culture plates (VWR, UK) for 24 h before transfection. The cells were conditioned for 2 h in Opti-MEM serum free-media (Gibco-Thermo Fisher Scientific, UK) after which RALA/pDNA NPs were added to each well in triplicate. Following incubation for 4.5 h, the media was removed and replaced with serum-supplemented media. Cells were maintained in complete media for 48 h before analysis.
2.7. Cytotoxicity assay
The in vitro cytotoxicity of RALA/pDNA NPs was analysed using the Alamar blue reduction test. NCTC-929 fibroblasts and DC2.4 dendritic cells were transfected in a 96-well tissue culture plate. At different time points (24 h, 48 h) of transfection, the Alamar Blue™ Cell viability Reagent (Invitrogen, Thermo Fisher Scientific, USA) at a final concentration of 10% was added to the cells which were then incubated in the dark for 2 h. The plate was then read in a FLUOstar Omega microplate reader (BMG Labtech, Germany) with excitation at 560 nm and emission at 590 nm.2.8. Flow cytometry analysis
Following transfection, the media was aspirated off and cells were rinsed with PBS, trypsinised, resuspended serum-supplemented media and centrifuged at 200g, 4 °C for 10 min. The cell pellets were then permeabilized by adding ice-cold 100% methanol for a minimum of 20 min at −20 °C and were centrifuged at 750g for 5 min at 4 °C. The pellet was resuspended in 1× PBS and centrifuged again. The cell pellet was resuspended in 100 μL of diluted (1:50 dilution) PRAME primary antibody (Cell Signalling Technology, USA) and incubated for 1 h at room temperature in the dark. The samples were washed twice by centrifugation in PBS. The cell pellet was resuspended in PBS and analysed via the FACScalibur system (BD Biosciences, UK).
2.9. Quantification of in vitro expression of PRAME protein
To quantify the in vitro expression of PRAME in cell lines Human PRAME SimpleStep ELISA® Kit (ab234561, Abcam, UK) was used after 48 h of transfection according to the manufacturer's protocol. The sample protein concentration in the cell extract was quantified using a Nanodrop 2000. The OD was recorded at 450 nm using a FLUOstar Omega microplate reader (BMG Labtech, Germany).2.10. In vivo animal experiments
All in vivo studies used 6 to 8-week-old C57BL/6 female mice purchased from Charles River Laboratories, UK. All the in vivo experiments in this study were undertaken with guidelines at Queen's University Belfast's Biological Services Unit. The experimental protocols were approved with the Animal (Scientific Procedures) Act of 1986 and were carried out under project licence PPL 2903.2.11. In vivo lymph node tracking of the RALA/pDNA nanoparticles
pPRAME was tagged with Cy5 DNA staining fluorophore using a Label IT® Tracker™ Intracellular Nucleic Acid Localisation Kit (Mirus Bio, USA) according to the manufacturer's protocol. Nanoparticles at N:P 8 were prepared with Cy-5 labelled pPRAME and injected via intradermal (ID) injection into the ear of female C57BL/6 mice. Intraperitoneal (IP) injection of Rompun/ketaset (100 μL/20 g mouse weight) was used to facilitate ID injections. The mice were culled using Schedule 1 methods of euthanasia after the appropriate time point and were imaged under the Bruker imaging machine for fluorescence.
2.12. Lymph node isolation and cell surface staining
The mice were sacrificed at the pre-defined time points. The right axillary lymph node from the C57BL/6 mouse was then removed aseptically. Lymphocytes were obtained by passing lymph nodes through a 70 μm cell strainer with ice-cold PBS. Cells were centrifuged at 350g for 5 min at 4 °C, resuspended in PBS and centrifuged again. The pellet was resuspended in 50 μL of viability stain, with incubation on ice for 20 min. Cells were washed with 1 mL of PBS media (PBS + 5% FBS + 2 mM EDTA) and then centrifuged. Each cell pellet was resuspended in an Fc block and stained with an antibody cocktail or FMO antibody cocktail and incubated for 30 min to facilitate antibody binding. 1 mL of PBS media was added, and centrifuged at 350g for 5 min at 4 °C. All samples were resuspended in 500 μL staining buffer and analysed on Attune NxT flow cytometer.2.13. In vivo tumour development study
The growth dynamics of E0771-PRAME orthotopic tumours were determined in immune-competent C57BL/6 mice. Briefly, under isoflurane anaesthesia mice were implanted in the right mammary fat pad with E0771 cells (2 × 105). Tumour volume, mouse weight and general health were monitored three times weekly. Mice were sacrificed via schedule 1 methods if they lost >20% of their initial body weight or when tumour volume reached 12 GMD (Geometric Mean Diameter).2.14. In vivo analysis of immune response
Immunocompetent female C57BL/6 mice were immunised with a 3-dose regimen of vaccine (20 μg) either ID or IV on days 0, 7, and 11 followed by a 21 day incubation period. GM-CSF was administered in plasmid form, similar to PRAME, as an adjuvant. The pGM-CSF was complexed with RALA at N:P ratio of 8 to form nanoparticles, mixed with RALA/pPRAME nanoparticles before lyophilisation. This formulation was reconstituted before injections. To facilitate ID injections, intraperitoneal (IP) injection of Rompun/ketaset (100 μL/20 g mouse weight) anaesthesia was used. On day 42, mice were sacrificed and culled using a CO2 hypoxia box (Schedule 1). Upon confirmation of death, a cardiac bleed was performed and spleens harvested and processed for downstream applications.
2.15. Intracellular staining
At the study endpoint, the spleens were aseptically resected from each mouse and placed in 5 mL sterile MACS Tissue Preservation Solution and stored briefly on ice. Spleens were processed to single cell suspensions and were seeded with the corresponding media master mixes for unstimulated media only, Con A + LPS positive stimulation and PRAME overlapping peptide (PepTivator®PRAME, human-Miltenyi Biotech, USA). Brefeldin A 1000× (Invitrogen, UK) was diluted in a 1:5 ratio and 10 μL was added into each well and mixed. Plates were incubated for 18 h in an incubator at 37 °C, 5% CO2. The splenocytes were subsequently stained with a panel of antibodies specific to key markers of T-cell activation and maturation. The different T-cell populations, including effector and memory T cells, were then analysed using flow cytometry on an Attune NxT flow cytometer. These included T helper (Th) CD4 and cytotoxic T cells (CTLs) CD8 antibodies, CD44 to identify activated or memory T cells, and CD62L to determine the lymphocyte homing potential. Additionally, cytokine production was measured using antibodies against interferon-gamma (IFN-γ) and interleukin-2 (IL-2), which are indicative of T cell activation and immune response. These antibodies allowed for the identification and quantification of specific T-cell subsets, providing detailed insights into activation status and functional characteristics.
