
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntimicrobial and anti-inflammatory effects of polyethyleneimine-modified polydopamine nanoparticles on a burn-injured skin model†
Sadman
Sakib‡
 a,
Nesha May O.
Andoy‡
b,
Jessica Y. C.
Yang
b,
Anna
Galang
bc,
Ruby May A.
Sullan
*bc and
Shan
Zou
*a
a,
Nesha May O.
Andoy‡
b,
Jessica Y. C.
Yang
b,
Anna
Galang
bc,
Ruby May A.
Sullan
*bc and
Shan
Zou
*a
aMetrology Research Centre, National Research Council Canada, 100 Sussex Drive, Ottawa, Ontario K1N 5A2, Canada. E-mail: shan.zou@nrc-cnrc.gc.ca
bDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, 1065 Military Trail, Toronto, ON, M1C 1A4 Canada
cDepartment of Chemistry, University of Toronto, 80 St. George St., Toronto, ON M5S 3H6, Canada. E-mail: ruby.sullan@utoronto.ca
First published on 20th February 2025
Abstract
Chronic infections involving bacterial biofilms pose significant treatment challenges due to the resilience of biofilms against existing antimicrobials. Here, we introduce a nanomaterial-based platform for treating Staphylococcus epidermidis biofilms, both in isolation and within a biofilm-infected burn skin model. Our approach leverages biocompatible and photothermal polydopamine nanoparticles (PDNP), functionalized with branched polyethyleneimine (PEI) and loaded with the antibiotic rifampicin, to target bacteria dwelling within biofilms. A key innovation of our method is its ability to not only target planktonic S. epidermidis but also effectively tackle biofilm-embedded bacteria. We demonstrated that PDNP–PEI interacts effectively with the bacterial surface, facilitating laser-activated photothermal eradication of planktonic S. epidermidis. In a 3D skin burn injury model, PDNP–PEI demonstrates anti-inflammatory and reactive oxygen species (ROS)-scavenging effects, reducing inflammatory cytokine levels and promoting healing. The rifampicin-loaded PDNP–PEI (PDNP–PEI–Rif) platform further shows significant efficacy against bacteria inside biofilms. The PDNP–PEI–Rif retained its immunomodulatory activity and efficiently eradicated biofilms grown on our burn-injured 3D skin model, effectively addressing the challenges of biofilm-related infections. This achievement marks a significant advancement in infection management, with the potential for a transformative impact on clinical practice.
Introduction
With the rise of multidrug-resistant bacteria, developing improved antibacterial strategies to treat challenging infectious diseases is crucial. Common microbial resistance mechanisms include reduced drug absorption, increased efflux pump activity, and the emergence of resistance genes.1 However, the formation of biofilms—protective communities of bacterial cells encased in a matrix of polysaccharides and extracellular DNA—also enhances bacterial virulence.2 Bacteria within biofilms can tolerate significantly higher doses of antibiotics than their planktonic counterparts.3 Due to their inherent resistance to antimicrobial agents, bacterial biofilms contribute to chronic infections through persistent inflammation and tissue damage.4Bacterial biofilms such as those found in chronic wounds significantly impede healing and occur in 80% of chronic wounds, compared to only 6% in acute wounds.5,6 While acute wounds progress through stages of inflammation, proliferation, and maturation,4 chronic wounds often stall in the inflammation stage due to persistent biofilm formation.6 Therefore, chronic wounds such as burns, diabetic foot ulcers and pressure injuries pose significant public health and economic challenges.7,8 Eradicating biofilms in chronic wounds is challenging due to their ability to inhibit proinflammatory cytokine production and growth factor assembly essential for wound healing. These survival strategies, influenced by the host environment, render biofilm-related diseases highly persistent and transiently responsive to antimicrobial therapy.9,10 Common pathogens like Staphylococcus aureus and Pseudomonas aeruginosa, prevalent in chronic leg ulcers, exhibit resistance to antibiotics, contributing to resistance development.11,12 Furthermore, polymicrobial infections in wounds alter virulence, delay healing, and affect the susceptibility of biofilms to antimicrobials.13 Burn wounds, in particular, are highly susceptible to biofilm formation due to the extensive tissue damage and the nutrient-rich environment that promotes bacterial growth.14 The presence of biofilms in burn injuries delays the healing process and increases the risk of systemic infections and sepsis.15,16 Addressing biofilm-related complications in burn wounds is, therefore, crucial for enhancing healing outcomes and reducing morbidity and mortality associated with severe burn injuries. However, the resilience of biofilms and their complex defense mechanisms make them difficult to eradicate with conventional treatments. It is thus critical to create novel antibacterial strategies and treatments that target the numerous defense mechanisms of biofilms.
Nanomaterial-based therapeutics offer a versatile approach to combating biofilm-dwelling bacteria by leveraging the unique physicochemical properties of nanomaterials to enhance treatment efficacy.17 These materials can carry out various bactericidal processes, making it difficult for bacteria to adapt to treatments.18 Among these nanomaterials, polydopamine films and polydopamine nanoparticles (PDNPs) have gained recognition in biomedical and materials applications due to their excellent qualities.19–21 Inspired by the adhesive properties of mussels, these nanomaterials are among the few that exhibit intrinsic biocompatibility and biodegradability.6,20,22,23 Moreover, PDNPs have been shown to modulate different pro- and anti-inflammatory cytokines, which can help ameliorate inflammation at wound sites and promote healing.24,25 Additionally, PDNPs demonstrate potent free radical (e.g. reactive oxygen species (ROS)) scavenging ability that contributes to their anti-inflammatory effects.26–28 PDNPs also serve as efficient drug carriers because their catechol/quinone moieties can anchor molecules onto their surface via physical or chemical bonding.20,29 Furthermore, the photothermal activity of PDNPs has been applied in photothermal (PTT) and photodynamic (PDT) therapies for both anticancer and antimicrobial applications.20,29–31 This photothermal property has also been leveraged to control the release of drugs in various systems.32–35
The inherent negative surface charge of PDNPs poses a disadvantage when targeting bacteria and bacterial biofilms due to the high negative charge densities found on bacterial surfaces and matrices of biofilms.17,36 However, the presence of catechol groups on the surface of PDNPs allows for facile surface functionalization with –NH2-containing molecules, like polyethyleneimine (PEI), to make the surface of PDNPs positively charged.37 The high positive charge density on the branched PEI has been utilized for diverse biomedical applications, such as the delivery of nucleic acids for gene therapy38–40 and improved uptake of nanomaterials for testing their cytotoxicity.41 PEI also exhibits antimicrobial properties and acts as an adjuvant for other biocidal/bacteriostatic agents,42–44 and it has been used to disperse bacterial biofilms and enhance the susceptibility of drug-resistant biofilm-dwelling bacteria to antibiotics.45–49 PEI has also been used to decorate the surface of other nanomaterials to facilitate cell penetration.50,51 Our group recently demonstrated the use of branched PEI to coat the photothermally active PDNPs (PDNP–PEI) to target bacterial surfaces.52 While various nanoparticles such as antibacterial nanozymes, polymeric particles, metal particles and liposomes have been reported to facilitate drug delivery as well as bacterial and cancer cell elimination, PDNP–PEI may possess certain properties that can result in better health outcomes.53–55 Unlike enzyme mimicking metal based nanoparticles (e.g., iron oxide, zinc-based zeolitic-imidazolate-framework (ZIF-8)-derived carbon nanomaterials), which generate ROS for bacterial elimination but risk damaging host tissues, PDNP–PEI would mediate antimicrobial action via localized heating, ROS scavenging and PEI's cationic properties.54,56,57 Polymeric nanoparticles and liposomes function by primarily working as a drug delivery vector, whereas PDNP–PEI can intrinsically facilitate bacterial elimination, reduce inflammation and promote healing in addition to acting as a nanocarrier.53,58,59
In the current work, we combine the ability of branched PEI (bPEI) to target bacterial surfaces, along with PDNPs’ ability to deliver small molecule antibiotics and to locally generate heat via laser activation—to create PEI-coated PDNPs that target biofilms of S. epidermidis and eliminate biofilm-dwelling bacteria.
The unique structure of the skin, characterized by varying degrees of thickness, folds, and moisture content, as well as differences in temperature and pH, provides a unique microenvironment for bacterial growth.60–63 Skin cells such as epidermal keratinocytes, dermal fibroblasts and different skin appendages produce various factors that can either promote or prevent bacterial growth. For example, the hydrolysis of lipids in skin sebum lowers the pH of the skin (pH = 5.6), inhibiting bacterial growth.64 Moreover, human skin keratinocytes and fibroblasts have immune functions, releasing antimicrobial peptides upon interaction with microbes, which disrupt bacterial walls and can kill bacteria.65,66 As a result, bacteria grown on the skin may exhibit distinctive characteristics compared to those grown in vitro.64 Thus, in vitro culture of S. epidermidis in isolation, without the presence of a skin equivalent, may lack physiological relevance.67
Here, we have successfully generated a three-dimensional (3D) skin model of burn injury that closely recapitulates the composition, morphology, and burn injury phenotype of skin in vivo. This injury model effectively simulates the opportunistic biofilm infection of S. epidermidis. We assessed the compatibility of PEI-coated PDNPs (PDNP–PEI) and antibiotic loaded PDNP–PEIs (PDNP–PEI–Rif) in a 3D burn-injured skin model and determined the optimal working concentrations that eradicate biofilms while preserving the viability and functionality of the skin models. We also evaluated and established the anti-inflammatory properties of PDNP in both our PDNP–PEI and PDNP–PEI–Rif platforms, which may have the potential to aid the wound healing response.
Experimental
Polydopamine nanoparticles (PDNP): synthesis, surface functionalization and drug loading
PDNPs were synthesized by adding 600 μL of 100 mg mL−1 dopamine hydrochloride solution (Sigma-Aldrich) to 200 mL 10 mM Tris buffer pH 9, to make a final concentration of 0.3 mg mL−1 dopamine. The dopamine solution was then placed on a 60 rpm shaker for a 24 h incubation in the dark. The nanoparticles were then collected by centrifugation (Centrifuge Avanti J301, Beckman Coulter) at 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 1 h at 4 °C. Supernatant was discarded, and the pellet was washed 3× with 10 mM Bicine buffer pH 8.5. The solution of PDNPs was stored in Bicine buffer at 4 °C until use.
000g for 1 h at 4 °C. Supernatant was discarded, and the pellet was washed 3× with 10 mM Bicine buffer pH 8.5. The solution of PDNPs was stored in Bicine buffer at 4 °C until use.
