
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of microfluidic modulation spectroscopy for simultaneous structural and thermal stability analysis of commercial mAbs under varying formulation conditions†
Yalan
Yang‡
,
Chuanfei
Yu‡
,
Maoqin
Duan‡
,
Chunyu
Liu
,
Meng
Li
,
Gang
Wu
,
Jialiang
Du
,
Gangling
Xu
,
Xiaojuan
Yu
and
Lan
Wang
*
National Institutes for Food and Drug Control, China
First published on 23rd April 2025
Abstract
The secondary structure of proteins is extremely important because it contains some of the most fundamental information about how a protein folds into its tertiary and quaternary structures. The most widely used biophysical tools for analyzing protein secondary structures, however, suffer from major limitations such as low resolution, poor reproducibility, and a narrow concentration range. This study utilized Microfluidic Modulation Spectroscopy (MMS), a novel automated protein structural characterization technique, to analyze the secondary structure of nine commercial monoclonal antibodies (mAbs) under various concentrations and buffer conditions. The results revealed that diluting the mAbs in formulation buffer did not affect the protein structure, whereas reconstituting the mAbs from formulation buffer to PBS altered the protein structure by a small but detectable margin. In addition, properties such as the antibody subtype and target seem to have little relation to the secondary structure of the protein, based on the 9 mAbs tested in this study. However, differences or changes in the secondary structure of antibodies led to discrepancies in their thermal stability and melting temperature. This study shows that MMS can detect the secondary structure of monoclonal antibodies reproducibly and reliably, based on which we can derive the factors affecting the determination of the secondary structure from the experimental results of small peak shifts in the spectrum.
Introduction
The structural characterization of therapeutic proteins is necessary due to the close relationship between the protein structure and function. The secondary structure refers to the local folding of polypeptide chains into regular, repeating patterns through non-covalent interactions, which serve as the foundation for the formation of the three-dimensional conformation of the entire protein.1 The secondary structure is highly conserved in the native conformation of proteins. The most common forms of the protein secondary structure are the α-helix and β-sheet. In addition to these two predominant structures, other conformations such as turns and random coils also exist. The diversity in higher-order protein structures arises from the varying proportions and combinations of these different conformational forms, enabling proteins to achieve more complex biological functions. Some influencing factors (e.g. temperature and pH) can affect the stability of the protein secondary structure. For example, high temperature could break hydrogen bonds and lead to the unfolding of the secondary structure, which in turn affects the protein's function.2 Thus, it is critical to ref. 1 accurately assess the correct relative abundance of the different secondary structural forms for protein HOS analysis.Two of the most commonly used biophysical techniques for assessing higher order structural differences in proteins are Fourier-transform infrared (FTIR) and circular dichroism (CD) spectroscopy. FTIR and CD spectroscopy utilize characteristic absorption features at specific wavenumbers and wavelengths to evaluate the protein secondary structure. Both FTIR and CD apply multivariate analysis techniques to perform spectral deconvolution either against reference spectral datasets of proteins or using curve fitting algorithms based on specific secondary structural elements that have been assigned to discrete wavenumbers. CD is especially useful for investigating α-helical-rich proteins due to the intense signal in the far-UV CD region provided by α-helix structures. FTIR on the other hand is equally sensitive to α-helix and β-sheet structures and provides extra information compared to CD, such as signals from β-turn structures. Additionally, FTIR is excellent for analysis of high concentration samples, adding great advantages to analyzing formulated drug products such as mAbs.
Although both techniques provide reliable assessments of protein secondary structural information, the sample requirements are drastically different between CD and FTIR. CD measurements require low sample concentrations, typically <5 mg mL−1, with 0.1–1 mg mL−1 being the most commonly measured protein concentration. The low concentration requirement is due to an optimal absorbance to obtain good CD data.3 Furthermore, certain components or excipients in the buffer can cause interference with the protein signal. Histidine, for example, is a common excipient in mAb formulations and has a strong interference signal in CD.4 Compared to CD, FTIR has better buffer compatibility. However, it requires complicated workflows such as manual background subtraction and suffers from low reproducibility.