2.16. ELISpot analysis
ELISpot plates for INF-γ and IL-2 were prepared as per the manufacturer's instructions. 100 μL of splenocyte cell suspension was added to each well along with 100 μL of respective 2× master mix media. 100 μL of splenocyte cell suspension was added to each well and then incubated for 48 h in a humidified incubator at 37 °C, 5% CO2.2.17. Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, USA). Statistically significant differences were computed using an unpaired t-test when only one variable was studied, and a one-way analysis of variance (ANOVA) when two variables were studied simultaneously. A p ≤ 0.05 was considered significant. All data in this study was obtained from three independent repeats and is given as mean values ± SEM, unless otherwise specified.3. Results and discussion
3.1. Selection of HGSC-associated overexpressed tumour antigen candidate for vaccine delivery in available datasets
Preferentially expressed antigen in melanoma (PRAME) is a non-X autosomal CTA human tumour antigen which was first discovered in melanoma patients by Ikeda et al. in 1997.31 The results in this study showed a significant increase (p < 0.05) of PRAME gene expression across all datasets and at all stages of OC compared to the normal fallopian tube cells (NFT) and STIC (Fig. 2Ai–iv). The GENT2 gene expression database showed significant overexpression of PRAME between the normal cells and any type of cancerous ovary (p < 0.001) and HGSC subtype (p < 0.001) across 247 samples (Fig. 2Biv). Investigation of PRAME in The Human Protein Atlas online platform showed negligible or zero expression in both protein level and RNA levels in normal cells except for the testis. PRAME is not expressed in brain tissue or immune cells. The predicted location is intracellular with the subcellular location of the protein in the nucleoplasm or in the plasma membrane (Fig. 2Bi and ii). The summary of the results found from The Human Protein Atlas is attached in ESI (Table S1†). | ||
| Fig. 2 (A) Gene expression of PRAME in patient cohort datasets in (i) QUB cohort; (ii) GSE69429; (ii) GSE69428 & (iv) GSE10971. The data were analysed using Kruskal–Wallis one-way ANOVA on ranks and unpaired t-test to find the significant differences between the groups at alpha level 0.05 and the differences are shown with ‘*’. * < 0.01, ** < 0.008, **** < 0.0001. OSE: ovarian surface epithelium; NFT: normal fallopian tube; STIC: serous tubal intraepithelial carcinoma; HGSC: high-grade serous carcinoma. The results are represented as box and whisker plots using Tukey's method. (B) Analysis of PRAME using bioinformatics web platforms (i) and (ii) results for the PRAME protein from The Human Protein Atlas. (i) PRAME Protein expression in the human body and (ii) subcellular expression of PRAME in the nucleoplasm and Plasma membrane. These diagrams were created in BioRender.com and are based on the results of the PRAME protein query in The Human Protein Atlas. (iii) Genetic alterations of PRAME analysed through cBioPortal TCGA (Pan-Cancer Atlas study, n = 585); (iv) expression of PRAME according to GENT2 for Normal ovary vs. Ovarian Cancer (all types) and the HGSC ovarian cancer subtype. The data were analysed statistically using the Kruskal–Wallis test for significant differences between the groups at alpha level 0.05 and the differences are shown with **** < 0.0001. HGSC: high-grade serous carcinoma. The results are represented as box and whisker plots using Tukey's method. (C) Predicted cellular pathways associated with PRAME overexpression. This figure depicts the predicted cellular pathways associated with PRAME overexpression, which suggest that PRAME may influence various signalling cascades relevant to tumour progression. Activation of cellular pathways—the NF-κB signalling pathway and Wnt/β-catenin pathway are significantly impacted by PRAME overexpression and plays a crucial role in inflammation and tumour progression in HGSC. (D) Table of antigens of OC analysed in datasets and expression between NFT and HGSC. The table includes a list of the analysed antigens in ovarian cancer datasets, comparing the expression levels between normal fallopian tube (NFT) tissue and high-grade serous carcinoma (HGSC). | ||
PRAME also known as Cancer/Testis Antigen 130 (CT130) or Melanoma Antigen Preferentially Expressed in Tumours (MAPE) or Opa-Interacting Protein (OIP-4/OIP4) has been reported to be over-expressed in all stages of epithelial OC, most notably in HGSC by other researchers.32–34 Zhang et al. observed PRAME mRNA overexpression using RT-qPCR in 60% of patients with primary epithelial OC in comparison to normal ovarian epithelial cells.32 PRAME gene overexpression was also observed in established solid tumour cell lines by Pankov et al.34 The ovarian cancer cell line OVCAR showed a 20-fold change in PRAME gene expression through RT-PCR when normalised to the BT-474 breast cancer cell line with low PRAME expression.34 The actual functionality of PRAME in cancer and in tumourigenesis has not been extensively studied, however, it can be associated with being a repressor of the retinoic acid receptor (RAR) which has a negative effect on the antiproliferative and cytotoxic effects of retinoic acid (Fig. 2C). Epping et al. reported that PRAME also mediates transcriptional repression and shows dose-dependent (0–3 μg) inhibition of RA-induced activation of a reporter gene (RARE-Luc).35 Knockdown of PRAME via shRNA in A375 Melanoma cells enhanced RAR signalling in human melanoma cells such as A375, FM6 and SK23, and MCF-7 human breast cancer cells indicating an inverse correlation.35 The results of PRAME overexpression in HGSC, corroborates previously reported results and predicts it to be a potential vaccine target for HGSC.
3.2. Nanoparticle characterisation
The mean particle diameter ranged from 90–150 nm across all N:P ratios except N:P 0 which was the pDNA-only control. This decrease in the size is attributed to the electrostatic interaction between the anionic naked plasmid cargo and cationic RALA peptide. An increase in the positive charge correlated with N:P ratio with values ≥10 mV at ≥N:P 6. All N:P ratios >8 had a PDI <0.3 (Fig. 3A), which is regarded as acceptable to enter cells via clathrin-mediated endocytosis.36,37 The spectra obtained for N:P > 8 demonstrate a tighter population of NPs indicating complete condensation (Fig. 3B). TEM confirmed the spherical morphology of RALA/pPRAME NPs had between 60–120 nm (Fig. 3C). The overall distribution of the size of the NPs appeared to be smaller than the mean hydrodynamic diameters measured using the DLS zetasizer. This was attributed to the presence of the dispersant medium in the DLS measurements which was absent for TEM imaging of NPs.38 At N:P ratio 2, DNA bands were visible at the same distance as the pPRAME DNA-only control although at a lower intensity on the agarose gel. At N:P 4, low levels of fluorescence can be observed in the well indicating the neutralisation of pDNA by RALA but not full condensation (Fig. 3D). At N:P > 2 ratios, the fluorescence ceases and is not observed indicating that the charge of RALA is sufficient to neutralise the pDNA. These results corroborate the findings by Jain et al. (2015) where RALA formed NPs at N:P ratios >3.39 The distance of migration is also dependent on the conformation of the pDNA. The different bands in the gel represented the linear, open circular and supercoiled conformation of the pDNA, respectively. The higher rate of supercoiled conformation is also indicative of greater biological functionality.40 The PicoGreen assay to quantify the encapsulation efficiency of the RALA/pDNA NPs at varying N:P ratios encapsulation of the pPRAME DNA by RALA increased from 27.39 ± 4.26 at N:P 2 to 81.89 ± 4.32 at N:P 12 after which there was minimal change in encapsulation (Fig. 3E). Quantification of eluants for the presence of unencapsulated free pDNA through IEC analysis showed that at N:P ≥ 8 there was <3% free DNA which is indicative of 97% encapsulation efficiency and this value did not increase at N:P 10 or 12 (Fig. 3F). The physical characteristics for RALA/pDNA NPs previously reported by McCarthy et al. (2014) and Cole et al. (2018) had similar characteristics with cationic NPs >10 mV and a mean hydrodynamic size <100 nm, encapsulation >80%.21,23,41,42 Kudsiova et al. (2021) studied the particle size of the clinically approved Pfizer-BioNTech BNT162b2 covid-19 vaccine by DLS and found the average particle size of the vaccine to be ≈90 nm.43 These vaccines were, however, developed using lipid nanoparticles. The results obtained for the RALA NP suggest optimum characteristics for cellular uptake.