Surface functionalization of PDNPs with bPEI was done by mixing 1% (w/v) 10 kDa bPEI with 1 mg mL−1 PDNPs in 10 mM Bicine buffer pH 9 with 0.1 M KCl (total volume is 1 mL). The solution was then covered with a foil and placed in a rotator at 20 rpm for 14–16 h. PEI-coated PDNPs were collected by centrifugation at 20800g for 30 min and washed 3× with 10 mM Bicine pH 7.5. PDNP–PEI stock was stored in 10 mM Bicine pH 7.5 at 4 °C until use.
A 30 mg mL−1 rifampicin stock solution was prepared by dissolving the antibiotic in dimethyl sulfoxide (DMSO). A 1 mg mL−1 rifampicin solution was then mixed with 1 mg mL−1 PDNP–PEI in 10 mM Bicine buffer pH 7.5 (i.e., the absorbance of PDNP–PEI solution at 800 nm was used to determine nanoparticle concentration). The solution was then placed in a rotator and mixed at 20 rpm overnight at RT. Drug loaded PDNP–PEI (i.e., PDNP–PEI–Rif) was collected via centrifugation at 20800g for 30 min and washed 3× with 10 mM Bicine buffer pH 7.5 to remove loosely bound antibiotics. To estimate the drug loading capacity of PDNP–PEI, UV-Vis absorption spectroscopy (Cary 60 UV–Vis, Agilent Technologies) was done on (i) the rifampicin solution added to PDNP–PEI (Initial Rif) and (ii) the supernatant after PDNP–PEI–Rif was collected (final Rif), as well as on (iii) the solutions obtained after the three washes (wash1, wash2, and wash3). The concentration of rifampicin was estimated from the absorbance at 475 nm. The drug loading capacity was calculated using:
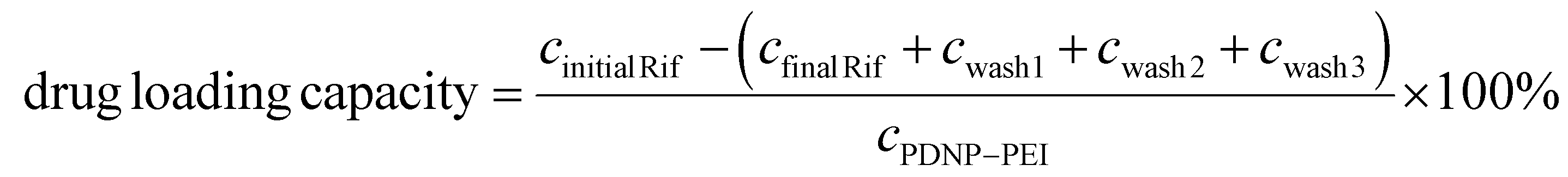
Characterization of nanoparticles
The size of nanoparticles (before and after surface modification) was monitored using DLS (NanoBrook Omni, Brookhaven Instruments). The surface zeta potential was determined using PALS (NanoBrook Omni, Brookhaven Instruments). Dried samples of the nanoparticles were further characterized using a Fourier transform infrared spectrometer (Bruker Alpha-P FTIR). To visualize the nanoparticles, both TEM (Hitachi H7500) and AFM (Nanowizard 4, JPK Instruments) were used. For TEM imaging, a freshly UV-O3-treated copper grid was placed on top of a drop of aqueous solution of PDNP–PEI and further incubated for 5 min before drying and imaging.For the photothermal characterization of PDNP–PEI solution, a 490 mW CW 808 nm laser with a diameter ≈3 mm (MDL-III-808, CNI Laser, China) was used to irradiate 50 μL aqueous solutions of PDNP–PEI. A thermal imager (Compact Spot Finder IR camera Xi 80, Optris, Germany) was used to determine the solution temperature during laser irradiation.
Bacterial strains and growth conditions
The bacterial strain of Staphylococcus epidermidis (S. epidermidis, 35984) was purchased from American Type Culture Collection (ATCC). Cryostocks were preserved in 20% glycerol at −80 °C. One colony from a bacterial plate was cultured in 5 mL of tryptic soy broth (TSB, Millipore) at 37 °C overnight.PDNP–PEI and PDNP–PEI–Rif treatment of S. epidermidis
Overnight liquid cultures of S. epidermidis were washed with 0.85% NaCl, then 180 μL OD = 0.01 were mixed with increasing concentrations of PDNP–PEI (0, 25, 50, 100, 200 μg ml−1) in 10 mM Bicine pH 7.5. After 30 min incubation at RT, the bacteria–nanoparticle mixture (50 μL) was treated with a CW 808 nm laser (7 W cm−2) for 30 min. Colony forming units (CFU) quantification was then done on tryptic soy agar (TSA) plates using 10 μL of the treated sample and subsequent 10-fold dilutions.For AFM characterization, bacteria solutions with and without PDNP–PEI were filtered using a PEI-coated polycarbonate membrane filter (0.1 μm). 0.85% NaCl solution was then used to wash the membrane to remove unbound bacteria and nanoparticles. The membrane immobilized bacteria were then fixed with 2.5% glutaraldehyde in PBS for 2 h and then washed with 0.85% NaCl. The quantitative imaging (QI) modality of AFM (JPK Nanowizard 4) was used so bacteria and nanoparticle-bound bacteria could be imaged without needing to dry the sample. The following parameters were used in QI imaging using SNL cantilevers (nominal spring constants 0.28–0.35 N m−1, Bruker): 1 nN relative force setpoint, z-range of 1000 nm, cantilever speed of 100 μm s−1. AFM topography and elasticity data were analyzed using JPK Image Processing software.
For the drug loaded nanoparticles, S. epidermidis (OD = 0.01) were mixed with PDNP–PEI–Rif (0, 1, 5, 25, 50 μg ml−1) in 10 mM Bicine pH 7.5 for 10 min. The nanoparticle–bacteria solution was then centrifuged and washed to remove unbound nanoparticles. The nanoparticle treated bacteria was then added to TSB and allowed to grow for 18 h at 37 °C to evaluate the antimicrobial activity of PDNP–PEI–Rif.
The ability of PDNP–PEI–Rif to prevent biofilm formation was also evaluated and compared to rifampicin only. S. epidermidis (OD = 0.01) were mixed with PDNP–PEI–Rif (0, 1, 5, 25, 50 μg ml−1) and rifampicin (0.001, 0.01, 0.1, 1, 10 μg ml−1) in 10 mM Bicine pH 7.5 for 10 min. The treated bacteria solution was then filtered using 0.8 μm PC membrane and washed to remove the unbound drugs and nanoparticles. The PC membranes were then placed on TSA plates and incubated at 37 °C to form colony biofilms. After 20 h of growth, the biofilm that formed on the PC membrane was quantified using crystal violet (CV) staining.
Cell lines and cell culture
Primary human dermal fibroblast cells (HDFCs) (PCS-201-010, ATCC) and human epidermal keratinocytes (HEdKs) (PCS-200-010, ATCC) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and in Dermal Cell Basal Medium (PCS-200-030, ATCC) supplemented with Keratinocyte Growth Kit (PCS-200-030, ATCC), respectively. Human monocytes (THP1) (TIB-202, ATCC) were cultured in RPMI-1640 medium (ATCC 30-2001) supplemented with 10% fetal bovine serum (FBS) and 0.05 mM 2-mercaptoethanol. All cell cultures were carried out in a humidified incubator with 5% CO2 at 37 °C and maintained at low passage numbers (<10).Generation of 3D skin model
3D skin models were generated as previously described with minor modifications.68 1 mL of an ice-cold acellular matrix pre-gel solution composed of 10% FBS, 1.48 mM L-glutamine, 0.21 mg mL−1 sodium bicarbonate (Thermofisher), and 0.84 mg mL−1 rat tail collagen (Gibco A1048301) were cast onto a 6-well 3 μm PET transwell insert (Corning 353091) and allowed to gel for 10 min in a 37 °C incubator. Then, a suspension of 450 × 103 HDFCs and 50 × 103 THP1 monocytes in 2 mL of the same pre-gel solution was dispensed on top of the acellular matrix. The cellular layer was then allowed to gel at 37 °C. After 2 h, HDFC media was added to the apical and basal chambers, and the cells were cultured for 24 h to allow the collagen gel to contract. Next, media from both chambers were removed and approximately 500 × 103 HEdKs resuspended in 50 μL of epidermalization media 1 (EPM1) were seeded on top of the collagen matrix.68 The plates were incubated in a humidified cell culture incubator for 2–3 h to allow the HEdKs to attach to the collagen matrix, and then EPM1 media was added to both the apical and basal wells. After 48 h of incubation, the media from the basal chamber was replaced with epidermalization media 2 (EPM2).68 In contrast, the media in the apical chamber was removed to create an air–liquid interphase (ALI) culture. The HEdKs were allowed to differentiate for 14 days with complete media changes every 48 h in a dehumidified incubator at 37 °C.Cytotoxicity of PDNP-PEI on the skin model
The skin model was treated with 12.5, 25, and 50 μg mL−1 of PDNP–PEI. Samples with no treatments were used as controls. After 48 and 72 h of treatment, the skins were harvested to prepare frozen sections (10–12 μm) for staining with Click-iT plus TUNEL Assay Kit with Alexa Flour 647. The number of apoptotic cells (TUNEL+ve and DAPI+ve cells) and total cells (DAPI+ve) was quantified to determine the percentage (%) of TUNEL+ve apoptotic cells.Generation of 3D skin model of burn injury
Skin models were cultured for 14 days, and then burn injury was inducted by placing a metal rod (2 mm diameter), which was heated to 90 °C on the epidermal layer for approximately 10 s. Care was taken to let the metal rod rest on the skin model without the application of additional pressure. The control group was treated in the same fashion with an RT metal rod.Formation of biofilm on skin models
Overnight suspension culture of S. epidermidis was performed in TSB media and approximately 5 μL of the bacterial suspension (OD = 0.475 ± 0.009) was dispensed on the burned epidermal layer. The bacteria were then allowed to form a biofilm in a 37 °C incubator for 16 h.Treatment of biofilms grown on skin models
Biofilms grown on burned skin models were treated with 25 and 50 μg mL−1 of PDNP–PEI and 25 μg mL−1 of PDNP–PEI–Rif. The treatment was carried out for 2 h. The skin samples were then washed with PBS to remove any unbound particles and irradiated with laser (808 nm CW, 490 mW) for 30 min and placed into culture for another 24 h to facilitate bacterial removal. The quantification of bacterial load was performed via CFU calculation and RT-qPCR, as described in the following sections.Immunohistology analysis of 2D and 3D skin
The morphology of skin, skin burn model, and biofilm was characterized using immunohistochemistry. Frozen sections (10–12 μm) were prepared and blocked with 10% goat serum. The sections were then incubated overnight at 4 °C with anti-CK14, anti-S. epidermidis, anti-E-cadherin, anti-CD14, anti-CD163, anti-CD68, and anti-Vimentin primary antibodies. Fluorescence labelling was performed with secondary antibodies conjugated with Alexa Fluor 488 and 555. Hoechst was used for labelling the nuclei. Imaging was performed using laser scanning confocal microscopy.Reverse transcription quantitative polymerase chain reaction
Relative quantification of bacterial load was assessed via reverse transcription quantitative polymerase chain reaction (RT-qPCR) for expression of S. epidermidis gene. Skin models with biofilms were harvested, and DNA was isolated using a QIAamp DNA Kit (Qiagen 56304) and RT-qPCR was carried out with a Microbial DNA qPCR Assay Kit specific for S. epidermidis (Qiagen BBID00316AR) as per the manufacturer's instructions. mRNA expression of pro- and anti-inflammatory factors was determined via RT-qPCR using QuantiTect Primer Assays (Qiagen) for CD68, CD163, IL6, TNFA, IL1B, MMP9, TGFB and IL10. RNA from the skin models was isolated using RNeasy Mini Kit, and RT-qPCR was carried out with Power SYBR Green RNA-to-CT 1-Step Kit. The expression levels were presented relative to GAPDH. Statistical analysis was performed on the mean of ΔCt.Measurement of ROS