Microfluidic Modulation Spectroscopy (MMS) is an automated IR technology for protein HOS analysis to address the limitations of conventional CD and FTIR techniques. MMS utilizes a tunable quantum cascade laser (QCL) to generate an attenuated optical signal ∼100 times brighter than conventional light sources for ultra-high sensitivity, along with a microfluidic flow cell that allows real-time buffer subtraction to produce drift-free and robust measurements. The highly sensitive and automated platform of MMS allows samples to be run over a wide concentration range of 0.1–200 mg mL−1 in most buffers and excipients without interference.5 MMS delivers high-quality data for HOS analysis of mAbs in complex formulation buffers and shows superior sensitivity for low-concentration samples (1 mg mL−1) in a side-by-side comparison with FTIR.6,7 Furthermore, it has been shown that the limit of quantitation (LOQ) of MMS is significantly lower than that of CD and FTIR when analyzing the structural impurities of IgG spiked with BSA.8
In this study, MMS is used to accurately determine the secondary structures of 9 commercial mAbs both in their formulation buffers and in phosphate buffered saline (PBS) (Table 1). A typical mAb formulation contains an antibody, a buffer, a surfactant, and an excipient to adjust pH or osmolality for solutions or a lyoprotectant for lyophilized powders. MMS results show that there is a subtle but detectable change in the protein secondary structure when the mAbs are dissolved in PBS instead of their formulation buffers. These small changes in the secondary structure result in differences in thermal stability and unfolding patterns.
| Sample name | Formulation | Antibody subtype | Source |
|---|---|---|---|
| Trastuzumab | 21 mg mL−1 trehalose dihydrate (20 mg mL−1) | lgG1 | Humanized |
| Polysorbate 20 (0.08 mg mL−1) | |||
| Histidine HCl (0.49 mg mL−1) | |||
| Histidine (0.32 mg mL−1) | |||
| pH 6.0 | |||
| Pertuzumab | 30 mg mL−1 | lgG1 | Humanized |
| Sucrose (120 mM) polysorbate 20 (0.2 mg mL−1) | |||
| Histidine acetate (0.020 M) | |||
| pH 6.0 | |||
| Atezolizumab | 60 mg mL−1 sucrose (40 mg mL−1) | lgG1 | Humanized |
| Polysorbate 20 (0.4 mg mL−1) | |||
| Acetic acid (0.8 mg mL−1) | |||
| Histidine (3 mg mL−1) | |||
| pH 5.8 | |||
| Durvalumab | 50 mg mL−1 trehalose dihydrate (104 mg mL−1) | lgG1κ | Humanized |
| Polysorbate 80 (0.2 mg mL−1) | |||
| Histidine HCL (2.7 mg mL−1) | |||
| Histidine (2 mg mL−1) | |||
| Cetuximab | 5 mg mL−1 sodium chloride (8.5 mg mL−1) | lgG1 | Chimeric |
| Sodium phosphates (0.015 M) | |||
| pH 7.0–7.4 | |||
| Bevacizumab | 25 mg mL−1 trehalose dihydrate (60 mg mL−1) | lgG1 | Humanized |
| Polysorbate 20 (0.4 mg mL−1) | |||
| Sodium phosphates (0.05 M) | |||
| pH 6.2 | |||
| Basiliximab | 4 mg mL−1 mannitol (16 mg mL−1) | lgG1κ | Humanized |
| Sucrose (4 mg mL−1) | |||
| Sodium chloride (0.32 mg mL−1) | |||
| Glycine (8 mg mL−1) | |||
| Monobasic potassium phosphate (1.4 mg mL−1) | |||
| Disodium hydrogen phosphate (0.2 mg mL−1) | |||
| pH 6.5 | |||
| Pembrolizumab | 25 mg mL−1 sucrose (70 mg mL−1) | lgG4 | Humanized |
| Polysorbate 80 (0.2 mg mL−1) | |||
| Histidine (1.55 mg mL−1) | |||
| pH = 5.5 | |||
| Tocilizumab | 20 mg mL−1 sucrose (0.146 M) | lgG1κ | Humanized |
| Polysorbate 80 (0.5 mg mL−1) | |||
| Sodium phosphates (0.015 M) | |||
| pH 6.5 |
Experimental section
Materials and reagents
The mAbs that were used in this study are listed in Table 1. PBS (pH 7.4) was purchased from Gibco.Sample preparation
The samples at their original concentration in formulation buffer were analyzed as is using their respective matching buffer. The samples at 5 mg mL−1 in formulation buffer were diluted directly from the original concentration using the formulation buffer before analysis. The samples at 5 mg mL−1 in PBS were prepared via buffer exchange using ultrafiltration spin columns (Millipore, 3KD). Briefly, a calculated amount of the sample at its original strength was added to the spin column, together with a sufficient amount of PBS. Then, the spin column was centrifuged at 4500 rpm for 30 minutes. This centrifugation step was repeated 5–6 times until the original buffer was replaced with PBS. After the final centrifugation, a calculated amount of PBS was added so that the final concentration of the sample was 5 mg mL−1.Isothermal microfluidic modulation spectroscopy (MMS)