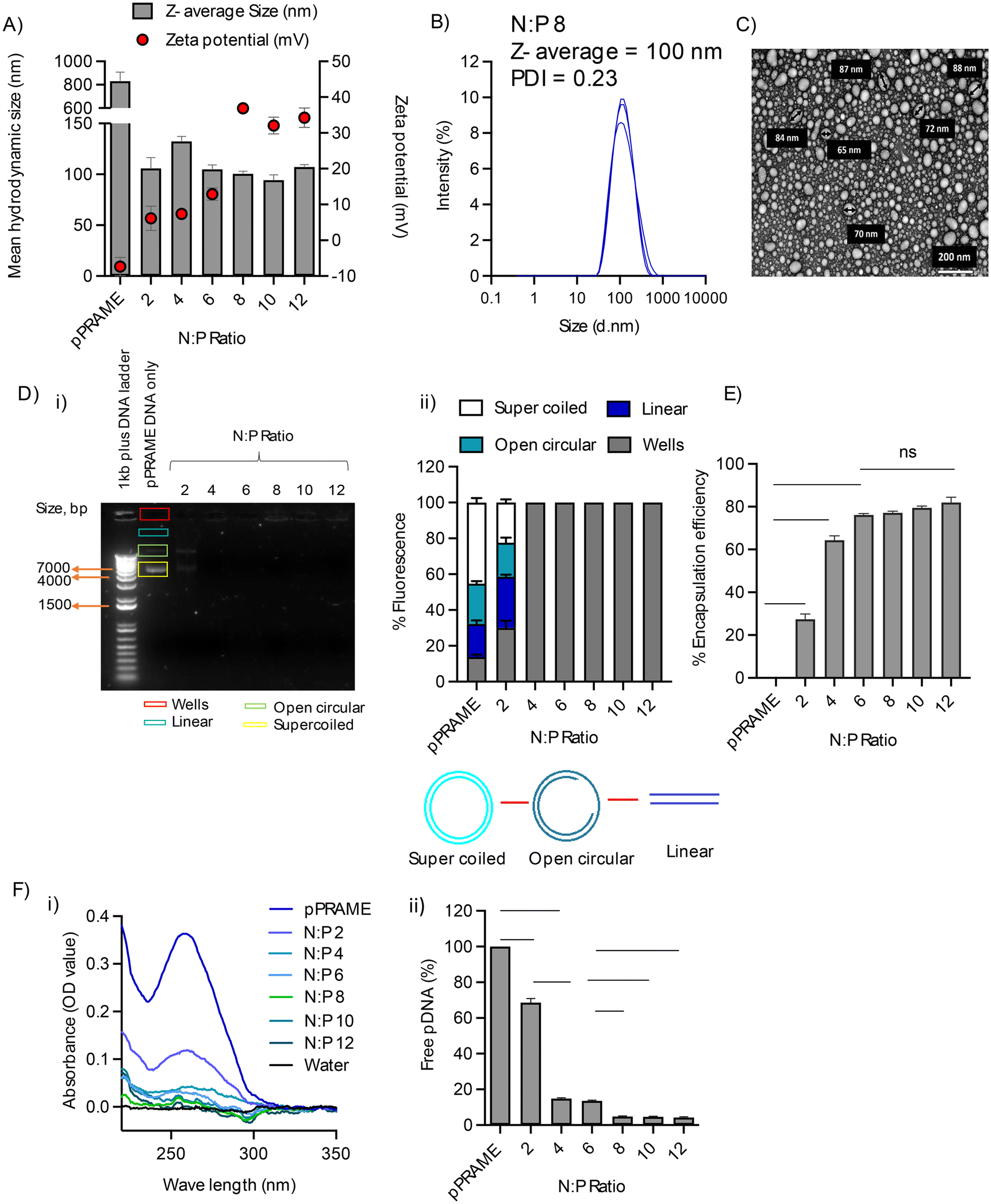 | ||
| Fig. 3 Characterisation of RALA/pPRAME NPs: (A) Z-average size and Zeta potential of RALA/pPRAME NP; (B) DLS spectra for RALA/pPRAME NP. All the complexes were formed with 0.5 μg of pDNA in a total volume of 50 μL made up of DNAase/RNAase-free distilled water and incubated for 30 min at room temperature prior to measurement in the Malvern Zetasizer NanoZS instrument. (C) TEM analysis for RALA/pPRAME NP captured at ×40 K magnification using a JEOL-JEM 1400 plus TEM (JEOL, USA) at 80 kV. Fresh NPs were formulated at a concentration of 0.1 μg μL−1 and placed in a copper mesh grid. The grids were then imaged using TEM after staining overnight with an Uranyless stain. The results displayed were analysed using the Image J software. (D) Gel Retardation Assay (1.5% w/v): (i) RALA/pPRAME NPs were formulated at a range of N:P ratios and placed in the wells. Naked pDNA was used as a control and a 1 kb DNA ladder was used in Lane 1. The gel was run at 100 V for up to 60 min and visualised under the UV light. The image shown is representative of the three replicates & (ii) densitometric analysis via Image J software to quantify fluorescence within each lane. The intensity of each band corresponds to the DNA concentration at each N:P ratio. (E) Encapsulation efficiency of RALA/pPRAME NPs. NPs were formulated at a range of N:P ratios with 1.5 μg of pDNA to a total volume of 150 μL and placed in the 96 well plate in triplicate to which Quanti-iT PicoGreen Reagent was added. The samples were read in a plate reader with an excitation at 480 nm and fluorescence at 520 nm. (F) IEC analysis for RALA/pPRAME NP: (i) spectra analysed using the nanodrop & (ii) percentage of unencapsulated free DNA. Fresh NP was formulated at a concentration of 0.1 μg μL−1 and passed through a negatively charged SP Sephadex resin column. The eluents were then quantified in the nanodrop. Results are displayed as mean ± SEM, N = 3. The data were analysed statistically using a one-way ANOVA with ‘*’. * < 0.05, ** < 0.01, **** < 0.0001, ns = not significant. | ||
3.3. In vitro functionality and cytotoxicity of RALA/pDNA NP
A dose-dependent transfection rate was observed in both cells with the highest transfection rate of approximately 17% transfection in NCTC-929 cells and 12.5% transfection in DC 2.4 cells with 1.5 μg cargo which was significantly higher (p < 0.05) when compared to the untreated cells. Lower rates of transfection were observed with lower cargo amounts, i.e., 1 μg and 0.5 μg. ELISA quantification of the concentration of PRAME protein in RALA/pPRAME NP treated cells measured high expression of PRAME with >20 ng mL−1 compared to the untreated cells where levels were <1.5 ng mL−1. Thus, RALA was able to deliver pPRAME to the cells and it was successfully translated into the PRAME protein inside the murine fibroblasts and immune cells, indicating functionality of the nanoparticles (Fig. 4B). | ||
| Fig. 4
In vitro characterisation of RALA/pPRAME NP: (A) cytotoxicity of the nanoparticles (i) assessed in NCTC-929 murine fibroblasts cells & (ii) assessed in DC 2.4 murine dendritic cells; (B) functionality of RALA/pPRAME NP (i) flow cytometry analysis assessed in NCTC-929 cells; (ii) flow cytometry analysis assessed in DC2.4 cells & (iii) PRAME concentration via ELISA analysis in NCTC-929 cells. The cells were transfected in a 96-well plate with 20000 cells per well at N:P 8 for 4.5 h in Opti-MEM media and were trypsinised after 48 h of transfection. To measure the transfection efficiency with flow cytometry the cells were then stained with Alexa Fluor 488 conjugated PRAME primary antibody and the transfection efficiency was analysed via flow cytometry. For PRAME protein concentration NCTC-929 cells were transfected in a 24-well plate with 80000 cells per well at N:P 8 for 4.5 h in Opti-MEM media and were collected for total protein 48 h post transfection. To measure the PRAME protein, the cells were then analysed using an ELISA kit. Results are displayed as mean ± SEM, N = 3. The data were analysed statistically using a one-way ANOVA with ‘*’. * < 0.05, ** < 0.01, **** < 0.0001, ns = not significant. | ||
According to the ISO 10993-5:2009 standards, biotoxicity can be categorised under the following categories: weak cytotoxicity (80–60%), moderate cytotoxicity (60–40%) and <40% is highly toxic.44 The results showed cell viability >80% at all N:P ratios at both 24 h and 48 h time points, apart from N:P 12 at 24 h in DC2.4 cells, although this was above 80% at 48 h indicating recovery. There was no statistically significant difference in cell viability of treated cells compared with the naked pDNA (N:P 0) and untreated cells (Fig. 4A). RALA NP has previously been reported to show no cytotoxicity (>80% cell viability by Trypan blue Exclusion assay) in PC-3 prostate cancer cells after 72 h of transfection by Massey et al. (2016).45
3.4. In vivo lymph node tracking of the RALA/pDNA nanoparticles
The NPs formed a depot effect on the injection site (right ear) which decreased over time. The fluorescence sum intensity of the injection site was significantly (p < 0.05) higher with NPs compared to naïve and mice receiving pPRAME only. It was observed as ∼3 × 107 counts at 2 h which reduced to ∼1 × 107 counts at 96 h. It indicates the localised and sustained release of vaccine antigens at the site of administration which mimics a natural infection. No fluorescence (with negligible sum intensity) was observed at the injection site for pPRAME control and naïve mice. When the mice were dissected and imaged, fluorescence was observed in the axillary lymph nodes on the injection side (right) of the mice receiving the NPs. The fluorescence sum intensity of the mice receiving NPs was sustained throughout the different time points and was significantly higher (p < 0.05) compared to the mice receiving pPRAME only or naïve mice. The lymph nodes were excised and processed via surface staining of markers to determine which cell types had taken up the NPs. There was no significant difference between mice receiving the NPs and the pPRAME for Langerhans cells and lymphoid resident cDC1 and cDC2 cells at different time points. However, migratory cDC1 were 2-fold higher in RALA/pPRAME treated mice at 2 h ∼10% compared to the mice receiving pPRAME only and naïve mice with ∼5% migratory cDC1s present (Fig. 5). Migratory cDC1 (CD103+ CD11b−) are potent immune cell target capable of acquiring the antigens from periphery, undergo maturation and then migrate to the lymphoid site to interact with naïve T-cells to generate CTL effector cells.46 cDC1 cells also have a stronger affiliation to cross present antigens and activate CD8+ cytolytic T cells than cDC2 cells. This can be associated to expression of receptors such as DNGR-1 (Dendritic Cell Natural Killer Lectin Group Receptor-1), facilitating cross presentation presenting antigens to CD8+ T-cells via MHC I pathway. Thus, enhanced uptake by cDC1 cells can amplify the robust activation of CTL, enhancing tumour antigen recognition and cancer cell apoptosis. However, their presence at the injection site diminishes as they complete their migration process which is evident from the results observed at 24 h and 96 h timepoint. | ||