The ROS levels were measured using the ROS–Glo H2O2 assay (Promega G8820). Briefly, after the appropriate treatment, skin models were treated with H2O2 substrate for 6 h. Then, 50 μL of media was collected from each well and mixed with an equal volume of ROS–Glo detection solution in an opaque white plate. The plate was then incubated at room temperature for 20 min, and average luminescence, or relative luminescence units, were recorded using a plate reader.Calculation of colony forming units of bacteria grown on skin model
Skin models with bacterial biofilms (treatment and controls) were collected and homogenized with PBS by running them through a 20 g syringe. The number of colony forming units (CFU) was then quantified by adding 10 μL of the homogenized sample directly and subsequent 10-fold dilutions on TSA plates.Statistical analysis
All the results presented here are from at least three independent experiments. Data were analyzed using GraphPad Prism 8 software. Unpaired two-tailed t-tests were done for single comparisons between two groups. For more than two groups, one-way ANOVA with Tukey's multiple comparison tests were performed. The threshold for statistical significance was set at P < 0.05.Results and discussion
Polyethyleneimine (PEI) decorates polydopamine nanoparticles (PDNP–PEI) and imparts laser-activated antimicrobial activity
Branched polyethyleneimine (bPEI) can be immobilized onto polydopamine nanoparticles (PDNP) through the formation of an amine-catechol adduct, facilitated by Michael-type addition or Schiff-base formation between the primary amine groups on the bPEI and the catechol groups on the nanoparticle surface.37 Dynamic light scattering (DLS), phase analysis light scattering (PALS) (Fig. S1A†), transmission electron microscopy (TEM) (Fig. S1B†), and Fourier-transform infrared (FTIR) spectroscopy (Fig. S1C†) confirmed successful surface functionalization. These analyses showed an increase in hydrodynamic size from 85 ± 23 nm (PDNPs) to 205 ± 43 nm (PDNP–PEI) and a shift in zeta potential from −33 ± 4 mV to +31 ± 4 mV. FTIR spectroscopy also showed additional peaks corresponding to aliphatic ν(C–H) stretching modes at 2940 and 2850 cm−1.Similar to our previous observation of PDNPs decorated with bPEI binding to the surface of E. coli,52 PDNP functionalized with 10 kDa bPEI could efficiently target the surface of S. epidermidis (Fig. 1). Fig. 1A shows AFM images of S. epidermidis before (left panel) and after incubation with PDNP–PEI (right panel). The AFM image of S. epidermidis with PDNP–PEI distinctly shows tiny spherical bumps, which contrasts with the otherwise smooth surface of bare bacteria. This indicates that PDNP–PEI effectively binds to and decorates the surface of S. epidermidis, forming a nanoparticle coating. This coating extends over nearly the entire bacterial surface when incubated with a concentration of 25 μg mL−1 nanoparticles and ≈106 colony forming units (CFU) per mL (OD600 nm = 0.01), as can be seen in the right panel of Fig. 1A and Fig. S2.†
 | ||
| Fig. 1 Antimicrobial activity of PDNP–PEI against planktonic S. epidermidis. (A) Atomic force microscopy (AFM) topography images comparing S. epidermidis alone (left) and when incubated with PDNP–PEI (right). (B) Quantification of colony forming units (CFU) after 30 min incubation with PDNP–PEI, w/ and w/o laser. The limit of detection is 102 CFU mL−1. | ||
Given that bPEI has been shown to exhibit biocidal activity,46,69 we tested how PEI-coated PDNPs impact the viability of S. epidermidis after pre-exposure to increasing concentrations of PDNP–PEI. In Fig. 1B (without laser irradiation), PDNP–PEI exhibits slight toxicity to S. epidermidis due to its binding to the bacterial surface. This results in a decreasing number of CFU with increasing nanoparticle concentrations. For instance, when bacteria were pre-exposed to 200 μg mL−1 PDNP–PEI, a 200-fold reduction in CFUs was observed from 8 × 105 to 4 × 103. Moreover, as a photothermal nanomaterial, PDNP solutions can be heated when irradiated with a laser (Fig. S3†). Fig. 1B (with laser irradiation) shows that after a 30 min laser exposure, there was a 4000-fold decrease in CFUs (from 8 × 105 to 2 × 102) for bacteria pre-incubated with only 50 μg mL−1 PDNP–PEI. We posit that laser-induced heating of nanoparticles, particularly those bound directly to the bacterial surface, significantly contributes to the biocidal effects of PDNP–PEI, despite the lower nanoparticle concentration (i.e., 50 μg mL−1). At this concentration, laser-induced heating of the nanoparticles only increased the overall solution temperature from an initial 23 °C to 38 °C (Fig. S3†). Unlike at higher concentrations, where solution temperature reached 46 °C (200 μg mL−1) and 58 °C (200 μg mL−1), leading to complete bacterial viability loss, the temperature increase to 38 °C is insufficient to kill S. epidermidis by hyperthermia alone. This is evident from the fact that bacterial colony formation remained unaffected even after a 30 min heat treatment at 42 °C using a water bath (Fig. S4†). Our data, therefore, suggest that laser-induced heating of PDNP–PEI bound to the bacterial surface is the main factor in the enhanced killing of S. epidermidis. This is consistent with our previous results where antimicrobial peptide (AMP)-functionalized PDNP was effective in low temperature photothermal killing of a drug-resistant E. coli, showcasing the modularity and general applicability of this approach.70
PDNP–PEI showed time- and dose-dependent toxicity in a 3D skin model
Bacteria grown on conventional cell cultures may differ widely compared to those growing on human skin during chronic infections.64 To create a physiologically relevant model, 3D skin models composed of epidermal keratinocytes, dermal fibroblasts and THP1 monocytes were generated to mimic the native skin (Fig. 2A). Briefly, a collagen layer with fibroblasts and macrophages was cultured and then seeded with keratinocytes, which, under air–liquid interface culture, underwent differentiation and maturation. The extracellular matrix (ECM) protein collagen provides a structural scaffold for the cells to co-localize and migrate through, in order to generate a physiologically relevant skin microenvironment that supports keratinocyte stratification and macrophage polarization.64,71–74 After 14 days of culture, the keratinocytes formed striated epidermis-like structures (Fig. 2B). 3D skins were further characterized with immunohistochemistry. E-cadherin, a basal epithelial cell marker, was used to delineate the dermis and epidermis. CK14+ve keratinocytes were observed at the epidermal layer, while Vimentin+ve dermal fibroblasts and CD14+ve THP1 monocytes were in the dermal layer. This indicated the formation of a 3D skin model that bore similarities to in vivo skin (Fig. 2C). This skin model builds upon existing designs by prioritizing reproducibility, scalability, and clinical relevance. While traditional models primarily rely on keratinocytes alone75 or in combination with fibroblasts,76 our model integrates THP1-derived macrophages to incorporate innate immune responses, enabling a more comprehensive study of immune interactions within a physiologically relevant microenvironment. Recent advances in skin organoid models derived from pluripotent stem cells have demonstrated the ability to recapitulate developmental processes and generate skin appendages such as hair follicles, melanocytes, sebaceous glands, adipocytes, and sensory neurons.77 However, the generation of these organoids requires an intricate, multi-stage differentiation process spanning several months, involving precisely timed exposure to growth factors.77 These technical and temporal constraints limit their scalability and reproducibility. In contrast, our model utilizes primary human keratinocytes and fibroblasts, ensuring greater translational relevance while maintaining a practical, high-throughput approach for laboratory studies. | ||
| Fig. 2 Cultured keratinocytes, fibroblasts and monocytes organize into an organotypic 3D skin model, which displays time- and dose-dependent cytotoxicity in response to PDNP–PEI treatment. (A) Schematic representation of skin model generation. Generated using Biorender.com. (B) Images of the whole skin model (top panel) and a brightfield image of the surface of the skin model (bottom panel) after 14 days of culture. Scale bar = 360 μm. (C) Morphology of the skin model after 14 days of culture. Top panel shows a schematic representation of skin morphology (brown cells–keratinocytes, green cells–fibroblasts and purple cells–THP1 monocytes). Generated using Biorender.com. The middle and bottom panels show immunofluorescence imaging of the skin model. E-cadherin (red), CK14 (green), CD14 (red) and Vimentin (green) show the presence and organotypic localization of keratinocytes, dermal fibroblasts and monocytes. Hoechst (blue) is used as a nuclear stain. The white dashed line delineates the dermis from the epidermis. Scale bar = 100 μm. (D) Quantification of % of TUNEL+ve apoptotic cells in the skin model treated with PDNP–PEI. Blue bars represent 48 h and red bars represent 72 h treatments. Values on each graph are shown as mean ± SD of three independent experiments (n = 3). Statistical significance was determined with one-way ANOVA. P > 0.05 was considered not significant (ns). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
To evaluate the cytotoxicity of PDNP–PEI, the 3D skin models were then treated with 12.5, 25, and 50 μg mL−1 of PDNP–PEI for 48 and 72 h (Fig. 2D). For both time points, concentrations of 25 μg mL−1 and below showed no negative effects (20% for 48 h and 27% for 72 h treatments) on cell viability compared to their respective untreated controls (Fig. 2D). Treatment with the highest minimally cytotoxic concentration of 25 μg mL−1 led to 20% and 27% apoptotic cells after 48 h and 72 h exposure, respectively. In contrast, the higher dose of 50 μg mL−1 resulted in approximately 50% of the cells exhibiting elevated levels of apoptosis after 72 h (Fig. 2D).