All samples were analyzed on an AQS3®pro MMS production system (RedShift BioAnalytics, Boxborough, MA, US) at 25 °C using a modulation rate of 1 Hz and a backing pressure of 5 psi with analysis at 29 discrete wavenumbers across the amide I band. Three replicates were collected for samples. To calculate the percent similarity, the area of overlap (AO) was used on the baseline-subtracted second derivative spectrum for each sample using the baseline setting type “Rubberband,” which fixes both ends of the second derivative spectrum to the baseline. HOS structural elements were calculated using the same baseline-corrected plot and Gaussian curve fit settings shown in Table 2.| Wavenumber (cm−1) | HOS structural element |
|---|---|
| 1618 | β-Sheet |
| 1637 | β-Sheet |
| 1642 | β-Sheet |
| 1650 | Unordered |
| 1656 | α-Helix |
| 1660 | Turn |
| 1666 | Turn |
| 1672 | Turn |
| 1680 | Turn |
| 1688 | Turn |
| 1690 | β-Sheet |
| 1695 | β-Sheet |
Thermal ramping MMS of Trastuzumab and Pembrolizumab
Trastuzumab and Pembrolizumab in their respective formulations (Table 1) and PBS at 0.5 mg mL−1 were studied using the thermal ramping capability of MMS. An Aurora TX (RedShift BioAnalytics, Boxborough, MA) was used to capture the differential spectral data of sample buffer across a range of temperatures. Spectra were captured from 1712 to 1588 cm−1 at 1 cm−1 resolution. The flow rate was calibrated to 1 μL min−1 at 25 °C and each fluid exchange lasted 0.4 s to remove carryover effects in the MMS cell. The thermal ramping was carried out continuously at 1 °C min−1 from 25 to 90 °C. Raw MMS spectra were captured approximately every 17 s along with cell temperature as measured using a thermistor attached to the MMS flow cell. Dark scan, sample channel, and reference channel measurements were captured 3 s apart. Due to the continuous temperature ramp, dark scan and reference detector signals were phase-corrected to align them in time and temperature with sample measurements. The sample and reference channel signals were dark offset corrected, and water vapor was subtracted to yield differential absorbance.Results and discussion
Data repeatability of MMS
Nine commercial mAbs were analyzed under 3 different conditions: formulated concentration in formulation buffer, 5 mg mL−1 protein in formulation buffer, and 5 mg mL−1 protein in PBS (except for Basiliximab, where the original strength is 4 mg mL−1). For all samples, MMS measurements were taken in triplicate for statistical analysis and repeatability information. All data generated exhibited extremely high repeatability (spectral similarity within replicates), with the lowest being 99.5% and the highest being 99.98%. A complete list of the data repeatability of all samples is shown in Table S1 in the ESI.† Such high repeatability achieved by MMS is a significant improvement compared to the 94–96% repeatability achieved using FTIR or the 97–99% repeatability achieved using CD (Table S2†) for protein structural analysis.10–12Fig. 1 shows the MMS data output for Trastuzumab, including the raw differential absorbance spectra, the absolute absorbance, and the second derivative plots of the absolute absorbance. Due to the high repeatability, the spectra of all replicates in each sample are visually identical (Fig. 1A). The absolute absorbance spectra were obtained by normalizing the concentration and path length, resulting in highly overlayed plots for the samples under the three conditions (Fig. 1B). The spectral discrepancies of each sample start to manifest in the second derivative plots (Fig. 1C), especially for the sample in PBS, as a small shift of the 1639 cm−1 peak can be observed (Fig. 1C inset). This subtle but detectable change in the spectrum demonstrates a possible structural change in Trastuzumab in PBS versus in its formulation. All other samples showed similar results, i.e. no change between the formulation concentration and diluted samples, but a visible change between the samples in PBS and in formulation buffer (Fig. S1–S7,† ESI).