| Fig. 5 (A) Cy5 fluorescence at the injection site: (i) images from the Bruker machine & (ii) sum intensity of fluorescence. (B) Cy5 fluorescence in the draining lymph node: (i) the images obtained through Bruker machine for naïve mice and mice 2 h, 24 h and 96 h post injection (ii) sum intensity of fluorescence & (iii) flow cytometry analysis of lymphocyte phenotype from draining lymph nodes showing uptake by migratory cDC1. Female C57BL/6 immunocompetent mice were injected with Cy5 labelled pPRAME (n = 3) and RALA/pPRAME-Cy5 N:P 8 NPs (n = 4) intradermally in the right ear. NPs were lyophilised and reconstituted to a concentration of 1 mg mL−1 before injecting. Mice were culled after 2 h, 24 h, and 96 h time points using schedule 1 methods and imaged for Cy-5 fluorescence using the Bruker imaging machine. The injected ear was removed placed in a 24-well transparent plate and imaged for Cy5 fluorescence. The images were processed using the Bruker Molecular Imaging software (v.7.5.2.22464) and the sum intensity was calculated. The intensity scale was set at 3000–10000 counts. Naïve (N = 3) mice were used as controls. Results are displayed as mean ± SEM. The data was analysed statistically using two-way ANOVA to find the significant differences between the groups at alpha level 0.05 and the differences are shown with ‘*’. * < 0.05, *** < 0.001. | ||
In contrast, the prolonged depot effect of NPs over four days was observed which can be attributed to the physicochemical properties of the RALA formulation with uptake at the site of injection. This depot effect is advantageous for prolonged antigen exposure and improved immune activation. A depot effect, achieved by localised and sustained antigen release at the injection site, can enhance vaccine immunogenicity and efficacy by mimicking a natural infection and prolonging immune cell stimulation. Vaccine adjuvants such as alum are known to improve this effect via slow antigen release and immune activation for up to 14 days.47 However, alum promotes a Th2 response and robust antibody production which is beneficial for prophylactic vaccines,48 but not for therapeutic vaccines where a Th1 response is needed. The RALA/pPRAME vaccine produced a depot effect in C57BL/6 mice evidenced via Cy-5 fluorescence for up to 4 days, but further studies are needed to determine the duration of this effect with respect to cellular response. While depot effects can be advantageous, they are not always critical for therapeutic vaccines49 aimed at rapidly activating antigen-presenting cells, as excessive depot formation might delay the antigen release impairing the activation of T-cells and counteracting cellular immunity, which is crucial for cancer immunotherapy.
3.5. In vivo analysis of the immune response
Flow cytometric analysis of the splenocytes extracted from the vaccinated mice showed elevated CD8+ and CD4+ response in all the vaccinated groups (both ID and IV) compared to the control groups. The flow cytometry gating strategy used for the identification of the different subset of population of T-cells based on the activation markers has been included in the ESI figures (ESI Fig. 6†). The CD8+ response was significantly (p < 0.05) higher in the groups vaccinated with the GM-CSF adjuvant following both ID and IV delivery routes (∼22%) compared to all control groups (∼16%). The CD4+ response was significantly higher (p < 0.05) in the RALA/pPRAME + pGM-CSF IV group (∼35%) compared to the naïve (∼28%) and RALA ID (25%) control groups. When the CD4+ and CD8+ T-cells were further analysed based on the expression of CD44 and CD62L memory markers, significantly higher (p < 0.05) T-cell memory responses was observed in all the vaccinated groups compared to naïve control mice. All vaccinated groups also showed elevated levels of both TEM and TCM memory cells compared to all control groups (Fig. 6A and B). Significantly (p < 0.05) higher responses were attributed to the RALA/pPRAME + pGM-CSF ID group for CD8+TEM (∼15%) compared to all the control groups (∼5%). For CD8+TCM as well RALA/pPRAME + pGM-CSF ID group showed the highest response (∼14%) when compared to all the controls (∼6%) (p < 0.05). | ||
| Fig. 6 Immune response of primary splenocytes against PRAME vaccination: (A) effector memory T-cell response (i) effector memory CD8+ T-cells (CD62L− CD44+) & (ii) effector memory CD4+ T-cells (CD62L− CD44+); (B) central memory T-cell response (i) central memory CD8+ T-cells (CD62L+ CD44+) & (ii) central memory CD4+ T-cells (CD62L+ CD44+. (C) IFN-γ expression by primary CD8+ memory T-cells in RALA/pPRAME vaccinated mice immune memory response of primary splenocytes against PRAME vaccination assessed in (i) effector memory CD8+ T-cells & (ii) central memory CD8+ T-cells; (D) IFN-γ expression by primary CD4+ memory T-cells (i) effector memory CD4+ T-cells & (ii) central memory CD4+ T-cells. (E) Cytokine expression by primary Splenocyte T-cells in RALA/pPRAME vaccinated mice via ELISpot analysis of primary splenocytes extracted from vaccinated mice: (i) representative images of IFN-γ and IL-2 ELISpot wells containing splenocytes stimulated with PRAME overlapping peptides; (ii) expression of IFN-γ with PRAME antigen-specific overlapping peptides & (iii) expression of IL-2 with PRAME antigen-specific overlapping peptides. The PRAME antigen-specific immune response of splenic T-cells was analysed in C57BL/6 female immunocompetent mice. The mice were vaccinated either through intradermal or intravenous route with 3 doses of RALA/pRAME vaccine on days 0, 7, and 11. The NPs were prepared at N:P ratio 8 (20 μg), lyophilised with Trehalose cryoprotectant (10% in trehalose in reconstituted volume), and reconstituted to 1 mg mL−1 concentration prior to injecting. pGM-CSF was used along with pPRAME as an adjuvant for the vaccine. The spleens of the mice were extracted after 42 days of the 1st injection dose and the splenocytes were isolated, cultured and stained for intracellular markers of the immune cells. The stained splenocytes were analysed using flow cytometric analysis and the results are represented as the mean value in each group (N = 5). Following ex vivo splenocyte stimulation, IFN-γ and IL-2 expression were measured via ELISpot. Data represents the mean value (N = 5) and are statistically evaluated by one-way ANOVA and the statistical difference is represented with ‘*’. * < 0.05, ** < 0.001, *** < 0.0001. | ||
Elevated response of in CD8+ T-cells reflects the efficacy of the vaccine to induce robust CTL responses, a critical mechanism for targeting PRAME-expressing tumour cells. The increased memory T cell populations of both TCM and TEM suggest the potential for long-term immune protection, enabling rapid responses upon antigen re-exposure. TEM (CD62L− CD44+) cells are found in the peripheral tissues and respond rapidly to the antigen re-exposure producing effector cytokines IFN-γ, IL-2 and TNF-α. TCM (CD62L+ CD44+) on the other hand are found in the lymphoid tissues and provide long term protection and can differentiate into effector cells upon re-exposure of antigen producing cytokines such as IL-2 and IFN-γ. Expression of surface marker chemokine receptor 7 (CCR7 also known as CD197) and CD62L differentiates TCM from TEM.50 Both the subsets of memory cells are important for T-cell activation upon re exposure of the antigen to target cancer cells.