PDNP–PEI shows anti-inflammatory and ROS scavenging effects on 3D skin model of burn injury
PDNPs exhibit potent ROS scavenging effects mediated by the abundant phenolic hydroxyl groups on their surface, which can attenuate oxidative damage and inflammation.25,59,78,79 Previous work has shown that PDNPs can reduce inflammation in acute lung injury,25 peritonitis,25 inflammatory bowel disease,59 and lipopolysaccharide (LPS)-induced murine model of depression.79 In order to validate the ROS scavenging properties and anti-inflammatory effects of our PDNP platform, we generated a burn wound skin model. This was achieved by placing a 2 mm metal rod, preheated to 90 °C, on 3D skin model for 10 s without applying any additional force (Fig. 3A). The induction of burn resulted in the disruption of CK14+ve keratinocytes and the accumulation of damaged DNA after 48 h, as illustrated by condensed Hoechst staining (in blue) on the epidermal side (Fig. 3B, top panel). The extent of the injury appeared to be localized primarily in the epidermal layer, as the dermal region, characterized by Vimentin+ve fibroblasts and CD14+ve THP1 cells, was largely unaffected (Fig. 3B, middle panel). The loss of epidermis at the site of thermal injury signifies compromised keratinocyte attachment and viability, mirroring the pathological features of a second-degree partial burn in native skin.80 Phuphanitcharoenkun et al. reported epidermal loss and detachment when skin equivalent models are exposed to ultraviolet radiation.81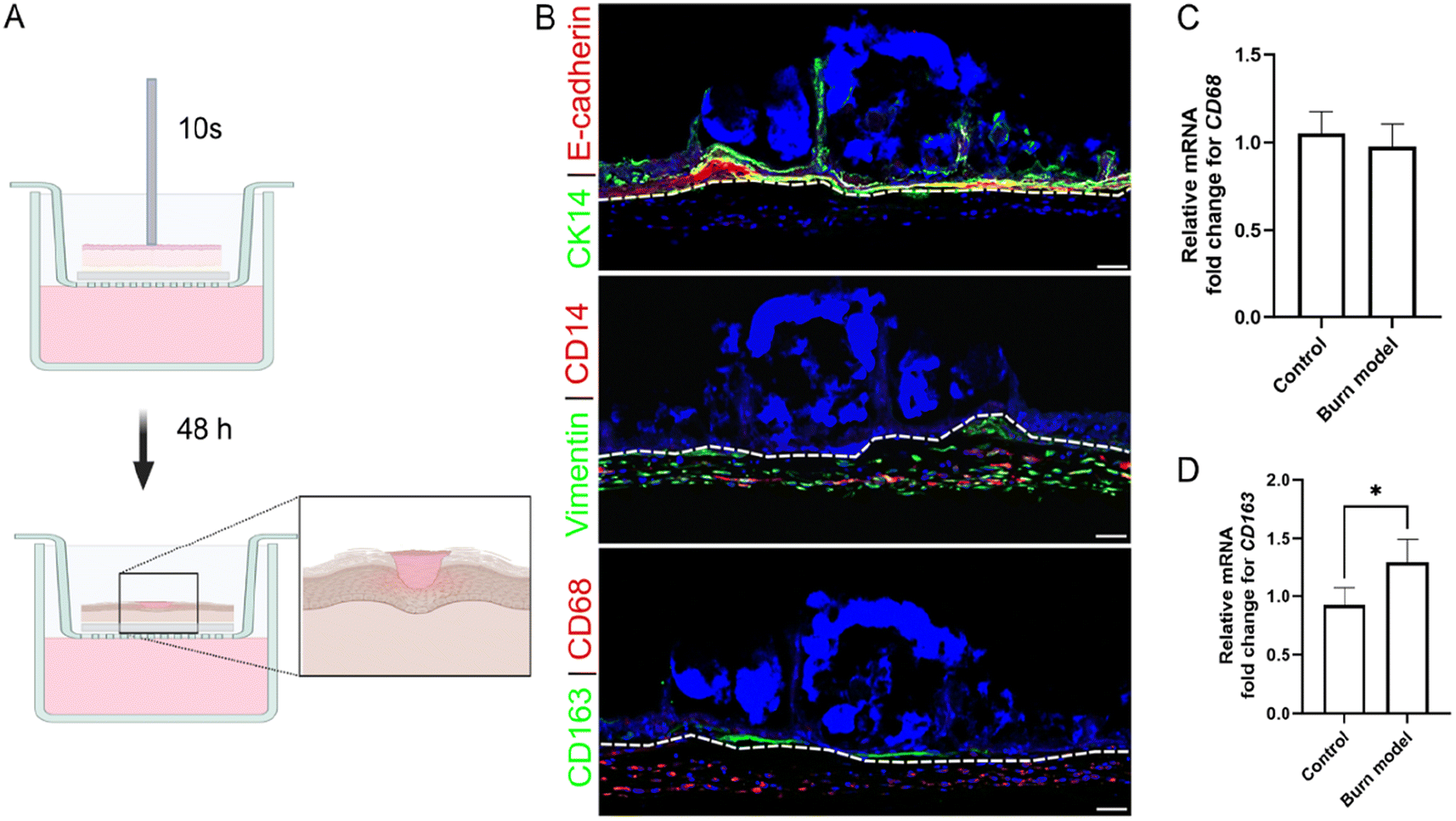 | ||
| Fig. 3 Induction of burn injury results in disruption of epidermis and differentiation of monocytes. (A) Schematic representation of skin model of burn injury generation. Generated using Biorender.com. (B) Immunofluorescence imaging of burn wound skin model 48 h post induction. Top panel, E-cadherin+ve keratinocytes (red) and white dash line delineate the epidermis and dermis; and CK14+ve keratinocytes (green) show disruption of epidermis. Middle panel, CD14+ve THP1 monocytes (red), Vimention+ve fibroblast (green) show dermal residents. Bottom panel, CD68 (red) and CD163 (green) show differentiated monocytes. Hoechst (blue) is used as a nuclear stain. Scale bar = 100 μm. (C and D) Relative fold change of mRNA expression for CD68 (C) and CD163 (D) 48 h after burn induction. Values on each graph are shown as mean ± SD of three independent experiments (n = 3). Statistical significance was determined with t test. P > 0.05 was considered not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
Since stimuli such as tissue damage or inflammatory cues in tumor microenvironment have been shown to induce differentiation of monocytes into macrophages, we evaluated the expression of CD68, a macrophage marker.82–84 CD68+ve macrophages were observed evenly distributed throughout the dermal compartment (Fig. 3B, bottom panel). No significant difference in the expression of CD68 gene was observed between the control and burn models (Fig. 3C), indicating THP1 monocytes in the controls were also undergoing differentiation into macrophages. This is likely driven by being embedded in the collagen matrices. In vivo, the differentiation of monocytes involves a transition from blood to tissue. Bhattacharya et al. demonstrated that a transition from fluid-like to gel-like physical environment (such as agarose, alginate, collagen and Matrigel) is sufficient to induce differentiation of THP1 cells, even in the absence of chemical inducers.85 Monocytes embedded in collagen show a marked downregulation of the monocyte markers and an upregulation of macrophage markers.86 Similar observations have been reported in 3D skin equivalent as well.81 In addition, a small population of CD163+ve macrophages were found localized around the site of injury (Fig. 3B, lower panel). CD163 is a marker of pro-healing macrophages. In response to injury or stimuli, monocytes can undergo differentiation into these macrophages, which help propagate the healing response.83,87
In order to evaluate the therapeutic effects of PDNP–PEI, the burn injury model was treated with 25 μg mL−1 PDNP–PEI for 7 days, and the level of ROS, expression of pro-inflammatory markers interleukin 6 (IL6),88,89 tumor necrosis factor alpha (TNFA),88 interleukin 1 beta (IL1B)90 and matrix metalloproteinase-9 (MMP9);91 and anti-inflammatory markers interleukin 10 (IL10) and transforming growth factor beta (TGFB)88 were measured. Compared to the control samples, the burn model showed higher expression (approximately 2-fold increase) of pro-inflammatory markers IL6, TNFA, IL1B and MMP9 (Fig. 4A–D). The gene expressions of pro-inflammatory markers IL6, TNFA and IL1B were downregulated by the treatment when compared against the untreated burn injury models (Fig. 4A–C), which is consistent with previous findings.92
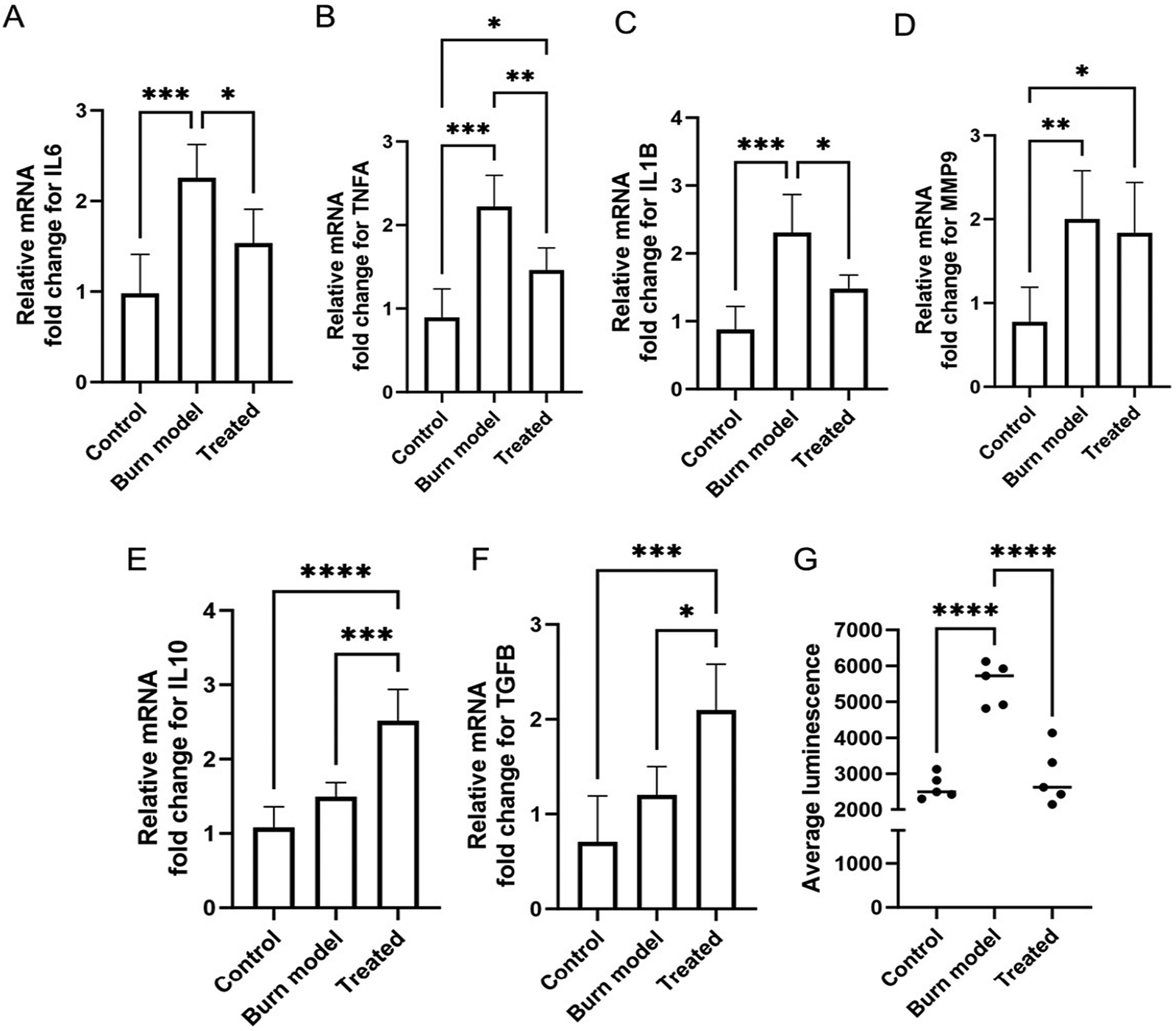 | ||
| Fig. 4 PDNP–PEI treatment results in elevated levels of anti-inflammatory factors and downregulation of ROS and pro-inflammatory factors. (A–F) Relative fold change of mRNA expression for IL6 (A), TNFA (B), IL1B (C), MMP9 (D), IL10 (E) and TGFB. (G) Quantification of ROS levels shown as average luminescence using ROS–Glo assay. Values on each graph are shown as mean ± SD of five independent experiments (n = 5). Statistical significance was determined with one-way ANOVA. P > 0.05 was considered not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
The expression of MMP9, however, was not affected by the PDNP–PEI treatment (Fig. 4D). MMP9 is a matrix metalloproteinase, and its primary role is to degrade extracellular matrix to aid in tissue remodelling and repair.91 MMP9 is locally and systemically increased in burn trauma and remains elevated until the progression of tissue repair and resolution of inflammation.91 It is likely that the degree of repair was insufficient during the short (7 days) PDNP–PEI treatment, and as a result, MMP9 levels remained unaffected.