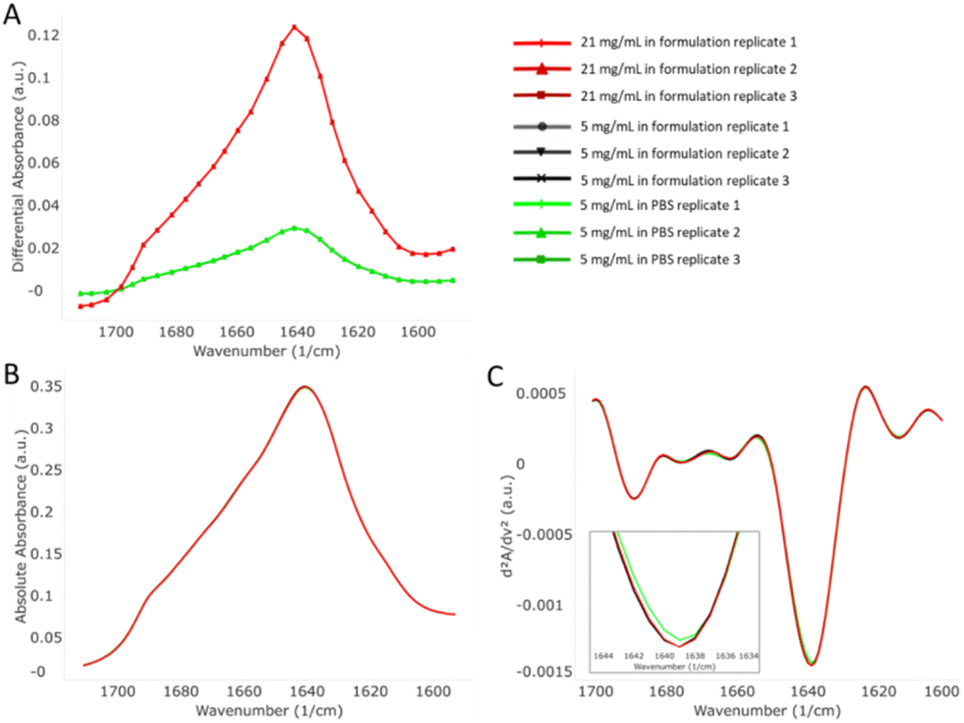 | ||
| Fig. 1 MMS data output for Trastuzumab. (A) Raw differential absorbance spectra of 21 mg mL−1 sample in formulation buffer, 5 mg mL−1 sample in formulation buffer, and 5 mg mL−1 sample in PBS. Triplicates are shown in the graph. (B) Absolute absorbance of the 3 samples. (C) Second derivative plots of the 3 samples. | ||
Effect of dilution on the mAb structure
To accurately assess the HOS of proteins, CD is normally preferred over FTIR for mAbs due to better sensitivity and repeatability. However, due to the underlying limitations of CD, namely the narrow concentration range and signal interference with certain excipients such as histidine and arginine, sample dilution from formulation strength and/or reconstitution in simple buffers such as PBS is a common practice. It is therefore essential to evaluate the effect of dilution on the protein structure so that the CD results remain valid.MMS covers a wide range of concentrations from 0.1 to 200 mg mL−1 and thus allows a direct structural comparison of protein samples before and after dilution. The second derivative plots for the mAbs at the original concentration and at 5 mg mL−1 in formulation buffer are shown in Fig. 2. The spectra of samples before and after dilution are highly overlayed among all mAbs tested. Sample-to-sample similarity scores indicate that there is no significant structural difference between the samples at the original concentration and at 5 mg mL−1 in formulation (Table S3,† ESI).