The CD8+ and CD4+ naïve T-cells, TEM and TCM were further analysed for the expression of the pro-inflammatory cytokines essential for T-cell activation, differentiation, and propagation. IFN-γ, IL-2 and TNF-α are also markers of Th1 immune response and are necessary for activation of CTLs for an antitumour effect. The vaccinated groups showed significantly higher (p < 0.05) expression of IFN-γ in the CD8+TEM and TCM groups compared to the controls. The highest IFN-γ expression was found in the RALA/pPRAME + pGM-CSF ID group for the CD8+ cells with ∼8% expression in TEM and ∼10% in TCM respectively (Fig. 6Ci and ii). IFN-γ expression was also observed to be elevated in CD4+TEM and TCM cells for all the vaccinated groups compared to control naïve and RALA groups. IFN-γ expression was highest in the RALA/pPRAME + pGM-CSF IV group for CD4+TEM (∼22%) and in RALA/pPRAME IV group for CD4+TCM (∼40%) compared to all the control groups with ∼4% and ∼9% expression respectively (p < 0.05) (Fig. 6D). IFN-γ and IL-2 expression were also analysed via ELISpot analysis. Both IFN-γ and IL-2 were found to be elevated with higher Spot-forming Units (SFU) in the vaccinated group compared to the unvaccinated controls. Significantly high (p < 0.05) responses for IFN-γ were found in the RALA/pPRAME ID group with a mean value of ∼150 SFU/106 and in the RALA/pPRAME IV group with a mean value of ∼190 SFU/106 for IL-2 response (Fig. 6E).
The differentiation of naïve CD4+ cells into Th1 cells is driven primarily by the production of cytokines such as IFN-γ. Activated Th1 cells produce IFN-γ which in turn produces a positive feedback loop for Th1 differentiation. Th1 cytokines, such as IFN-γ and IL-2, promote the development of CTLs which can directly lyse tumour cells. For a cancer vaccine, a Th1 skewed immune pathway is preferred for an optimal antitumour effect and tumour rejection.51,52
A PRAME-specific recombinant protein vaccine elicited higher CD4+ responses in female CB6F1 mice assessed after 2 weeks (∼2%) and 2 months (∼1%) after compared to PBS control (0%) via intramuscular injections on days 0, 14, 28, and 42.53 But this response was generated only in combination with immunostimulant AS15 (liposomal adjuvant containing CpG oligodeoxynucleotides, monophosphoryl lipid and a saponin based adjuvant QS-21) which makes it difficult to determine the PRAME protein response. On the other hand, RALA/pPRAME vaccine was able to generate a robust immune response even in the absence of any adjuvant irrespective of the route of administration.
3.6. In vivo tumour growth study
The E0771 breast cancer cell line was modified to overexpress PRAME, and its tumour growth was studied in immune-competent female C57BL/6 mice. The group of mice with the highest tumour cell number (2 × 105) cells implanted had 100% tumour uptake where all the tumours were growing exponentially after 14 days post-implantation (n = 6). The survival analysis comparing the E0771-PRAME overexpressing group and the E0771 control group revealed a significant reduction in overall survival in animals with PRAME overexpression (p < 0.01) (Fig. 7A). Tumour xenograft models are extensively used in preclinical research to study ovarian cancer biology and develop new treatments. However, most ovarian cancer xenografts are intraperitoneal and highly metastatic, making orthotopic implantation technically challenging and requiring advanced bioimaging techniques for monitoring. The heterogeneity of high-grade serous ovarian carcinoma further complicates the development of suitable models. Connolly et al. (2003) reported the first transgenic mouse model for epithelial ovarian carcinoma, created by inducing SV40 expression driven by the Müllerian inhibitory substance type II receptor gene promoter (MISRII).54 This model had a 50% tumour development rate and associated peritoneal ascites and metastasis.55 MOVCAR cell lines, derived from ascites of C57BL/6 TgMISIIR-TAg transgenic mice, mimic human high-grade serous ovarian carcinoma but developed disseminated carcinomatosis only in severely immunocompromised mice.56 The E0771 cell line, a well-established murine breast cancer model, was used for PRAME overexpression studies. E0771 cells, when injected into the mammary fat pad, show high tumour uptake (∼100% at cell numbers >105).57,58 Ewens et al. (2005) achieved 97% tumour uptake with E0771 cells in C57BL/6 mice, consistent with the 100% observed in this study with E0771-PRAME cells.59 However, PRAME overexpression significantly decreased mouse survival (p = 0.01) compared to non-PRAME-expressing E0771 tumours. PRAME has been associated with tumour growth; Zhang et al. (2021) reported that PRAME downregulation reduced tumour growth in stable H1299 cell lines in nude mice.60 | ||
| Fig. 7 In vivo tumour growth and regression study (A) tumour growth analysis of PRAME overexpressing cell lines (i) survival rate & (ii) overexpression of PRAME in modified cell line via ELISA. E0771 breast cancer cells were modified to overexpress PRAME and were injected into the mammary fat pad of female C57BL/6 mice to characterize the growth. The volume of the tumour was monitored 3 times weekly and tumour-bearing mice were culled once the tumour reached approximately 12 GMD in size using schedule 1 methods. Tumour-free mice were monitored until 50 days and were culled thereafter. (B) Tumour volume in different treatment groups of tumour regression study; (C) survival data of tumour regression study of the RALA/pPRAME vaccine; (D) table of tumour free mice for different treatment group 85 days post implantation; (E) relative change in tumour volume per day & (F) representative images of tumours in tumour regression study. The tumours growing in the vaccinated mice showed signs of necrosis and tumour coring (highlighted with yellow arrow) which was not observed in unvaccinated naïve mice. Immunocompetent C57BL/6 female mice were injected with 2 × 105 E0771-PRAME cells and were subjected to 3 doses of vaccination on day 4, 7, and 11 post-tumour implantations either through intradermal or intravenous route. The tumour volume and weight were monitored 3 times weekly throughout the study. The mice without tumour uptake were monitored for 85 days post-implantation and were culled thereafter using schedule 1 methods. Following ex vivo splenocyte stimulation, surface memory markers were measured via flow cytometry. Data represents mean value ± SEM and are statistically evaluated using Graph pad prism version 10. | ||
3.7. In vivo tumour regression study
The treatments (all 3 doses of vaccine) were well tolerated by female C57BL/6 tumour-bearing mice. Vaccinated mice showed higher rates of survival compared to unvaccinated mice. The tumour uptake in the vaccinated mice was observed to be lower (Fig. 7D) with lower rates of growth per day (Fig. 7E) compared to the naïve mice (not significant at p = 0.085). The growth rate was almost reduced to <10 mm3 day−1 in RALA/pPRAME ID and RALA/pPRAME + pGM-CSF IV groups compared to unvaccinated naïve mice with a ∼40 mm3 day−1 tumour growth (Fig. 7E). 100% of all naïve mice were dead by 32 days post implantation vs. 60% of vaccinated mice survived >85 days post implantation. The one exception was the RALA/pPRAME IV group with 40% survival (Fig. 7C). Necrosis and tumour coring were also observed in vaccinated mice which were not observed in unvaccinated mice (Fig. 7F). Tumour coring is often associated with significant infiltration of CTL into the tumour leading to central collapse and cavitation in the tumour. This is also indicative of decreased rate of angiogenesis disrupting the formation of new blood vessels which are essential for tumour growth and survival.61 Tumour-free mice were monitored until 85 days post-implantation and were culled thereafter when no tumour uptake was observed.The therapeutic efficacy of a vaccine in a tumour syngeneic model is assessed based on the immune response, tumour uptake, and survival of vaccinated mice. Yang et al. (2019) achieved complete tumour eradication (TC-1, 5 × 105 cells) with a peptide vaccine (50 μg E7 + 20 μg CpG adjuvant) in cervical cancer models, whereas control mice exhibited 100% tumour uptake.62 Similarly, Chiriva-Internati et al. (2010) found that 100% of mice vaccinated with Sp17 protein + CpG (50 μg + 20 μg, intramuscular) remained tumour-free 91 days post-ID8 cell injection (2 × 106), with 79% and 80% survival rates in prophylactic and therapeutic studies, respectively.63 In a therapeutic study with RALA/pPRAME in C57BL/6 mice, vaccinated groups showed fewer tumours and improved survival (∼40%), though differences compared to unvaccinated controls were not statistically significant, possibly due to the low sample size. Although RALA/pPRAME administered intradermally (ID) showed significant tumour growth inhibition, survival outcomes are influenced by multiple factors beyond tumour size reduction alone. Notably, in some vaccinated mice, extensive tumour necrosis was observed (37.5% in RALA/pPRAME ID and 25% in the RALA/pPRAME + pGM-CSF IV) which, while indicative of an immune response, also led to ulceration and severe tissue damage. As per ethical guidelines, these animals had to be humanely culled, which impacted the overall survival data. This explains why tumour growth inhibition was evident, yet the survival benefit did not correspond proportionally. This suggests that while the treatment elicited potent anti-tumour responses, it may also have provoked local immune-related adverse effects. Future studies will focus on optimising dose and administration route to minimise such side effects while preserving therapeutic efficacy. The inclusion of GM-CSF was intended to enhance dendritic cell recruitment and antigen presentation, thereby boosting immune responses. However, as GM-CSF is a pleiotropic cytokine, its immunomodulatory effects can be complex, and its impact on tumour progression may depend on the tumour microenvironment. It can have dual role in cancer immunotherapy. The observed increase in tumour volume in the RALA/pPRAME + pGM-CSF group compared to RALA/pPRAME alone suggests that, while GM-CSF enhanced immune activation, it may have also influenced other immune or stromal components in a manner that facilitated tumour growth. Although GM-CSF is generally known to promote antitumour immunity via APC activation and recruitment, it has also been reported in some cases to modulate the tumour microenvironment in complex ways, inducing potential recruitment of myeloid-derived suppressor cells (MDSCs) and polarizing tumour-associated macrophages (TAMs) towards an M2 phenotype, which secrete IL-10 and TGF-β to suppress cytotoxic T-cell activity. GM-CSF also induces PD-L1 expression on neutrophils and macrophages, enabling tumour immune evasion. This can contribute to an immunosuppressive tumour microenvironment and counteract anti-tumour immunity.64 Although our current dataset does not directly assess such populations, this may partially explain the observed increase in tumour volume despite enhanced immune activation. Further in vivo studies would however, be needed to substantiate this. No significant differences in immune responses were observed between intradermal and intravenous routes at 20 μg doses, suggesting future studies should consider doses and regimens.
4. Conclusion and future perspectives
This study identified that PRAME is a valid antigen from an available clinical data set and highlighted the significant immunogenic potential of RALA/PRAME vaccination in HGSC, but further studies are warranted. The effect of GM-CSF as an adjuvant could be analysed further by optimising the dose levels in future in vivo experiments. Future experiments can also be designed to study the prophylactic effect of the vaccine and the effect of multiple antigenic vaccines for HGSC along with PRAME. Given the recent success of mRNA vaccines (Nobel prize 2023 awarded to Weissman & Kariko) by Pfizer and Moderna to combat the global pandemic SARS-CoV-2 (COVID-19), enhancing the immune response of the RALA/pPRAME vaccine through the use of mRNA encoding PRAME may hold promise. mRNA does not need to overcome the nuclear membrane barrier for protein translation and has been proven efficient in a previous study with a CPP to generate a CD8+ CTL response.65 Nucleoside-modified mRNA (pseudo uridine incorporation) has higher translation and stability (∼10 times) compared to unmodified mRNA in mammalian cells or when injected IV into mice (0.012–0.15 mg kg−1). Peptide-nucleic acid NPs could provide a better vaccination platform with modified mRNA to induce a better and elevated CTL immune response for PRAME overexpressing HGSC tumours as compared to the pDNA vaccine investigated in this study. The efficacy of the RALA/pPRAME vaccine could also be improved by exploring multivalent vaccine options over it being a monovalent vaccine. Thus, the inclusion of more than one antigenic along with PRAME could aid in the development of a multivalent vaccine therapy for HGSC OC patients providing a larger spectrum of antigenic and ultimately broader patient coverage stimulating a more robust immune response.Author contributions
CS drafted the initial version and produced all the figures and tables. AE, BJ and YS helped with the in vivo experimentation. EG helped in the development of the PRAME overexpression cell line. HM, NB, ND and RD supervised CS throughout this study. HM, NB and ND rewrote the final drafts, made edits and polished the manuscript.Data availability
Data sets for Development of a Nano-Vaccine for High-Grade Serous Ovarian Cancer can be found at https://pure.qub.ac.uk/en/datasets/.These will have a publicly available DOI once the manuscript has been accepted. At the moment these files are restricted due to sensitivities around animal data.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
This study was carried out as a part of the PhD project under the MoU signed between Queen's University Belfast, UK and Tezpur University, Assam, India.References
- K. Xie, C. Fu, S. Wang, H. Xu, S. Liu, Y. Shao, Z. Gong, X. Wu, B. Xu, J. Han, J. Xu, P. Xu and X. Jia, J. Ovarian Res., 2019, 12, 1 CrossRef CAS PubMed.
- R. J. Kurman and I. Shih, Am. J. Surg. Pathol., 2010, 34(3), 433–443 CrossRef PubMed.
- H. Qi, S. Xiao, R. Shi, M. O. Ward, Y. Chen and W. Tu, et al. , Nature, 2018, 539–547 Search PubMed.
- C. Kreuzinger, A. Geroldinger, D. Smeets, E. Braicu, J. Sehouli, J. Koller, A. Wolf, S. Darb-Esfahani, K. Joehrens, I. Vergote, A. Vanderstichele, B. Boeckx, D. Lambrechts, H. Gabra, G. B. A. Wisman, F. Trillsch, G. Heinze, R. Horvat, S. Polterauer, E. Berns, C. Theillet and D. Cacsire Castillo-Tong, Clin. Cancer Res., 2017, 23(24), 7621–7632 CrossRef CAS PubMed.