The anti-inflammatory cytokine IL10 plays a critical role in wound repair by negatively downregulating pro-inflammatory cytokines TNFA, IL6 and IL1B.93,94 TGFB regulates inflammation and contributes to tissue remodelling. It has been shown to reduce the expression of TNFA expression in microglia95 and astrocytes.96 Thus, IL10 and TGFB serve as anti-inflammatory and pro-healing factors. Expression of both markers was upregulated after 7 days of treatment with PDNP–PEI compared to control and injured models. The expression of TGFB increased two-fold, while IL10 expression was upregulated by 2.5-fold relative to controls (Fig. 4E and F). This is in agreement with findings from Li et al., who reported a modulatory effect of PDNP on IL10 and TGFB expression.92
In addition, to assess the ROS-scavenging properties of the PDNP–PEI platform, H2O2 levels were measured. Thermal injuries often lead to tissue and cellular degradation, causing an overproduction of ROS that can drive oxidative damage and further propagation of inflammation.97,98 Polydopamine is a melanin mimicking substance that exhibits antioxidant properties due to its polyphenol structure.26,28 We found ROS levels increased by greater than two-fold after thermal injury but were efficiently downregulated by PDNP–PEI in our skin model, indicating that PDNP–PEI successfully retains the ROS-scavenging properties of PDNP (Fig. 4G).
Altogether, this indicates that our 3D skin burn model closely recapitulates the burn injury phenotype seen in vivo, which responds to treatment by the PDNP–PEI platform, as evidenced by the upregulation of pro-healing factors and downregulation of ROS and pro-inflammatory factors.
Antibiotic loading of PDNP–PEI retains anti-inflammatory activity and has enhanced anti-biofilm activity on 3D skin model
S. epidermidis is one of the most abundant commensal bacteria found on human skin, and can help promote defense against more harmful pathogens.99 However, it can also act opportunistically and contribute to infection and inflammation if the skin barrier integrity is compromised due to injuries such as burns.100 In order to test the antimicrobial effects of PDNP–PEI in the context of skin, the 3D skin model of burn injury was challenged with an overnight planktonic culture of S. epidermidis to promote bacterial adhesion and generate biofilm (Fig. 5A). These biofilms were then treated with 25 and 50 μg mL−1 PDNP–PEI and irradiated with laser (808 nm CW, 490 mW, 30 min) to test the antibiofilm activity. Although 50 μg mL−1 of PDNP–PEI had been shown to negatively impair cell viability in the 3D skin model, particularly after 72 h of exposure (Fig. 2D), we reasoned that skin model may still tolerate this concentration since the biofilm eradication treatment would require 2 h of exposure time and removal of any unbound particle via washes with phosphate buffered saline (PBS). Quantification of bacteria with both CFU counts and bacterial DNA load showed no observable difference between the control and the treatment with 25 μg mL−1 PDNP–PEI (Fig. S5B and C,†Fig. 5B–C). The treatment with 50 μg mL−1 PDNP–PEI showed a significant decrease in bacterial load (Fig. S5A–C†). This indicated that the biofilm on 3D skin models was susceptible to a low concentration of 50 μg mL−1 of PDNP–PEI, whereas no antibiofilm activity was observed when PDNP–PEI was used to treat S. epidermidis biofilms (without skin model), even at concentrations as high as 100 μg mL−1 (data not shown). This is likely mediated by the immune functions of skin cells and the THP1 macrophages, which contain pattern recognition receptors such as Toll-like receptors (TLRs), dectin-1 and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) that can facilitate the release of antimicrobial peptides such as cathelicidins, defensins, S100 proteins, ribonuclease 5 and 7, which can prevent bacterial colonization and elimination.66,101–103 These immune properties may render S. epidermidis more susceptible to lower concentrations of PDNP–PEI. However, degeneration of the skin model was observed upon extended culture (7 days) of skin models treated with 50 μg mL−1 PDNP–PEI and laser irradiation. In 3D culture systems, limited nutrient and oxygen diffusion challenges can lead to reduced viability and loss of integrity over time.104,105 However, no such degeneration and loss of structural integrity were observed in control skin models (untreated) or those treated with 25 μg mL−1 PDNP–PEI and irradiation when cultured for an equivalent amount of time. This indicated that it is the relatively high concentration of 50 μg mL−1 PDNP–PEI that is leading to loss of the skin models, and the removal of PDNP–PEI post 2 h exposure may be insufficient to counter the cytotoxicity of this (50 μg mL−1) concentration. Thus, to enhance PDNP–PEI with an additional biocidal mechanism, we loaded the nanoparticles with rifampicin, a highly effective antibiotic against S. epidermidis (Fig. S6†) and other Gram-positive bacteria.106,107 The total amount of rifampicin loaded into the nanoparticle yielded a drug capacity of 19 ± 9% (w/w) for PDNP–PEI (Fig. S7 and ESI section II† for detailed characterization of rifampicin-loaded PNP–PEI, i.e., PDNP–PEI–Rif).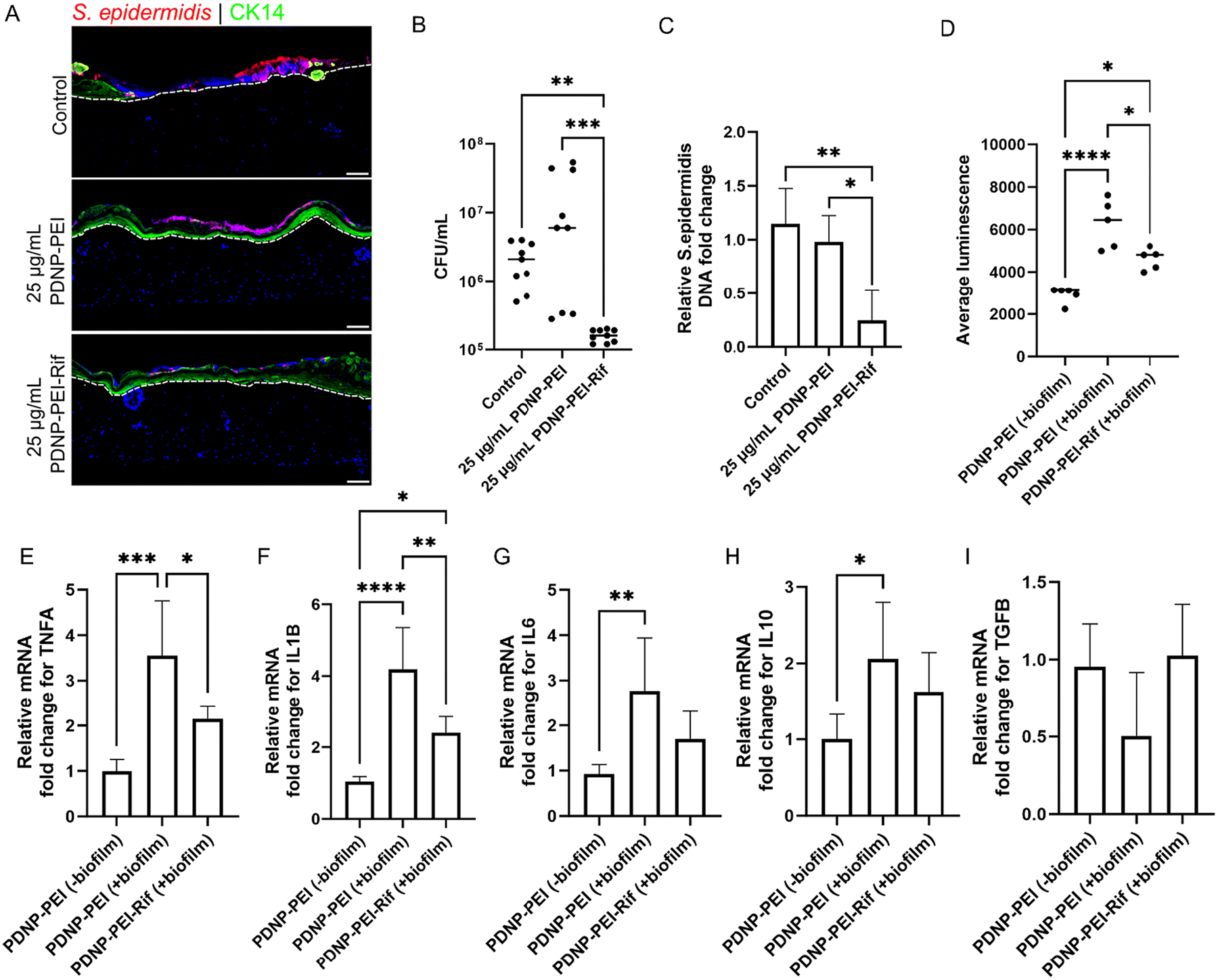 | ||
| Fig. 5 Loading of PDNP–PEI with antibiotic enhances its bactericidal activity and do not impair its immunomodulatory functions post laser assisted bacterial clearance. (A) Immunofluorescence imaging of biofilm shown using immunohistochemistry for S. epidermidis (red) on CK14+ve keratinocytes (green). Dotted lines delineate the dermis and epidermis. Hoechst (blue) is used as a nuclear stain. Scale bar = 100 μm. (B) Number of viable bacteria on skin after treatment with PDNP–PEI–Rif. (C) Relative fold change of S. epidermidis DNA expression after PDNP–PEI–Rif treatment. (D) Quantification of ROS levels shown as average luminescence using ROS–Glo assay 7 days post laser mediated bacterial clearance. (E–I) Relative fold change of mRNA expression for TNFA (E), IL1B (F), IL6 (G), IL10 (H) and TGFB (I) after laser-mediated bacterial clearance. Values on each graph are shown as mean ± SD of at least five independent experiments (n = 5). Statistical significance was determined with one-way ANOVA. P > 0.05 was considered not significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. | ||
Burn skin models were challenged with S. epidermidis and treated with the non-cytotoxic concentration of 25 μg mL−1 PDNP–PEI–Rif, which resulted in a significant reduction of bacterial load compared to PDNP–PEI alone at the same concentration (Fig. 5A–C). Absolute quantification with CFU counts showed around a hundred-fold reduction in the number of viable bacteria when treated with PDNP–PEI–Rif (Fig. 5B). Unsurprisingly, no difference in bacterial load was seen between the samples treated with PDNP–PEI and untreated controls (Fig. 5B and C). The improved bacterial clearance observed with PDNP–PEI–Rif is likely due to the combined antimicrobial activity of rifampicin and the photothermal properties of PDNP–PEI. Rifampicin inhibits bacterial transcription106 by binding to RNA polymerase, preventing RNA synthesis and halting bacterial proliferation. Meanwhile, PDNP–PEI converts near-infrared light to localized heat,108,109 compromising bacterial membrane integrity. This photothermal disruption increases cell wall permeability, which may facilitate greater intracellular rifampicin uptake, thereby amplifying its bactericidal efficacy. The dual-mode action of antibiotic interference and thermal damage creates a synergistic effect that leads to more effective bacterial eradication than either component alone.109 In addition, to evaluate if the loading of rifampicin impairs the anti-inflammatory effects of PDNP–PEI, the skin models were cultured post-laser irradiation and bacterial removal to assess ROS levels and the expression of pro- and anti-inflammatory effectors.