 | ||
| Fig. 2 Second derivative plots comparing the mAbs at their original concentration formulation and 5 mg mL−1 in formulation. The two plots in each mAb are highly overlayed, indicating a strong similarity in structure between the mAbs in formulation strength and diluted samples. Note: Cetuximab and Basiliximab are not shown because their formulation strength is 5 mg mL−1 or below. | ||
Effect of change in formulation on the mAb structure
All the diluted samples were buffer exchanged with PBS while maintaining the same concentration of 5 mg mL−1. The MMS second derivative plots, comparing the formulated and PBS samples, are shown in Fig. 3. Subtle but detectable spectral changes can be observed for every mAb. Specifically, the most noticeable changes occurred in the β-turn (1660–1680 cm−1) and the β-sheet region (1638–1640 cm−1). It was established earlier that the high data repeatability resulted in the replicate spectra visually overlaying, and thus these noticeable spectral variations represent actual changes in the protein secondary structure. The sample-to-sample similarity scores calculated from the area of overlap are shown in Table S4.† | ||
| Fig. 3 Second derivative plots comparing the mAbs at 5 mg mL−1 in formulation and 5 mg mL−1 in PBS. Spectral discrepancies can be observed in the turn region (1660–1680 cm−1) and the β-sheet region (1638–1640 cm−1). Note: Basiliximab is not shown because its formulation strength is below 5 mg mL−1. Tocilizumab in PBS data is not included. | ||
In all cases, the similarity score of the PBS sample compared to the formulation sample is significantly lower than the repeatability of the PBS triplicate itself. This observation can be attributed to the absence of excipients such as polysorbate 20 or 80 leading to protein structural changes.13 A similar comparison of formulated and PBS samples at 5 mg mL−1 was performed using CD. The sample-to-sample similarity and replicate repeatability are listed in Table S5.† Again, it showed a structural difference between the PBS samples and the formulated samples as indicated by the similarity scores.
The composition of HOS structural elements was calculated using the baseline-corrected second derivative plot by Gaussian curve fitting based on the HOS structural element designations shown in Table 2. The fractional contributions of the HOS structural elements, β-sheet, α-helix, turn, and unordered structures are shown in Fig. 4. Although all mAbs showed structural changes after reconstitution with PBS, not all of them were affected in the fractional contribution of the structural elements. In particular, the unordered structure increased and the β-sheet/turn structures decreased in Trastuzumab, Atezolizumab, and Durvalumab. These changes indicate possible unfolding of β-sheets turning into unordered structures when the buffer environment changed from formulation to PBS. It is important to note that differences in the HOS fractional contribution may or may not indicate a change in function or activity. One must be cautious drawing conclusions from the HOS readout without further assessment of the function or activity of the protein in relation to its function. In this study, one thing in common among these three mAbs is that histidine is used as the buffer in their formulations. A previous study demonstrated that an antibody conjugated with ruthenium stored in PBS exhibited an increased level of aggregates compared to that stored in histidine-sucrose buffer.14 Based on our results, this suggests that the absence of histidine may lead to instability of the secondary structure of Trastuzumab, Atezolizumab, and Durvalumab. Nevertheless, the structural differences may be caused by ultrafiltration in the process of buffer displacement, which needs to be further verified by MMS.
 | ||
| Fig. 4 HOS analysis comparing each mAb at its original concentration in formulation, 5 mg mL−1 in formulation, and 5 mg mL−1 in PBS. An increase in unordered structures in the 5 mg mL−1 PBS samples can be observed for Trastuzumab, Atezolizumab, and Durvalumab. | ||
The HOS for Pertuzumab, Bevacizumab, and Pembrolizumab remained unchanged. The unchanged HOS with actual spectral change suggests that there were subtle peak shifts, particularly in the β-sheet and turn regions, in these mAbs when reconstituted in PBS, yet the shifts were not large enough to affect the fitting parameter in the Gaussian curve model. These three mAbs (Pertuzumab, Bevacizumab, and Pembrolizumab) with the unchanged HOS seem to be more stable and resistant to the process of formulation buffer displacement compared to the other three mAbs (Trastuzumab, Atezolizumab, and Durvalumab).
Secondary structure effect on the thermal stability of mAbs
To investigate the effect of small structural differences on the thermal stability of mAbs, we performed thermal ramping MMS experiments on Trastuzumab and Pembrolizumab, in their respective formulations and in PBS. Thermal ramping MMS is a novel method for real-time HOS alterations of biomolecules upon heat treatment, providing both structural and stability measurements simultaneously.15 Other thermoanalytical techniques such as differential scanning calorimetry (DSC) and differential scanning fluorimetry (DSF), while sufficiently providing the thermal unfolding and melting temperature (Tm) of proteins, are unable to assess the real-time structural change of the molecules during thermal treatment. MMS utilizes the change of the secondary structure during the thermal ramping to provide measurements of Tm of specific structural elements, such as the α-helix and β-sheet.At 25 °C, Trastuzumab and Pembrolizumab displayed apparent structural differences as reflected in the β-sheet and unordered regions (Fig. S8†). Within the same mAb but different buffers, there was also a consistent but small difference that can be quantified in the HOS bar graph. Specifically, Trastuzumab exhibited more β-sheet (2–3%) and less unordered (2–3%) content than Pembrolizumab regardless of the buffer condition; the formulation condition resulted in more β-sheet (0.5–1%) and less unordered (0.5–1%) content than the PBS condition.