- E. K. Lee and P. A. Konstantinopoulos, Trends Cancer, 2019, 524–528 CrossRef CAS PubMed.
- J. Hamanishi, M. Mandai, T. Ikeda, M. Minami, A. Kawaguchi, T. Murayama, M. Kanai, Y. Mori, S. Matsumoto, S. Chikuma, N. Matsumura, K. Abiko, T. Baba, K. Yamaguchi, A. Ueda, Y. Hosoe, S. Morita, M. Yokode, A. Shimizu, T. Honjo and I. Konishi, J. Clin. Oncol., 2015, 33(34), 4015–4022 CrossRef CAS PubMed.
- J. L. Tanyi, S. Bobisse, E. Ophir, S. Tuyaerts, A. Roberti, R. Genolet, P. Baumgartner, B. J. Stevenson, C. Iseli, D. Dangaj, B. Czerniecki, A. Semilietof, J. Racle, A. Michel, I. Xenarios, C. Chiang, D. S. Monos, D. A. Torigian, H. L. Nisenbaum, O. Michielin, C. H. June, B. L. Levine, D. J. Powell Jr, D. Gfeller, R. Mick, U. Dafni, V. Zoete, A. Harari, G. Coukos and L. E. Kandalaft, Sci. Transl. Med., 2018, 10(436), eaao5931 CrossRef PubMed.
- H. Fukuhara, Y. Ino and T. Todo, Cancer Sci., 2016, 107(10), 1373–1379 CrossRef CAS PubMed.
- E. A. Mittendorf, G. T. Clifton, J. P. Holmes, E. Schneble, D. van Echo, S. Ponniah and G. E. Peoples, Ann. Oncol., 2014, 25(9), 1735–1742 CrossRef CAS PubMed.
- B. Temizoz, E. Kuroda and K. J. Ishii, Int. Immunol., 2016, 28(7), 329–338 CrossRef CAS PubMed.
- K. R. Kalli, M. S. Block, P. M. Kasi, C. L. Erskine, T. J. Hobday, A. Dietz, D. Padley, M. P. Gustafson, B. Shreeder, D. Puglisi-Knutson, D. W. Visscher, T. K. Mangskau, G. Wilson and K. L. Knutson, Clin. Cancer Res., 2018, 24(13), 3014–3025 CrossRef CAS PubMed.
- E. A. Mittendorf, J. P. Holmes, S. Ponniah and G. E. Peoples, Immunotherapy, 2008, 1511–1521 CAS.
- A. A. Ali, C. M. McCrudden, J. McCaffrey, J. W. McBride, G. Cole, N. J. Dunne, T. Robson, A. Kissenpfennig, R. F. Donnelly and H. O. McCarthy, Nanomedicine, 2017, 13(3), 921–932 CrossRef CAS PubMed.
- T. A. Brown, K. Byrd, T. J. Vreeland, G. T. Clifton, D. O. Jackson, D. F. Hale, G. S. Herbert, J. W. Myers, J. M. Greene, J. S. Berry, J. Martin, J. C. Elkas, T. P. Conrads, K. M. Darcy, C. A. Hamilton, G. L. Maxwel and G. E. Peoples, Cancer Med., 2019, 8(10), 4678–4687 CrossRef CAS PubMed.
- D. L. Cecil, J. B. Liao, Y. Dang, A. L. Coveler, A. Kask, Y. Yang, J. S. Childs, D. M. Higgins and M. L. Disis, Clin. Cancer Res., 2021, 27(23), 6405–6412 CrossRef CAS PubMed.
- J. Lim, J. Kang, H. H. Park, J. K. Choi, J. H. Choi, S. Y. Jang, M. A. Kim, M. K. Park, M. L. Disis, E. Joung, A. Y. Kim and H. Jung, Cancer Res., 2024, 84(6), 4114 CrossRef.
- H. Shin and J. B. Liao, A Randomized Phase 2 Study to Evaluate the Efficacy and Safety for Adjuvant Therapeutic Cancer Vaccine (AST-201, pUMVC3-hIGFBP-2) in Patients With Advanced Ovarian Cancer- NCT05794659, 2023 Search PubMed.
- S. Chow, J. S. Berek and O. Dorigo, Vaccines, 2020, 8, 1–18 Search PubMed.
- J. Habault and J. L. Poyet, Molecules, 2019, 24(5), 927–944 CrossRef PubMed.
- L. Feni and I. Neundorf, Adv. Exp. Med. Biol., 2017, 1030, 279–295 CrossRef CAS PubMed.
- G. Cole, J. McCaffrey, A. A. Ali, J. W. McBridE, C. M. McCrudden, E. M. Vincente-Perez, R. F. Donnelly and H. O. McCarthy, Hum. Vaccines Immunother., 2017, 13(1), 50–62 CrossRef PubMed.
- J. W. Lee and R. S. Foote, Methods Mol. Biol., 2009, 544, 547–557 CrossRef PubMed.
- H. O. McCarthy, J. McCaffrey, C. M. Mccrudden, A. Zholobenko, A. A. Ali, J. W. McBride, A. S. Massey, S. Pentlavalli, K. H. Chen, G. Cole, S. P. Loughran, N. J. Dunne, R. F. Donnelly, V. L. Kett and T. Robson, J. Controlled Release, 2014, 189, 141–149 CrossRef CAS PubMed.
- G. Dranoff, Immunol. Rev., 2002, 188, 147–154 CrossRef CAS PubMed.
- A. Kumar, A. Taghi Khani, A. Sanchez Ortiz and S. Swaminathan, Front. Immunol., 2022, 13, 901277 CrossRef CAS PubMed.
- H. L. Kaufman, C. E. Ruby, T. Hughes and C. L. Slingluff, J. ImmunoTher. Cancer, 2014, 2, 11 CrossRef PubMed.
- M. A. Perales, J. Yuan, S. Powel, H. F. Gallardo, T. S. Rasalan, C. Gonzalez, G. Manukian, J. Wang, Y. Zhang, P. B. Chapman, S. E. Krown, P. O. Livingston, S. Ejadi, K. S. Panageas, M. E. Engelhorn, S. L. Terzulli, A. N. Houghton and J. D. Wolchok, Mol. Ther., 2008, 16(12), 2022–2029 CrossRef CAS PubMed.
- J. P. Beirne, D. G. McArt, A. Roddy, C. McDermott, J. Ferris, N. E. Buckley, P. Coulter, N. McCabe, S. L. Eddie, P. D. Dunne, P. O'Reilly, A. Gilmore, L. Feeney, D. L. Ewing, R. I. Drapkin, M. Salto-Tellez, R. D. Kennedy, I. J. G. Harley, W. G. McCluggage and P. B. Mullan, Gynecol. Oncol., 2019, 155(2), 305–317 CrossRef CAS PubMed.
- Y. Yamamoto, G. Ning, B. E. Howitt, K. Mehra, L. Wu, X. Wang, Y. Hong, F. Kern, T. S. Wei, T. Zhang, N. Nagarajan, D. Basuli, S. Torti, M. Brewer, M. Choolani, F. McKeon, C. P. Crum and W. Xian, J. Pathol., 2016, 238(4), 519–530 CrossRef CAS PubMed.
- A. A. Tone, H. Begley, M. Sharma, J. Murphy, B. Rosen, T. J. Brown and P. A. Shaw, Clin. Cancer Res., 2008, 14(13), 4067–4078 CrossRef CAS PubMed.
- H. Ikeda, B. Lethé, F. Lehmann, N. van Baren, J. F. Baurain, C. de Smet, H. Chambost, M. Vitale, A. Moretta, T. Boon and P. G. Coulie, Immunity, 1997, 6(2), 199–208 CrossRef CAS PubMed.
- W. Zhang, C. J. Barger, K. H. Eng, D. Klinkebiel, P. A. Link, A. Omilian, W. Bshara, K. Odunsi and A. R. Karpf, Oncotarget, 2016, 7(29), 45352–45369 CrossRef PubMed.
- M. Griffioen, J. H. Kessler, M. Borghi, R. A. van Soest, C. E. van der Minne, J. Nouta, S. H. van der Burg, J. P. Medema, P. I. Schrier, J. H. Falkenburg, S. Osanto and C. J. Melief, Clin. Cancer Res., 2006, 12(10), 3130–3136 CrossRef CAS PubMed.
- D. Pankov, L. Sjöström, T. Kalidindi, S. G. Lee, K. Sjöström, R. Gardner, M. R. McDevitt, R. O'Reilly, D. L. J. Thorek, S. M. Larson, D. Veach and D. Ulmert, Oncotarget, 2017, 8(39), 65917–65913 CrossRef PubMed.
- M. T. Epping, L. Wang, M. J. Edel, L. Carlée, M. Hernandez and R. Bernards, Cell, 2005, 122(6), 835–847 CrossRef CAS PubMed.
- H. O. McCarthy, J. McCaffrey, C. M. Mccrudden, A. Zholobenko, A. A. Ali, J. W. McBride, A. S. Massey, S. Pentlavalli, K. H. Chen, G. Cole, S. P. Loughran, N. J. Dunne, R. F. Donnelly, V. L. Kett and T. Robson, J. Controlled Release, 2014, 189, 141–149 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3(3), 145–150 CrossRef CAS PubMed.
- T. G. F. Souza, V. S. T. Ciminelli and N. D. S. Mohallem, J. Phys.: Conf. Ser., 2016, 733, 012039 CrossRef.
- A. K. Jain, A. Massey, H. Yusuf, D. M. McDonald, H. O. McCarthy and V. L. Kett, Int. J. Nanomed., 2015, 10, 7183–7196 CrossRef CAS PubMed.
- W. R. Bauer, F. H. C. Crick and J. H. White, Sci. Am., 1980, 243(1), 118–133 CAS.
- G. Cole, A. A. Ali, E. Mcerlean, E. J. Mulholland, A. Short, C. M. Mccrudden, J. McCaffrey, T. Robson, V. L. Kett, J. A. Coulter, N. J. Dunne, R. F. Donnelly and H. O. McCarthy, Acta Biomater., 2019, 96, 480–490 CrossRef CAS PubMed.
- G. Cole, A. A. Ali, C. M. Mccrudden, J. W. McBride, J. McCaffrey, T. Robson, V. L. Kett, N. J. Dunne, R. F. Donnelly and H. O. McCarthy, Eur. J. Pharm. Biopharm., 2018, 127, 288–297 CrossRef CAS PubMed.
- L. Kudsiova, A. Lansley, G. Scutt, M. Allen, L. Bowler, S. Williams, S. Lippett, S. Stafford, M. Tarzi, M. Cross and M. Okorie, BMJ Open Sci., 2021, 5(1), e100203 Search PubMed.
- ISO 10993-5:2009(en), Biological evaluation of medical devices—Part 5: Tests for in vitro cytotoxicity. Available from: https://www.iso.org/obp/ui/#iso:std:iso:10993:-5:ed-3:v1:en.
- A. S. Massey, S. Pentlavalli, R. Cunningham, C. M. McCrudden, E. M. McErlean, P. Redpath, A. A. Ali, S. Annett, J. W. McBride, J. McCaffrey, T. Robson, M. E. Migaud and H. O. McCarthy, Mol. Pharm., 2016, 13(4), 1217–1228 CrossRef CAS PubMed.
- R. Kushwah and J. Hu, Immunology, 2011, 133(4), 409–419 CrossRef CAS PubMed.
- J. Fan, S. Jin, L. Gilmartin, I. Toth, W. M. Hussein and R. J. Stephenson, Vaccines, 2022, 10(7), 1120 CrossRef CAS PubMed.
- S. Awate, L. A. Babiuk and G. Mutwiri, Front. Immunol., 2013, 4, 114 Search PubMed.
- S. Hutchison, R. A. Benson, V. B. Gibson, A. H. Pollock, P. Garside and J. M. Brewer, FASEB J., 2012, 26(3), 1272–1279 CrossRef CAS PubMed.
- M. Berard and D. F. Tough, Immunology, 2002, 106, 127–138 CrossRef CAS PubMed.
- M. L. Disis, W. C. Watt and D. L. Cecil, OncoImmunology, 2014, 3(9), e954971 CrossRef PubMed.
- T. Nishimura, K. Iwakabe, M. Sekimoto, Y. Ohmi, T. Yahata, M. Nakui, T. Sato, S. Habu, H. Tashiro, M. Sato and A. Ohta, J. Exp. Med., 1999, 190(5), 617–628 CrossRef CAS PubMed.
- C. Gérard, N. Baudson, T. Ory, L. Segal and J. Louahed, J. Immunother., 2015, 38(8), 311–320 CrossRef PubMed.
- D. C. Connolly, R. Bao and A. Y. Nikitin, et al. , Cancer Res., 2003, 63, 1389–1397 CAS.
- R. Pieretti-Vanmarcke, P. K. Donahoe, P. Szotek, T. Manganaro, M. K. Lorenzen, J. Lorenzen, D. C. Connolly, E. F. Halpern and D. T. MacLaughlin, Clin. Cancer Res., 2006, 12(5), 1593–1598 CrossRef CAS PubMed.
- B. A. Quinn, F. Xiao, L. Bickel, L. Martin, X. Hua, A. Klein-Szanto and D. C. Connolly, J. Ovarian Res., 2010, 3(1), 24 CrossRef PubMed.
- A. Le Naour, A. Rossary and M. P. Vasson, Cancer Med., 2020, 9(21), 8074–8085 CrossRef PubMed.
- R. G. Hoover, G. Gullickson and J. Kornbluth, Front. Immunol., 2012, 3, 393 Search PubMed.
- A. Ewens, E. Mihich and M. J. Ehrke, Anticancer Res., 2005, 25, 3905–3916 Search PubMed.
- W. Zhang, L. Li, L. Cai, Y. Liang, J. Xu, Y. Liu, L. Zhou, C. Ding, Y. Zhang, H. Zhao, J. Qin, Z. Shao, W. Wei and L. Jia, Cell Death Differ., 2021, 28(6), 1926–1940 CrossRef CAS PubMed.
- Y. G. Man, A. Stojadinovic, J. Mason, I. Avital, A. Bilchik, B. Bruecher, M. Protic, A. Nissan, M. Izadjoo, X. Zhang and A. Jewett, J. Cancer, 2013, 4(1), 84–95 CrossRef PubMed.
- Y. Yang, Y. Che, Y. Zhao and X. Wang, Int. Immunopharmacol., 2019, 69, 279–288 CrossRef CAS PubMed.
- M. Chiriva-Internati, Y. Yu, L. Mirandola, M. R. Jenkins, C. Chapman, M. Cannon, E. Cobos and W. M. Kast, PLoS One, 2010, 5(5), e10471 CrossRef PubMed.
- A. Kumar, A. Taghi Khani, A. Sanchez Ortiz and S. Swaminathan, Front. Immunol., 2022, 13, 901277 CrossRef CAS PubMed.
- V. K. Udhayakumar, A. De Beuckelaer, J. McCaffrey, C. M. McCrudden, J. L. Kirschman, D. Vanover, L. Van Hoecke, K. Roose, K. Deswarte, B. G. De Geest, S. Lienenklaus, P. J. Santangelo, J. Grooten, H. O. McCarthy and S. De Koker, Adv. Healthc. Mater., 2017, 6(13), 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01696c |
| This journal is © The Royal Society of Chemistry 2025 |