Three different groups were compared: (a) PDNP–PEI (−biofilm), burn models treated with PDNP–PEI without bacterial challenge; (b) PDNP–PEI (+biofilm), burn models treated with PDNP–PEI with bacterial challenge; and (c) PDNP–PEI–Rif (+biofilm), burn models treated with PDNP–PEI–Rif with bacterial challenge. Only the PDNP–PEI (−biofilm) and PDNP–PEI–Rif (+biofilm) were cultured for 7 days post-laser irradiation. Due to low bacterial clearance, PDNP–PEI (+biofilm) group was cultured for only 2 days post-laser irradiation. While ROS levels in both the PDNP–PEI–Rif (+biofilm) and PDNP–PEI (+biofilm) groups were higher than the PDNP–PEI (−biofilm), ROS levels were approximately 33% lower in PDNP–PEI–Rif (+biofilm) than in PDNP–PEI (+biofilm), indicating it retained ROS scavenging properties (Fig. 5D).
Macrophages have been reported to undergo an oxidative burst, which involves rapid oxygen consumption to produce large quantities of ROS when they encounter biofilms as part of their innate immune response.110 This likely accounts for the high levels of ROS seen in both PDNP–PEI (+biofilm) and PDNP–PEI–Rif (+biofilm) groups (Fig. 5D).
The expression of pro-inflammatory markers TNFA, IL1B and IL6 was unsurprisingly elevated (greater than two-fold) in the PDNP–PEI (+biofilm) group (Fig. 5E–G). However, except for IL6, both TNFA and IL1B expression was downregulated in the PDNP–PEI–Rif (+biofilm) groups, with TNFA showing greater than 1.5-fold and IL1B showing 2-fold downregulation (Fig. 5E–G). This is indicative of the anti-inflammatory activity of the PDNP–PEI–Rif (Fig. 5E–G). In the case of pro-healing factors, TGFB expression levels were largely unaffected between the three groups, with the PDNP–PEI (+biofilm) showing a slight trend towards reduced TGFB levels (Fig. 5I). The expression of IL10 was not significantly different between PDNP–PEI (−biofilm) and PDNP–PEI–Rif (+biofilm) (Fig. 5H). This indicates the IL10 modulatory effect of PDNP–PEI was intact post modification with rifampicin. However, IL10 gene expression was higher in the PDNP–PEI (+biofilm) compared to the control PDNP–PEI (−biofilm) (Fig. 5H). This may be explained by the presence of elevated levels of bacteria in this group. In mouse models of S. epidermidis biofilm infection of the central nervous system, it was found that IL10 cytokine levels tend to be increased in regions adjacent to the biofilm.111
Overall, our results demonstrate the adaptability of our 3D skin burn injury model for simulating infection mediated by S. epidermidis biofilm formation. This biofilm is highly susceptible to treatment with PDNP–PEI loaded with the antibiotic rifampicin. The combination of rifampicin and laser-induced localized heating of bacterial cells results in overall higher bacterial clearance. Importantly, the addition of rifampicin to the PDNP–PEI platform does not impair the ROS-scavenging and immunomodulatory effects of PDNP–PEI. These findings highlight the potential of our approach as a multifunctional therapeutic strategy for the treatment of biofilm-associated infections in burn injuries.
Conclusions
Polydopamine-based materials have emerged as promising agents for enhancing chronic wound treatments. Their inherent antioxidant properties can mitigate inflammation at wound sites and facilitate crucial cell migration necessary for wound healing. In this study, we present a PDNP-based platform for bactericidal and wound healing applications. By functionalizing PDNPs with the cationic polymer, branched polyethyleneimine, we achieved targeted binding to the surface of S. epidermidis. In addition, these bPEI-decorated PDNPs exhibited anti-inflammatory activity in biomimetic 3D model of skin burn injury. As an antibiotic nanocarrier, PDNP–PEI demonstrates a high loading capacity for rifampicin, efficiently eliminating both planktonic and biofilm-dwelling S. epidermidis. Addition of the antibiotic enabled laser-induced heating and eradication of biofilm on a skin model of S. epidermidis infection. It is worth noting that the 3D skin model used here, incorporating epidermal keratinocytes, dermal fibroblasts and THP1 monocyte-derived macrophages, provides greater biological relevance compared to monolayer cultures. Building upon our model, we can further enhance its complexity by incorporating skin appendages like hair follicles, vascular cells, and epidermal macrophages such as Langerhans cells.112 By integrating monocyte-derived cultured Langerhans-like cells or utilizing an ex vivo skin model with all relevant cells and appendages, we may create an even more biomimetic platform for testing nanotherapeutics in the future.Author contributions
R. M. A. S. and S. Z. were responsible for conceptualization; N. M. O. A., S. S., A. G. and J. Y. C. Y. were responsible for methodology; S. S. was responsible for skin models; N. M. O. A., A. G. and J. Y. C. Y. were responsible for nanomaterial synthesis and antimicrobial and antibiofilm evaluations; N. M. O. A., A. G., J. Y. C. Y. and S. S. were responsible for formal analysis and investigation; S. Z. and R. M. A. S. were responsible for resources; N. M. O. A., S. S., A. G. and J. Y. C. Y. were responsible for data curation; N. M. O. A., A. G. and S. S. were responsible for writing the original draft; S. Z. and R. M. A. S. were responsible for supervision, project administration and funding acquisition. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Drs. Jagdeep K. Sandhu and Qing Yan Liu from the Human Health Therapeutics Research Centre at the NRC Canada for facilitating immunohistology and gene expression analysis. We also would like to thank Drs. Durga Acharya and Bruno Chue of the CNS facility at UTSC for assistance in fluorescence and TEM imaging. This work was partially funded by the NRC New Beginning Ideation Funds.References
- W. C. Reygaert, AIMS Microbiol., 2018, 4, 482–501 CAS.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CrossRef CAS PubMed.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS PubMed.
- T. Bjarnsholt, APMIS, 2013, 121, 1–58 CrossRef PubMed.
- G. A. James, A. G. Zhao, M. Usui, R. A. Underwood, H. Nguyen, H. Beyenal, E. deLancey Pulcini, A. Agostinho Hunt, H. C. Bernstein, P. Fleckman, J. Olerud, K. S. Williamson, M. J. Franklin and P. S. Stewart, Wound Repair Regener., 2016, 24, 373–383 CrossRef PubMed.
- M. Malone, T. Bjarnsholt, A. J. McBain, G. A. James, P. Stoodley, D. Leaper, M. Tachi, G. Schultz, T. Swanson and R. D. Wolcott, J. Wound Care, 2017, 26, 20–25 CrossRef CAS PubMed.
- M. Olsson, K. Järbrink, U. Divakar, R. Bajpai, Z. Upton, A. Schmidtchen and J. Car, Wound Repair Regener., 2019, 27, 114–125 CrossRef PubMed.
- N. Kan, B. Tsend Ayush, L. Naiyou, W. S. Fagg, K. Ryuichiro, H. Thomas and E. Perenlei, Plast. Aesthetic. Res., 2024, 11, 54 Search PubMed.
- S. A. Eming, T. Krieg and J. M. Davidson, J. Invest. Dermatol., 2007, 127, 514–525 CrossRef CAS PubMed.
- R. G. Frykberg and J. Banks, Adv. Wound Care, 2015, 4, 560–582 CrossRef PubMed.
- H. C. Neu, Science, 1992, 257, 1064–1073 CrossRef CAS PubMed.
- R. Serra, R. Grande, L. Butrico, A. Rossi, U. F. Settimio, B. Caroleo, B. Amato, L. Gallelli and S. de Franciscis, Expert Rev. Anti-Infect. Ther., 2015, 13, 605–613 CrossRef CAS PubMed.
- J. L. Brown, E. Townsend, R. D. Short, C. Williams, C. Woodall, C. J. Nile and G. Ramage, npj Biofilms Microbiomes, 2022, 8, 19 CrossRef CAS PubMed.
- E. Maslova, L. Eisaiankhongi, F. Sjöberg and R. R. McCarthy, npj Biofilms Microbiomes, 2021, 7, 73 CrossRef PubMed.
- G. G. David, Plast. Aesthetic. Res., 2024, 11, 27 Search PubMed.
- F. N. Williams, D. N. Herndon, H. K. Hawkins, J. O. Lee, R. A. Cox, G. A. Kulp, C. C. Finnerty, D. L. Chinkes and M. G. Jeschke, Crit. Care, 2009, 13, R183 CrossRef PubMed.
- L.-S. Wang, A. Gupta and V. M. Rotello, ACS Infect. Dis., 2016, 2, 3–4 CrossRef CAS PubMed.
- A. Gupta, S. Mumtaz, C.-H. Li, I. Hussain and V. M. Rotello, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- V. Ball, Front. Bioeng. Biotechnol., 2018, 6, 109 CrossRef PubMed.
- R. Batul, T. Tamanna, A. Khaliq and A. Yu, Biomater. Sci., 2017, 5, 1204–1229 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- Y. Liu, K. Ai, J. Liu, M. Deng, Y. He and L. Lu, Adv. Mater., 2013, 25, 1353–1359 CrossRef CAS PubMed.
- R. Mrówczyński, ACS Appl. Mater. Interfaces, 2018, 10, 7541–7561 CrossRef PubMed.
- P. Jiang, A. Choi and K. E. Swindle-Reilly, Nanoscale, 2020, 12, 17298–17311 RSC.
- H. Zhao, Z. Zeng, L. Liu, J. Chen, H. Zhou, L. Huang, J. Huang, H. Xu, Y. Xu, Z. Chen, Y. Wu, W. Guo, J. H. Wang, J. Wang and Z. Liu, Nanoscale, 2018, 10, 6981–6991 RSC.
- J. Hu, L. Yang, P. Yang, S. Jiang, X. Liu and Y. Li, Biomater. Sci., 2020, 8, 4940–4950 RSC.
- K. R. Shakya, N. Mansoori, A. Anand, V. Sharma and V. Verma, ACS Appl. Bio Mater., 2024, 7, 6808–6822 CrossRef CAS PubMed.
- I. Zarkesh, F. Movahedi, H. Sadeghi-Abandansari, S. Pahlavan, M. Soleimani and H. Baharvand, Int. J. Biol. Macromol., 2024, 259, 129228 CrossRef CAS PubMed.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- Y. Fu, L. Yang, J. Zhang, J. Hu, G. Duan, X. Liu, Y. Li and Z. Gu, Mater. Horiz., 2021, 8, 1618–1633 RSC.
- P. Zheng, B. Ding and G. Li, Macromol. Biosci., 2020, 20, 2000228 CrossRef CAS PubMed.
- N. Falcone, N. M. O. Andoy, R. M. A. Sullan and H.-B. Kraatz, ACS Appl. Bio Mater., 2021, 4, 6652–6657 CrossRef CAS PubMed.
- A. GhavamiNejad, M. SamariKhalaj, L. E. Aguilar, C. H. Park and C. S. Kim, Sci. Rep., 2016, 6, 33594 CrossRef CAS PubMed.
- M. Patel, N. M. O. Andoy, S. M. Tran, K. Jeon and R. M. A. Sullan, J. Mater. Chem. B, 2023, 11, 11335–11343 RSC.
- B. Poinard, S. Z. Y. Neo, E. L. L. Yeo, H. P. S. Heng, K. G. Neoh and J. C. Y. Kah, ACS Appl. Bio Mater., 2018, 10, 21125–21136 CrossRef CAS PubMed.
- J. M. V. Makabenta, A. Nabawy, C.-H. Li, S. Schmidt-Malan, R. Patel and V. M. Rotello, Nat. Rev. Microbiol., 2021, 19, 23–36 CrossRef CAS PubMed.
- J. H. Ryu, P. B. Messersmith and H. Lee, ACS Appl. Bio Mater., 2018, 10, 7523–7540 CrossRef CAS PubMed.
- C. R. Dick and G. E. Ham, J. Macromol. Sci., Chem., 1970, 4, 1301–1314 CAS.
- U. Lungwitz, M. Breunig, T. Blunk and A. Göpferich, Eur. J. Pharm. Biopharm., 2005, 60, 247–266 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- J. Augustine, T. Cheung, V. Gies, J. Boughton, M. Chen, Z. J. Jakubek, S. Walker, Y. Martinez-Rubi, B. Simard and S. Zou, Nanoscale Adv., 2019, 1, 1914–1923 RSC.
- H. L. Alakomi, A. Paananen, M. L. Suihko, I. M. Helander and M. Saarela, Appl. Environ. Microbiol., 2006, 72, 4695–4703 CrossRef CAS PubMed.
- K. A. Gibney, I. Sovadinova, A. I. Lopez, M. Urban, Z. Ridgway, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2012, 12, 1279–1289 CrossRef CAS PubMed.
- I. M. Helander, K. Latva-Kala and K. Lounatmaa, Microbiology, 1998, 144, 385–390 CrossRef CAS PubMed.
- M. A. Foxley, A. W. Friedline, J. M. Jensen, S. L. Nimmo, E. M. Scull, J. B. King, S. Strange, M. T. Xiao, B. E. Smith, K. J. Thomas III, D. T. Glatzhofer, R. H. Cichewicz and C. V. Rice, J. Antibiot., 2016, 69, 871–878 CrossRef CAS PubMed.
- M. A. Foxley, S. N. Wright, A. K. Lam, A. W. Friedline, S. J. Strange, M. T. Xiao, E. L. Moen and C. V. Rice, ACS Med. Chem. Lett., 2017, 8, 1083–1088 CrossRef CAS PubMed.
- M. A. Hill, A. K. Lam, P. Reed, M. C. Harney, B. A. Wilson, E. L. Moen, S. N. Wright, M. G. Pinho and C. V. Rice, Biochemistry, 2019, 58, 3813–3822 CrossRef CAS PubMed.
- A. K. Lam, H. Panlilio, J. Pusavat, C. L. Wouters, E. L. Moen and C. V. Rice, ACS Infect. Dis., 2020, 6, 1085–1097 CrossRef CAS PubMed.
- A. K. Lam, C. L. Wouters, E. L. Moen, J. Pusavat and C. V. Rice, Biomacromolecules, 2019, 20, 3778–3785 CrossRef CAS PubMed.
- W.-J. Song, J.-Z. Du, T.-M. Sun, P.-Z. Zhang and J. Wang, Small, 2010, 6, 239–246 CrossRef CAS PubMed.
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS PubMed.
- N. M. O. Andoy, M. Patel, C. L. J. Lui and R. M. A. Sullan, Microorganisms, 2021, 9, 2176 CrossRef CAS PubMed.
- Y. Li, J. Wang and J. Xie, Smart Med., 2023, 2, e20230015 CrossRef PubMed.
- J. Shan, J. Che, C. Song and Y. Zhao, Smart Med., 2023, 2, e20220025 CrossRef CAS PubMed.
- K. Elumalai, S. Srinivasan and A. Shanmugam, Biomed. Technol., 2024, 5, 109–122 CrossRef CAS.
- B. Xu, H. Wang, W. Wang, L. Gao, S. Li, X. Pan, H. Wang, H. Yang, X. Meng, Q. Wu, L. Zheng, S. Chen, X. Shi, K. Fan, X. Yan and H. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS PubMed.
- S. Ghosh, B. Sarkar and T. J. Webster, in Medical Nanobiotechnology, ed. S. Ghosh and T. J. Webster, Woodhead Publishing, 2025, pp. 247–262 Search PubMed.
- Z. Xu, T. Wang and J. Liu, Int. J. Mol. Sci., 2022, 23, 7278 CrossRef CAS PubMed.
- M. Bao, K. Wang, J. Li, Y. Li, H. Zhu, M. Lu, Y. Zhang, Q. Fan, L. Han, K. Wang, D. Wang, Y. Gao, B. Peng, Z. Ming and W. Liu, Acta Biomater., 2023, 161, 250–264 CrossRef CAS PubMed.
- A. L. Byrd, Y. Belkaid and J. A. Segre, Nat. Rev. Microbiol., 2018, 16, 143–155 CrossRef CAS PubMed.
- C. L. Fischer and P. W. Wertz, in Skin Microbiome Handbook: From Basic Research to Product Development, ed. N. Dayan, Scrivener Publishing LLC, 2020, pp. 217–235 Search PubMed.
- E. A. Grice and J. A. Segre, Nat. Rev. Microbiol., 2011, 9, 244–253 CrossRef CAS PubMed.
- A. G. James, in Skin Microbiome Handbook: From Basic Research to Product Development, ed. N. Dayan, Scrivener Publishing LLC, 2020, pp. 79–129 Search PubMed.
- K. Skowron, J. Bauza-Kaszewska, Z. Kraszewska, N. Wiktorczyk-Kapischke, K. Grudlewska-Buda, J. Kwiecińska-Piróg, E. Wałecka-Zacharska, L. Radtke and E. Gospodarek-Komkowska, Microorganisms, 2021, 9, 543 CrossRef CAS PubMed.
- A. H. Benfield and S. T. Henriques, Front. Med. Technol., 2020, 2, 610997 CrossRef PubMed.
- C. Chessa, C. Bodet, C. Jousselin, M. Wehbe, N. Lévêque and M. Garcia, Front. Microbiol., 2020, 11, 1155 CrossRef PubMed.
- E. Jordana-Lluch, V. Garcia, A. D. H. Kingdon, N. Singh, C. Alexander, P. Williams and K. R. Hardie, Front. Microbiol., 2020, 11, 291 CrossRef PubMed.
- J. Kalabis, G. S. Wong, M. E. Vega, M. Natsuizaka, E. S. Robertson, M. Herlyn, H. Nakagawa and A. K. Rustgi, Nat. Protoc., 2012, 7, 235–246 CrossRef CAS PubMed.
- A. K. Lam, M. A. Hill, E. L. Moen, J. Pusavat, C. L. Wouters and C. V. Rice, ChemMedChem, 2018, 13, 2240–2248 CrossRef CAS PubMed.
- N. M. O. Andoy, K. Jeon, C. T. Kreis and R. M. A. Sullan, Adv. Funct. Mater., 2020, 30, 2004503 CrossRef CAS.
- C. S. Sobral, A. Gragnani, X. Cao, J. R. Morgan and L. M. Ferreira, J. Burns Wounds, 2007, 7, e6 Search PubMed.
- C.-W. Wong, C. F. LeGrand, B. F. Kinnear, R. M. Sobota, R. Ramalingam, D. E. Dye, M. Raghunath, E. B. Lane and D. R. Coombe, Sci. Rep., 2019, 9, 18561 CrossRef CAS PubMed.
- B. Russo, N. C. Brembilla and C. Chizzolini, Front. Immunol., 2020, 11, 648 CrossRef CAS PubMed.
- F. C. Simões, T. J. Cahill, A. Kenyon, D. Gavriouchkina, J. M. Vieira, X. Sun, D. Pezzolla, C. Ravaud, E. Masmanian, M. Weinberger, S. Mayes, M. E. Lemieux, D. N. Barnette, M. Gunadasa-Rohling, R. M. Williams, D. R. Greaves, L. A. Trinh, S. E. Fraser, S. L. Dallas, R. P. Choudhury, T. Sauka-Spengler and P. R. Riley, Nat. Commun., 2020, 11, 600 CrossRef PubMed.
- V. Schneider, D. Kruse, I. B. de Mattos, S. Zöphel, K. K. Tiltmann, A. Reigl, S. Khan, M. Funk, K. Bodenschatz and F. Groeber-Becker, Biomedicines, 2021, 9, 1153 CrossRef CAS PubMed.
- P. Gangatirkar, S. Paquet-Fifield, A. Li, R. Rossi and P. Kaur, Nat. Protoc., 2007, 2, 178–186 CrossRef CAS PubMed.
- J. Lee, W. H. van der Valk, S. A. Serdy, C. Deakin, J. Kim, A. P. Le and K. R. Koehler, Nat. Protoc., 2022, 17, 1266–1305 CrossRef CAS PubMed.
- J. Zhang, Y. Zhou, Z. Jiang, C. He, B. Wang, Q. Wang, Z. Wang, T. Wu, X. Chen, Z. Deng, C. Li and Z. Jian, J. Nanobiotechnol., 2023, 21, 354 CrossRef CAS PubMed.
- T.-T. Zhu, H. Wang, H.-W. Gu, L.-S. Ju, X.-M. Wu, W.-T. Pan, M.-M. Zhao, J.-J. Yang and P.-M. Liu, J. Nanobiotechnol., 2023, 21, 52 CrossRef CAS PubMed.
- M. G. Jeschke, M. E. van Baar, M. A. Choudhry, K. K. Chung, N. S. Gibran and S. Logsetty, Nat. Rev. Dis. Primers, 2020, 6, 11 CrossRef PubMed.
- S. Phuphanitcharoenkun, F. Louis, Y. Sowa, K. Uchida, M. Katsuyama, R. Waditee-Sirisattha, H. Kageyama, M. Matsusaki and T. Palaga, Commun. Biol., 2024, 7, 1284 CrossRef CAS PubMed.
- M. R. Scobie, A. Abood and C. D. Rice, Int. J. Mol. Sci., 2023, 24, 5115 CrossRef CAS PubMed.
- S. Bianconi, L. Leppik, E. Oppermann, I. Marzi and D. Henrich, Int. J. Mol. Sci., 2024, 25, 7272 CrossRef CAS PubMed.
- H. Jo, E.-Y. Lee, H. S. Cho, M. A. Rayhan, A. Cho, C.-S. Chae and H. J. You, Medicina, 2024, 60, 1009 CrossRef PubMed.
- A. Bhattacharya, M. Agarwal, R. Mukherjee, P. Sen and D. K. Sinha, Cell Death Dis., 2018, 9, 914 CrossRef PubMed.
- S. S. Jacob and P. R. Sudhakaran, Biochim. Biophys. Acta, Mol. Cell Res., 2001, 1540, 50–58 CrossRef CAS PubMed.
- P. P. G. Mulder, M. Vlig, E. Fasse, M. M. Stoop, A. Pijpe, P. P. M. van Zuijlen, I. Joosten, B. Boekema and H. Koenen, Front. Immunol., 2022, 13, 1034420 CrossRef CAS PubMed.
- M. Demircan and K. Gurunluoglu, J. Burn Care Res., 2019, 40, S154 CrossRef.
- M. Uehara, X. Li, A. Sheikhi, N. Zandi, B. Walker, B. Saleh, N. Banouni, L. Jiang, F. Ordikhani, L. Dai, M. Yonar, I. Vohra, V. Kasinath, D. P. Orgill, A. Khademhosseini, N. Annabi and R. Abdi, Sci. Rep., 2019, 9, 6535 CrossRef PubMed.
- J. E. DeJesus, J. J. Wen and R. Radhakrishnan, J. Pers. Med., 2022, 12, 1876 CrossRef PubMed.
- C. Sharma, G. P. Dobson, L. M. Davenport, J. L. Morris and H. L. Letson, Int. J. Burns Trauma, 2021, 11, 275–288 CAS.
- J. Li, W. Hou, S. Lin, L. Wang, C. Pan, F. Wu and J. Liu, Adv. Sci., 2022, 9, e2104006 CrossRef PubMed.
- M. Rossato, G. Curtale, N. Tamassia, M. Castellucci, L. Mori, S. Gasperini, B. Mariotti, M. De Luca, M. Mirolo, M. A. Cassatella, M. Locati and F. Bazzoni, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E3101–E3110 CrossRef CAS PubMed.
- Y. Sun, J. Ma, D. Li, P. Li, X. Zhou, Y. Li, Z. He, L. Qin, L. Liang and X. Luo, J. Neuroinflammation, 2019, 16, 66 CrossRef PubMed.
- K. Y. Ryu, G. S. Cho, H. Z. Piao and W. K. Kim, Exp. Neurobiol., 2012, 21, 151–157 CrossRef PubMed.
- E. N. Benveniste, L. P. Tang and R. M. Law, Int. J. Dev. Neurosci., 1995, 13, 341–349 CrossRef CAS PubMed.
- M. Cano Sanchez, S. Lancel, E. Boulanger and R. Neviere, Antioxidants, 2018, 7, 98 CrossRef PubMed.
- T. Lopez, M. Wendremaire, J. Lagarde, O. Duquet, L. Alibert, B. Paquette, C. Garrido and F. Lirussi, Biomedicines, 2022, 10, 2784 CrossRef CAS PubMed.
- M. M. Brown and A. R. Horswill, PLoS Pathog., 2020, 16, e1009026 CrossRef CAS PubMed.
- M. M. Severn and A. R. Horswill, Nat. Rev. Microbiol., 2023, 21, 97–111 CrossRef CAS PubMed.
- P. Ubanako, N. Xelwa and M. Ntwasa, PLoS One, 2019, 14, e0222614 CrossRef CAS PubMed.
- J. Wang, Z. Duan, X. Chen and M. Li, Exp. Dermatol., 2023, 32, 1326–1333 CrossRef CAS PubMed.
- J.-N. Wang and M. Li, Int. J. Dermatol. Venereol., 2020, 3, 231–238 CrossRef.
- P. Acharya, N. Y. Choi, S. Shrestha, S. Jeong and M.-Y. Lee, Biotechnol. Bioeng., 2024, 121, 489–506 CrossRef CAS PubMed.
- S. N. Patel, M. Ishahak, D. Chaimov, A. Velraj, D. LaShoto, D. W. Hagan, P. Buchwald, E. A. Phelps, A. Agarwal and C. L. Stabler, Sci. Adv., 2024, 7, eaba5515 CrossRef PubMed.
- L. Ferreira, E. Pos, D. R. Nogueira, F. P. Ferreira, R. Sousa and M. A. Abreu, Front. Microbiol., 2024, 15, 1435720 CrossRef PubMed.
- M. Beldman, C. Löwik, A. Soriano, L. Albiach, W. P. Zijlstra, B. A. S. Knobben, P. Jutte, R. Sousa, A. Carvalho, K. Goswami, J. Parvizi, K. A. Belden and M. Wouthuyzen-Bakker, Clin. Infect. Dis., 2021, 73, 1634–1641 CrossRef CAS PubMed.
- Z. Hou, M. Yang, L. Huang, S. Xin, H. Yang and J. Hou, Front. Chem., 2022, 10, 1111701 CrossRef CAS PubMed.
- H. Wang, J. Ma, S. Meng, H. Zou, H. Wang and M. Zhou, ACS Appl. Nano Mater., 2024, 7, 10429–10441 CrossRef CAS.
- W. S. da Cruz Nizer, M. E. Adams, K. N. Allison, M. C. Montgomery, H. Mosher, E. Cassol and J. Overhage, Biofilm, 2024, 7, 100203 CrossRef PubMed.
- Y. M. Gutierrez-Murgas, G. Skar, D. Ramirez, M. Beaver and J. N. Snowden, J. Neuroinflammation, 2016, 13, 271 CrossRef PubMed.
- M. Portugal-Cohen, D. Cohen, R. Kohen and M. Oron, Front. Physiol., 2023, 14, 1215266 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Surface modification of polydopamine nanoparticles (PDNP) with polyethyleneimine (PEI) to create PDNP–PEI; PDNP–PEI binds the surface of a Gram-positive bacteria; laser-induced photothermal activity of PDNP–PEI; antimicrobial activity of PDNP–PEI + heat treatment; PDNP–PEI displayed anti-biofilm activity only at 50 μg mL−1 post laser irradiation; antimicrobial activity of rifampicin; rifampicin loading, antimicrobial and antibiofilm activities of drug-loaded PDNP–PEI (NP–PEI–Rif); PDNP–PEI-Rif binds S. epidermidis biofilm. See DOI: https://doi.org/10.1039/d4bm01530d |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2025 |