These small differences in the secondary structure led to detectable changes in the mAb's thermal stability in formulation (Fig. 5). The differential absorbance spectra were recorded at every °C from 25 to 90 °C (Fig. 5A and B). At low temperatures (blue spectra), both mAbs exhibited a main peak at 1640 cm−1 representing the predominant β-sheet structure. Interestingly, as the temperature rose, the structures of Trastuzumab and Pembrolizumab changed differently. For Trastuzumab, the main peak shifted to 1660 cm−1 with a decrease in intensity, indicating the unfolding of β-sheets and turning into turn and unordered structures. For Pembrolizumab, the main peak shifted to 1624 cm−1, suggesting that the unfolding of β-sheets quickly turned into intermolecular β-sheets, potentially initiating aggregation. In contrast, the two mAbs in PBS gradually lost their β-sheet intensity during the thermal ramp (Fig. S9†), suggesting early unfolding and sedimentation of the molecules. These different unfolding pathways likely led to a difference in melting temperatures between these two mAbs (Fig. 5C, D, and S9†). Trastuzumab showed a higher Tm of losing its β-sheet structure at 83 °C, whereas Pembrolizumab showed a lower Tm of 75 °C. These results are consistent with previous melting temperature (Tm2) assessments using CD and DSC.16,17 It is important to note that a single transition sigmoid model is used for the fitting of these data. Thus, only one melting temperature was calculated for each thermal ramp in this study. Nonetheless, we demonstrated here that thermal ramping MMS allows accurate measurements of the melting temperature of mAbs by detecting the change in the secondary structure across a range of temperatures.
 | ||
| Fig. 5 Thermal stability of Trastuzumab and Pembrolizumab from 25 to 90 °C. (A) and (B) Differential absorbance spectra of Trastuzumab and Pembrolizumab in formulation. Blue spectra: at 25 °C. Red spectra: at 90 °C. (C) and (D) Thermal ramp heat maps of Trastuzumab and Pembrolizumab in formulation. The color of the heat map indicates the slope of the fitted sigmoid with respect to the change in absorbance across a range of temperatures. The red and blue regions indicate rapid increases and decreases in absorbance, respectively. The center of these regions indicates the point at which the rate of change in absorbance becomes the highest, which is the melting temperature of the corresponding secondary structure of this region. The green background indicates that the change in absorbance does not follow a bistable trajectory (i.e., no sigmoid is found). | ||
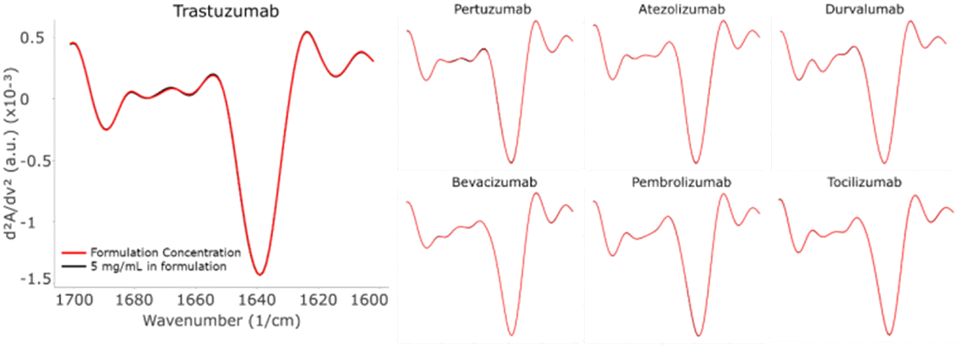
Secondary structure has little relation to targets or subtypes
Antibodies are often categorized by their subtypes while their functions vary significantly depending on the subset they belong to. As function is highly correlated with activity, it is important to learn whether the subtypes are also correlated to the secondary structures, i.e., whether antibodies with the same subtypes share similar structures. Table 3 shows the structural information in relation to the antibody subtype. When Trastuzumab was set as the control, all other mAbs had significantly lower similarity scores (given that the repeatability of all samples was above 99.5%), regardless of the subtype. The high-level structure of the monoclonal antibody is highly intricate, and the determinants of its three-dimensional morphology are multifaceted and complex. Based on the data of this study, the secondary structure of the monoclonal antibody seemingly has little relation to targets or to belonging to the same subtype. For example, Trastuzumab and Pertuzumab (targeting different epitopes of HER2) and Atezolizumab and Durvalumab (targeting the same target PD-L1) show lower similarity in secondary structures despite all of them belonging to the same subtype. Structural differences may lead to functional differences. A recent study compared the efficacy of Atezolizumab and Durvalumab in real world settings for extensive stage small cell lung cancer. The results showed that Durvalumab had a slight advantage in prolonging survival.18| Subtype | Sample name | Similarity (%) |
|---|---|---|
| IgG1 | Trastuzumab | 100 |
| IgG1 | Pertuzumab | 97.8 |
| IgG1 | Atezolizumab | 97.0 |
| IgG1κ | Tocilizumab | 96.4 |
| IgG1 | Bevacizumab | 96.2 |
| IgG1 | Cetuximab | 96.1 |
| IgG4 | Pembrolizumab | 95.7 |
| IgG1κ | Durvalumab | 95.5 |
Conclusion
The protein secondary structure is an important product quality attribute (PQA) that governs the structure–function relationship, safety, and efficacy in protein therapeutics.21 MMS is an emerging biophysical technology for the characterization of the protein secondary structure and provides the necessary accuracy, sensitivity, and reproducibility for high-quality structural assessment of mAbs in their formulation buffer and native concentration. MMS has been previously proven to detect very small structural changes enabling a level of characterization not achievable using conventional FTIR techniques. In this study, MMS was utilized to analyze the secondary structure of 9 commercial mAbs in their formulation buffers. A side-by-side comparison of HOS between mAbs at formulation strength, at low concentration, and in PBS was achieved using the MMS automated system. The results demonstrated that diluting the mAbs in formulation buffer did not affect the protein structure, whereas reconstituting the mAbs from formulation buffer to PBS altered the protein structure by a small but detectable margin, with different degrees. Moreover, using thermal ramping MMS revealed that small differences in the secondary structure between two mAbs led to drastically different unfolding pathways and thermal stability. The ability of MMS to analyze proteins in their native conditions without interference while providing this level of sensitivity makes MMS an ideal tool for structural characterization of protein therapeutics. Additionally, properties such as the antibody subtype and target seem to have little relation to the secondary structure of the protein, based on the 9 mAbs tested in this study.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
The manuscript was written through contributions from all authors. Yalan Yang: writing – original draft, data curation, and methodology. Chuanfei Yu: writing – review & editing, supervision, and conceptualization. Maoqin Duan: writing – review & editing. Chunyu Liu: writing – review & editing. Gang Wu: writing – review & editing. Jialiang Du: writing – review & editing. Gangling Xu: writing – review & editing. Xiaojuan Yu: writing – review & editing. Lan Wang: writing – review & editing, supervision, and conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Plan. (Grant number: 2021YFF0600804).References
- W. Kabsch and C. Sander, Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed.
- Y. Yao, B. Zhang and H. Pang, et al., The effect of radio frequency heating on the inactivation and structure of horseradish peroxidase, Food Chem., 2023, 398, 133875 CrossRef CAS PubMed.
- I. Tinoco and D. H. Turner, Fluorescence Detected Circular Dichroism. Theory, J. Am. Chem. Soc., 1976, 98(21), 6453–6456 CrossRef CAS.
- T. Shi, L. Yuan and J. Mu, et al., The effect of Arginine, Lysine and Histidine in the myosin secondary structure by circular dichroism and Raman spectroscopy, CyTA-Journal of Food, 2019, 17(1), 656–660 CrossRef CAS.
- V. A. Ivancic, H. L. Lombardo and E. Ma, et al., Advancing secondary structure characterization of monoclonal antibodies using Microfluidic Modulation Spectroscopy, Anal. Biochem., 2022, 646, 114629 CrossRef CAS PubMed.
- L. L. Liu, L. Wang, J. Zonderman, J. C. Rouse and H. Y. Kim, Automated, High-Throughput Infrared Spectroscopy for Secondary Structure Analysis of Protein Biopharmaceuticals, J. Pharm. Sci., 2020, 109(10), 3223–3230, DOI:10.1016/j.xphs.2020.07.030.
- D. Batabyal, L. Wang, J. Zonderman and M. Wikstrom, Shaping IR Spectroscopy into a Powerful Tool for Biopharma Characterizations, BioPharm Int., 2020, 33(5), 42–47 CAS.
- B. S. Kendrick, J. P. Gabrielson, C. W. Solsberg, E. Ma and L. Wang, Determining Spectroscopic Quantitation Limits for Misfolded Structures, J. Pharm. Sci., 2020, 109(1), 933–936, DOI:10.1016/j.xphs.2019.09.004.
- R. G. Strickley and W. J. Lambert, A Review of Formulations of Commercially Available Antibodies, J. Pharm. Sci., 2021, 2590–2608.e56, DOI:10.1016/j.xphs.2021.03.017.
- Y. Jiang, C. Li, X. Nguyen, S. Muzammil, E. Towers, J. Gabrielson and L. Narhi, Qualification of FTIR Spectroscopic Method for Protein Secondary Structural Analysis, J. Pharm. Sci., 2011, 100(11), 4631–4641, DOI:10.1002/jps.22686.
- M. van de Weert, P. I. Haris, W. E. Hennink and D. J. A. Crommelin, Fourier Transform Infrared Spectrometric Analysis of Protein Conformation: Effect of Sampling Method and Stress Factors, Anal. Biochem., 2001, 297(2), 160–169, DOI:10.1006/abio.2001.5337.
- J. P. Hennessey and W. C. Johnson Jr, Information Content in the Circular Dichroism of Proteins, Biochemistry, 1981, 20(5), 1085–1094 CrossRef CAS PubMed.
- R. Respaud, D. March, C. Parent, T. Pelat, P. Thullier, J. F. Tournamille, M. C. Viaud-Massuard, P. Diot, M. Si-Tahar, L. Vecellio and N. Heuzé-VourC’H, Effect of Formulation on the Stability and Aerosol Performance of a Nebulized Antibody, mAbs, 2014, 6(5), 1347–1355, DOI:10.4161/mabs.29938.
- R. J. Kubiak, N. Lee, Y. Zhu, W. R. Franch, S. v. Levitskaya, S. R. Krishnan, V. Abraham, P. F. Akufongwe, C. J. Larkin and W. I. White, Storage Conditions of Conjugated Reagents Can Impact Results of Immunogenicity Assays, J. Immunol. Res., 2016, 2016(1), 1485615, DOI:10.1155/2016/1485615.
- B. Wei, E. Ma, S. Tang, L. Cadang, V. Collins, S. Gorman, B. Chen, R. Huang, J. Wang, M. Ma and K. Zhang, Real-Time Monitoring of Higher-Order Structure of RNAs by Temperature-Course Size Exclusion Chromatography and Microfluidic Modulation Spectroscopy, Anal. Chem., 2025, 97(10), 5632–5642, DOI:10.1021/acs.analchem.4c06343.
- F. Sert, D. Hız, M. Gülmez, S. E. Cankurtaran, C. I. Kayalan, H. Kurt and M. Yüce, Temperature and pH-Dependent Behaviors of mAb Drugs: A Case Study for Trastuzumab, Sci. Pharm., 2022, 90, 21, DOI:10.3390/scipharm90010021.
- T. Ito and K. Tsumoto, Effects of subclass change on the structural stability of chimeric, humanized, and human antibodies under thermal stress, Protein Sci., 2013, 22(11), 1542–1551, DOI:10.1002/pro.2340.
- Y. Zou, X. Ren and H. Zhang, et al., Efficacy and safety of durvalumab+ chemotherapy vs. atezolizumab+ chemotherapy in the treatment of small cell lung cancer: a retrospective comparative cohort study, J. Thorac. Dis., 2023, 15(6), 3339 CrossRef PubMed.
- A. Dong, P. Huang and W. S. Caughey, Protein Secondary Structures in Water from Second-Derivative Amide I Infrared Spectra1, 1990, vol. 29, https://pubs.acs.org/sharingguidelines.
- A. Dong, J. D. Meyer, J. L. Brown, M. C. Manning and J. F. Carpenter, Comparative Fourier Transform Infrared and Circular Dichroism Spectroscopic Analysis of Α1-Proteinase Inhibitor and Ovalbumin in Aqueous Solution, Arch. Biochem. Biophys., 2000, 383(1), 148–155, DOI:10.1006/abbi.2000.2054.
- S. A. Berkowitz and D. J. Houde, The Complexity of Protein Structure and the Challenges It Poses in Developing Biopharmaceuticals, Biophys. Charact. Proteins Dev. Biopharm., 2020, 3–26, DOI:10.1016/B978-0-444-64173-1.00001-9.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay02324b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |