
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Validated LC-MS/MS methodology for the quantification of CBD, trace level THCA and UK controlled cannabinoids (Δ9-THC, Δ8-THC, CBN and THCV) in food samples†
T.
Hambidge
 *ab,
R.
Nash
a,
S.
Corless
a,
P.
Sanatcumar
c,
P.
Bowdery
c,
J.
Griffin
c,
P.
Sears
b and
C. J.
Hopley
a
*ab,
R.
Nash
a,
S.
Corless
a,
P.
Sanatcumar
c,
P.
Bowdery
c,
J.
Griffin
c,
P.
Sears
b and
C. J.
Hopley
a
aLGC, Queens Road, Teddington, TW11 0LY, UK. E-mail: tabatha.hambidge@lgcgroup.com
bSchool of Chemistry and Chemical Engineering, University of Surrey, Guildford, GU2 7XH, UK
cKent Scientific Services, Abbey Wood Road, Kings Hill, West Malling, Kent ME19 4YT, UK
First published on 13th January 2025
Abstract
Products containing cannabidiol (CBD) have become increasingly popular due to consumer-perceived benefits of improving health and well-being. More specifically in the United Kingdom (UK), CBD food products are categorised as novel foods. For these products to remain on the market, they must have authorisation from the Food Standards Agency on the basis that they are safe, correctly labelled, and do not contain substances classified under controlled drugs legislation in accordance with any existing or future Home Office guidance. This demands for analytical laboratories to be able to accurately measure the CBD concentration using validated methods to confirm correct labelling, as well as the controlled cannabinoid content to ensure the products comply with legislation. To address some of these challenges, this work describes two similar liquid chromatography tandem mass spectrometry (LC-MS/MS) methods which were developed and validated for measuring CBD and trace level controlled cannabinoids in CBD containing foods. The accuracy of the methods developed were tested for the first time through an interlaboratory comparison involving expert laboratories. The methods were applied to a comprehensive study of 148 CBD edible products. In 13 of the products tested (9% of the total) CBD was found below the limit of quantification. Of the remaining 135 products (91% of the total), 66% were found to have detectable amounts of one or more controlled cannabinoids. Of the 13 samples that did not contain detectable levels of CBD, two did contain quantifiable levels of controlled cannabinoids. The validation and sample analysis results reveal intriguing sets of data which help gauge the ongoing narrative surrounding CBD analysis and CBD products available in the UK. The research contributes to the global effort to keep unsafe products off the market.
Introduction
Products containing cannabidiol (CBD) have become increasingly popular. In the United Kingdom (UK), Cannabis sativa L is a scheduled illicit drug.1,2 Over 120 phytocannabinoids have been identified as components of Cannabis sativa L3 including tetrahydrocannabinolic acid (THCA) and cannabidiolic acid (CBDA) which are readily decarboxylated with light, air or heat to form tetrahydrocannabinol (THC) or cannabidiol (CBD).4 CBD is accepted as not having psychoactive properties and therefore is not controlled,5,6 however there are many reports of well-being or therapeutic effects when consuming CBD products.7,8 Nevertheless, there is a shortage of scientifically derived data that provide support for many of the benefits reported by consumers.9 CBD is either extracted from the Cannabis sativa L plant or less commonly synthesised in the laboratory and is often found as an ingredient in consumer products due to its therapeutic reputation.8 Food products containing CBD are available in a variety of matrices including drinks (with alcohol, tea, and coffee), gummy sweets, capsules, oils, chocolates, and tinctures.10–12 They are considered a novel food in the UK, regulated by the Food Standards Agency (FSA), and they must have an approved “novel food authorisation” application to remain on the market13,14 providing information on the composition of the food, proposed uses and intake, and safety information, among other things.15 The food products are usually enriched with high concentrations of isolated CBD and unintentionally contain trace levels of other cannabinoids, depending on the product. Unlike CBD, some of the other cannabinoids present in Cannabis sativa L can have potent psychoactive properties, a well-known example is THC.5 Conversely, some products, such as full spectrum CBD oils, contain other cannabinoids and terpenes on purpose for an “entourage effect”.16,17 The isolation and enrichment of cannabinoids in the food products means the concentrations can vary greatly to those in the inflorescences.CBD concentrations need to be tested in all products to ensure they contain CBD at appropriate levels for consumption and have correct labelling.15,18,19 CBD label compliance has been studied in many countries,20–34 with some studies also looking for additional cannabinoids or other toxic chemicals. UK Official Control Laboratories analyse retail samples taken by Trading Standards to measure CBD concentration for compliance with food labelling regulations and additionally determine whether there are controlled cannabinoids present in the product contrary to the 1971 Misuse of Drugs Act and 2001 Misuse of Drugs Regulations as advised by the Home Office.1,2,35 Manufacturers also require analysis to be carried out to ensure compliance before the products are placed on the market. Due to the structural similarities of cannabinoids, there is a high possibility for CBD products to contain controlled cannabinoids. The Advisory Council on the Misuse of Drugs (ACMD) identified 12 potentially psychoactive cannabinoids ((±) trans-Δ9-tetrahydrocannabinol-C5 (Δ9-THC), (±) cis-Δ9-tetrahydrocannabinol-C5, Δ9-tetrahydrocannabinol-C4 (THCB), Δ9-tetrahydrocannabinol-C3 (THCV), Δ9-tetrahydrocannabinol-C1, Δ8-tetrahydrocannabinol (Δ8-THC), Cannabinol-C5 (CBN), Cannabinol methyl ether-C5 (CBNM), Cannabinol-C4, Cannabinol-C3 (CBV), Cannabinol-C2, Cannabinol-C1) (ref. 36) therefore, laboratories would need to examine the products for all of these controlled cannabinoids to ensure they comply with the Home Office regulations. Historically, only four controlled cannabinoids were available as standards. Using their more conventional names, these four were: cannabinol (CBN), Δ9-tetrahydrocannabinol (Δ9-THC), Δ8-tetrahydrocannabinol (Δ8-THC), and tetrahydrocannabivarin (THCV) but there have been recent advances in more controlled cannabinoids becoming available (Cayman Chemicals, Michigan, United States of America).
A variety of analytical methods quantifying multiple cannabinoids in cannabis and cannabis products (not edible) have been published so far. The analytical techniques used in these methods were liquid chromatography tandem mass spectrometry (LC-MS/MS),37 gas chromatography mass spectrometry (GC-MS),38 high performance liquid chromatography (HPLC) with diode-array detection (HPLC-DAD)39,40 and HPLC with ultraviolet absorption detection (HPLC-UV).40,41 Specific methods have also been produced for CBD in edible products such as honey and olive oil.42 As reported in the Defence Science and Technology Laboratory (DSTL) report 2020 a – HPLC is preferred over GC, as the latter can cause conversion of THCA to THC due to the high temperature in the injection port. Furthermore, mass spectrometry is more sensitive than UV detection.43,44 Analysis for CBD and cannabinoids has been previously reviewed, concluding that there is a need for method validation in a range of edible matrices, better detection capability and, if possible, standardisation.4,45 Measuring CBD and controlled cannabinoids in foodstuffs presents a variety of challenges. These include:
I. The limited availability of all required standards and of isotopically labelled internal standards challenging identification and quantification of cannabinoids in samples. It is also worth noting it is difficult to source a CBD standard that does not contain at least trace amounts of the controlled cannabinoids.
II. The wide variety of complex matrices; foods such as oil drops, gummy sweets, drinks, spreads, and teas present potential challenges such as solubility and homogeneity during sample preparation and matrix effects during analysis.
III. Analytical challenges are compounded by CBD being typically present at high concentrations whereas other cannabinoids are at trace levels and the structural and molecular weight similarities of cannabinoids.
Other methods have been developed to measure CBD and occasionally other cannabinoids in CBD products, and while most methods are fit-for-purpose for measuring CBD, these methods are usually either not targeting UK controlled cannabinoids, or not sensitive enough to detect controlled cannabinoids at trace levels.21,23,24,27,28,31–33 The sensitivity of published methods for cannabinoids in foodstuff range dramatically, with a few methods measuring low ng mL−1 levels and most methods measuring μg mL−1 and even mg mL−1 concentrations.44 Many product certificates simply state the THC concentration is less then <0.2% which is 2 mg g−1. LC-UV methods reported limits of detection (LODs) were > 10 ng mL−1 in an interlaboratory comparison and only seven out of 29 labs could measure the cannabinoids at ≤ 1 ng mL−1 level, all seven using LC-MS/MS.46 Moreover, there is a lack of method validation data or wider performance assessment, for example through interlaboratory comparisons. To address the aforementioned challenges, this work describes validated LC-MS/MS methods that measures the trace levels (ng g−1) of THCA and controlled cannabinoids in highly enriched CBD (mg g−1) food samples.
The methods were developed as part of the UK Government Chemist programme to assess laboratory capability to measure these analytes in food matrices.4 In addition, this methodology was used to analyse 148 food samples, which is a much larger sample size than previous publications.20–34 This work provides valuable insights into cannabinoid analysis, which has benefits for analysts particularly from a UK perspective. Its application within a UK regulatory framework and the challenges it addresses in trace-level detection of a comprehensive panel of cannabinoids in complex food matrices provide added value. For the first time, full method validation for trace-level controlled cannabinoids in real matrices is described with method accuracy assessment through participation in an interlaboratory study.46 This comprehensive study was conducted to better understand the current state of CBD containing products available in the UK and help keep unsafe foods off the market.
Experimental
Materials and reagents
Cerilliant® analytical reference standards of CBD, CBN, THCV, Δ9-THC, Δ8-THC and THCA and labelled internal standards CBD-D3, CBN-D3, THCA-D3 and Δ9-THC-D3 were obtained from Merck (Gillingham, UK). Analytical grade formic acid was purchased from Merck (Gillingham, UK). Honeywell LC-MS grade methanol and acetonitrile were obtained from Fisher Scientific (Loughborough, UK). Propan-2-ol was acquired from Fisher Scientific (Loughborough, UK). Ultra-high purity water (≥18.2 mΩ) was prepared using an in-house Elga purification system.Standard and sample preparation
CBD and controlled cannabinoid (CBN, THCV, Δ9-THC, Δ8-THC and non-controlled THCA) analysis was performed using two complementary methods for two reasons. Firstly, due to the CBD being present in the samples at high concentrations and the controlled cannabinoids (including THCA) at trace levels. Secondly, all CBD standards tested contained trace levels of controlled cannabinoids, even when made synthetically. Having two separate methods prevented contaminating cannabinoids, present in the CBD standard, from biasing the controlled cannabinoid results. Although the controlled cannabinoid LC-MS/MS method could be used for quantifying CBD, the chromatography is longer and has a shallow gradient for the separation of Δ8-THC and Δ9-THC which is unnecessary when measuring CBD only. The methods also have different calibration concentrations to reflect the usual concentration ranges seen in the samples.Standards were prepared gravimetrically on a Mettler Toledo analytical balance (to four decimal places) and diluted from analytical standards in 100% methanol and then further diluted in 100% acetonitrile. All calibration curves were then prepared in acetonitrile/water 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v). The CBD calibration standards consisted of nominal concentrations at 500, 400, 300, 200, 100 and 5 ng g−1. 100 μL of CBD-D3 labelled internal standard was added to 500 μL of all calibration points at 500 ng g−1. The controlled cannabinoid (CBN, THCV, Δ9-THC, Δ8-THC including non-controlled THCA) mixed calibration line consisted of nominal concentrations at 250, 200, 150, 100, 50, 5, and 1 ng g−1. 50 μL of labelled internal standards CBN-D3, THCA-D3 and Δ9-THC-D3 were added to 200 μL of all calibration points at 500 ng g−1. Higher concentrations of internal standard were prepared, but smaller quantities were added into the LC-MS vial, resulting in in-vial internal standard concentrations within the calibration line (CBD-D3: 83 ng g−1 and deuterated controlled cannabinoids: 100 ng g−1).
:50 (v/v). The CBD calibration standards consisted of nominal concentrations at 500, 400, 300, 200, 100 and 5 ng g−1. 100 μL of CBD-D3 labelled internal standard was added to 500 μL of all calibration points at 500 ng g−1. The controlled cannabinoid (CBN, THCV, Δ9-THC, Δ8-THC including non-controlled THCA) mixed calibration line consisted of nominal concentrations at 250, 200, 150, 100, 50, 5, and 1 ng g−1. 50 μL of labelled internal standards CBN-D3, THCA-D3 and Δ9-THC-D3 were added to 200 μL of all calibration points at 500 ng g−1. Higher concentrations of internal standard were prepared, but smaller quantities were added into the LC-MS vial, resulting in in-vial internal standard concentrations within the calibration line (CBD-D3: 83 ng g−1 and deuterated controlled cannabinoids: 100 ng g−1).
Sample preparation for most edible matrices consisted of a series of dilutions in propan-2-ol, followed by acetonitrile and finally into acetonitrile/water 50:50 (v/v) to match the mobile phase starting conditions. In between each dilution the samples were sonicated and centrifuged for 10 minutes each. After centrifugation, if immiscible layers formed, then the top, translucent layer was taken by aspirating manually with a Gilson pipette. Gummy sweets were initially dissolved in warm water at 60 °C before the same dilution series as stated above.
In some of the later gummy sweet analysis an Agilent Technologies QuECheERS (Quick Easy Cheap Effective Rugged Safe) extract pouch method47 was used to improve recovery. The whole sample was cut and homogenised with water; taking the entire sample proved important as occasionally the concentration of CBD was not homogeneous within the sweets. Then acetonitrile containing 2% ammonium hydroxide was added and the sample was further homogenised until the gummy sweet was completely dissolved. An Agilent Technologies QuECheERS extract pouch was added and shaken. The sample was then centrifuged, and the acetonitrile supernatant transferred. In total this extraction was a 10-fold dilution. Subsequent dilutions into the mobile phase starting conditions of acetonitrile/water 50:50 (v/v) were required to dilute the samples into the calibration curve.
The CBD method required a dilution factor based on the stated food label concentration down into the calibration range; an example is shown in Fig. 1 (bottom). Whereas the controlled cannabinoids method required a dilution factor to reduce the CBD concentration to 0.1 mg g−1 to reduce loading of CBD onto the analytical instrumentation; an example is shown in Fig. 1 (top).
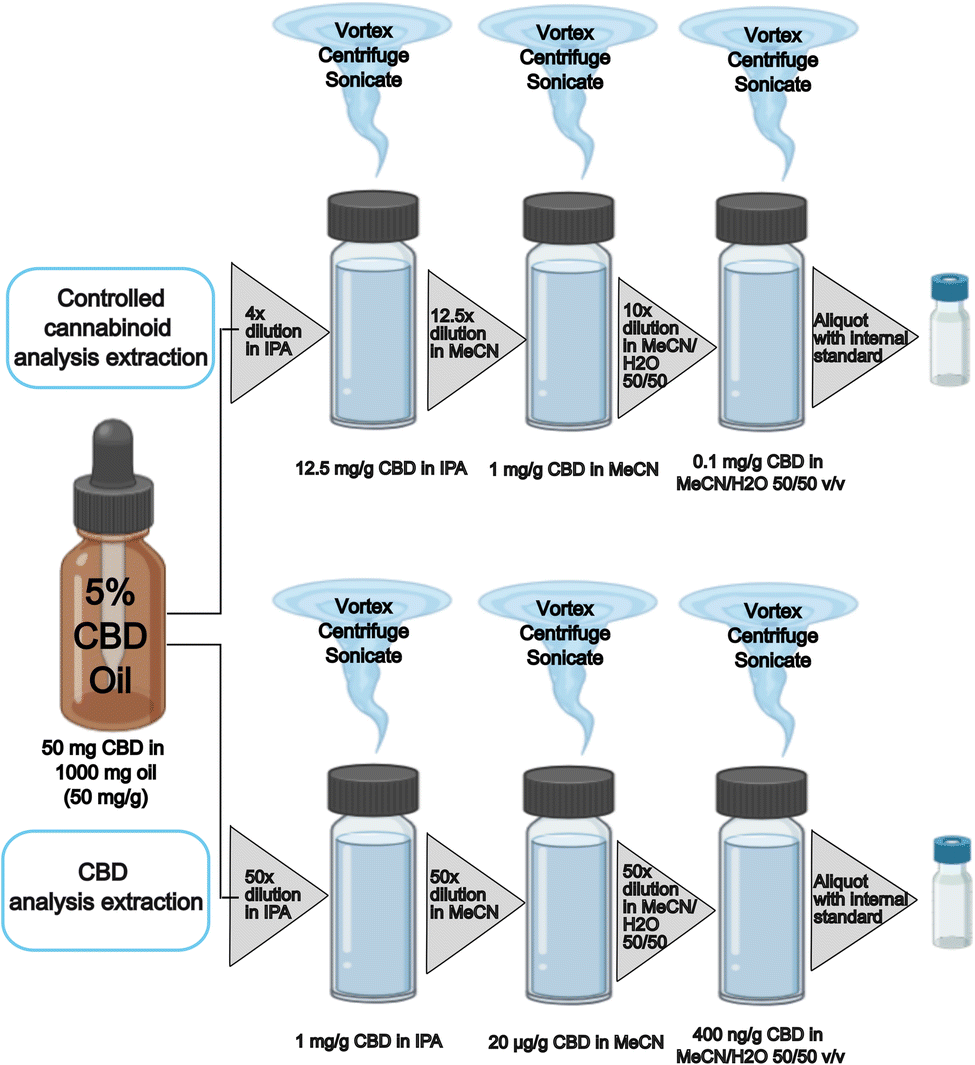 | ||
| Fig. 1 CBD and controlled cannabinoid dilution series examples according to label concentration of 5% CBD (50 mg g−1). For CBD analysis the sample is diluted to the calibration range of 400 ng g−1 (125000× dilution factor) and for controlled cannabinoid analysis the sample is diluted to 0.1 mg g−1 CBD (500× dilution factor). | ||
LC-MS/MS method
A Waters Acquity H-Class UPLC was coupled with a Sciex API 4000 triple quadrupole mass spectrometer for analysis of both CBD and controlled cannabinoid methods.The mobile phases consisted of A: deionised water with 0.1% formic acid (v/v) and B: acetonitrile with 0.1% formic acid (v/v) for both methods and both methods used a Waters Xbridge C8 BEH Column 2.5 μm 100 × 2.1 mm at 30 °C.
The CBD method used a gradient starting at 50% B for 1 minute, increasing to 100% B at 8 minutes, held at 100% B until 11 minutes and then equilibrated back to 50% B from 11.1 minutes to 15 minutes. The flowrate was 0.2 mL min−1. The system had a dwell volume of 500 μL. The multiple reaction monitoring (MRM) transitions are listed in Table 1.
| Analyte | Quantifier pre-cursor (m/z) | Quantifier product (m/z) | Qualifier pre-cursor (m/z) | Qualifier product (m/z) | Collision energy (V) | Retention time (mins) |
|---|---|---|---|---|---|---|
| CBD | 315.2 | 193.2 | 315.2 | 235.2 | 20 | 7.5 |
| CBD-D3 | 318.2 | 196.2 | 318.2 | 238.2 | 20 | 7.5 |
The controlled cannabinoid method (including non-controlled THCA) used a gradient starting at 50% B, increasing to 57% B at 12 minutes, to 80% B at 15.5 minutes, and 100% B at 17 minutes, held at 100% B until 20 minutes and reverting back to 50% B at 20.5 minutes and equilibrating until 28 minutes. The flowrate was 0.3 mL min−1 and the CBD peak was eluted to waste between 12 and 13 minutes, this is to reduce the chance of the detector getting contaminated, and additionally reduce the high baseline caused from loading a high concentration of CBD into the detector, which increases the limit of detection of Δ8-THC and Δ9-THC. The system had a dwell volume of 500 μL. Example chromatograms are included in ESI 1.† MRMs are listed in Table 2. The structures and fragmentation products for CBD, THC, THCV, THCA and CBN have been previously described by Mandolino et al.48
| Analyte | Quantifier pre-cursor (m/z) | Quantifier product (m/z) | Qualifier pre-cursor (m/z) | Qualifier product (m/z) | Collision energy (V) | Retention time (mins) |
|---|---|---|---|---|---|---|
| a CBD should not be observed in the chromatogram – the peak should elute when the flow is diverted to waste to help prevent contamination of the mass spectrometer. b THCA can be analysed in either positive ESI or negative ESI depending on instrument capability and sensitivity. This was performed in negative at LGC using a polarity switching method. | ||||||
| THCV | 287.2 | 165.1 | 287.2 | 231.2 | 30 | 9.7 |
| CBN | 311.0 | 223.5 | 311.0 | 241.5 | 30 | 13.7 |
| CBDa | 315.2 | 193.2 | 315.2 | 235.0 | 30 | 11 |
| Δ9-THC | 315.2 | 193.2 | 315.2 | 235.2 | 30 | 14.8 |
| Δ8-THC | 315.2 | 193.2 | 315.2 | 235.2 | 30 | 14.9 |
| THCAb | 359.5 Pos | 219.5 | 359.5 | 261.5 | 40 | 15.6 |
| 357.2 Neg | 313.2 | 357.2 | 245.0 | −35 | 15.6 | |
| CBN-D3 | 314.0 | 223.0 | 314.0 | 241.0 | 30 | 13.7 |
| Δ9-THC-D3 | 318.0 | 196.0 | 318.0 | 238.0 | 30 | 15.8 |
| THCA-D3b | 362.5 Pos | 264.5 | 362.5 | 222.5 | 40 | 15.6 |
| 360.0 Neg | 316.0 | 360.0 | 248.0 | −35 | 15.6 | |
The source temperature for both methods was 650 °C, the ion spray voltage was set at 5500 V and ion source gas 1 and 2 at 50. MRM windows were used to limit the number of analytes scanned for. Dwell times were adjusted to give a minimum of 13 points across the peaks. Collision energies were optimised by infusing the analytes and tuning on the instrument.
Additional checks were undertaken to assess whether THCA was converting to THC due to in source fragmentation by monitoring the THC transitions at THCA retention time in positive ESI mode. However, no THC peaks were observed at the retention time of THCA.
Method validation
The method was validated following the principles of the Eurachem method validation guidance where possible49 before it was used in an interlaboratory comparison organised by LGC to assess the capabilities of testing laboratories in measuring CBD and controlled cannabinoids.46 Subsequently, the method was shared with Kent Scientific Services (KSS) as part of the intercomparison exercise. Details of the method validation follows.The signal to noise (S/N) was greater than 10 at the LOQ for all primary transitions. At the LOQ, all secondary transitions had an S/N greater than 3. The instrumental limit of quantitation was 1 ng g−1 for THCV and Δ9-THC, 5 ng g−1 for CBD, Δ8-THC and CBN and 50 ng g−1 for THCA at LGC on all instruments. Some example LOQ peaks are shown in ESI 2 and 3.†
As the dilution factors were dependent on the labelled CBD concentration, the method quantified samples ranging from 100 ng g−1 to 500 mg g−1 CBD. The sensitivity of the method enabled detection of trace levels of controlled cannabinoids present in food samples from 5 ng g−1 to 15 mg g−1. This is more sensitive than typical supplier methods that state THC is <0.2% which would equate to 2 mg g−1.
A calibration standard and a blank were injected into the instrument after every six sample injections to confirm the accuracy of calculated concentrations. Calibration curves were plotted using Sciex Analyst® processing software. All lines of best fit were calculated with linear regression and 1/x weighting. An example calibration curve is shown in the ESI 4.† Test samples were diluted to fit CBD concentrations within this linear working range.
Each batch of samples analysed for controlled cannabinoids (and non-controlled THCA) were injected into the instrument along with seven gravimetrically prepared mixed cannabinoid calibration standards. For each batch calibration standards bracketed the samples and for a longer batch to account for instrument drift, additional calibration lines were added. A mid-point calibration standard and a blank were injected into the instrument after every six sample injections to confirm the accuracy of calculated concentrations. The calibration curves were plotted using Sciex Analyst® processing software. All lines of best fit were calculated with linear regression and 1/x weighting. An example calibration curve is shown in the ESI 5.† Test samples were diluted to approximately 0.1 mg g−1 CBD. Any samples with a concentration above the working range for specific cannabinoids were then further diluted in acetonitrile/water 50:50 (v/v) into the working range.
Chromatographic separation of Δ8-THC and Δ9-THC was challenging as they are isomers. On the quaternary method we could achieve fit for purpose separation (not baseline) but the binary pump systems needed further improvement of the elution conditions as it initially did not have chromatographic separation of Δ8-THC and Δ9-THC. By extending the elution gradient from 50% B to 57% B over 12 minutes and the final method performed well on all platforms. There was no statistically significant difference found between the two sets of data, generated by two analysts on different days, this is shown in the ESI 6.†
Although baseline separation was not achieved due to a trade-off between separating other interfering cannabinoids from the complex matrix and the high loading of CBD, Δ8-THC and Δ9-THC were easily integrated separately and met all calibration line and recovery criteria. Further information on chromatography and columns is included in the ESI 7.†
Results
Recovery and precision from sample matrix analysis
Due to a lack of certified reference materials for CBD-containing food matrices, recovery was calculated from in-house spiking experiments on CBD oil and gummy sweet matrices. Oil and gummy sweet matrices were chosen as they are most frequently sold on the market, but also are representative liquid and solid food matrices. Homogenous in vial concentrations of 300–400 ng g−1 after dilution were prepared from 100 mg g−1 or 10 mg g−1 CBD. Six analytical replicates, each of spiked and un-spiked samples were extracted following the same dilution steps (see sample preparation). Recovery was between 95 and 100% for each matrix type and the percent relative standard deviation (%RSD) of the 6 analytical replicates was below 10% for both matrices as shown in Table 3.| Sample | Un-spiked concentration of CBD (ng g−1) n = 6a | Spiked concentration of CBD (ng g−1) n = 6a | Expected spiked concentration of CBD (ng g−1) n = 6a | % recovery | Spiked sample standard deviation n = 6a | Spiked sample % RSD n = 6a |
|---|---|---|---|---|---|---|
| a n = 6 unless stated otherwise – n = 5 for oil 1, where only 5 aliquots of the sample were available. | ||||||
| Oil 1 | 53.1 (n = 5) | 350.2 | 368.8 | 95.0 | 21.0 | 6.0 |
| Oil 2 | 103.1 | 317.8 | 324.6 | 98.2 | 21.9 | 6.9 |
| Gummy bears | 0.8 | 396.3 | 422.7 | 93.8 | 17.7 | 4.5 |
Unlike CBD, high concentrations and quantities of the controlled cannabinoids are not available to purchase. Only 1 mL of 1 mg mL−1 could be purchased of each controlled cannabinoid (Δ8-THC, Δ9-THC, THCV, CBN) and THCA. Overall, this led to challenges with spiking high levels of cannabinoids into the test samples, thus recovery was difficult to calculate. However, in spiked samples where the cannabinoids were present above the limit of quantification, the %RSD of 6 analytical replicates were less than 15% as shown in Table 4. CBN was not over-spiked during this experiment so is not included and THCA was spiked below the LOQ of 50 ng g−1 and is therefore not included.
| Sample | Analyte | Average concentration (ng g−1) | Standard deviation | % RSD |
|---|---|---|---|---|
| n = 6 | n = 6 | n = 6 | ||
| Oil 2 | THCV | 4.3 | 0.2 | 3.8 |
| Oil 2 | Δ9-THC | 19.6 | 1.8 | 9.3 |
| Oil 2 | Δ8-THC | 6.8 | 0.8 | 11.7 |
Interlaboratory comparison
The interlaboratory comparison involved quantifying CBD and the four controlled cannabinoids (Δ9-THC, Δ8-THC, THCV and CBD) along with THCA in two food samples, a hemp oil and a medium chain triglyceride (MCT) based oil.46 The analysis consisted of three analytical replicates prepared on different days and run in three separate batches.The methods presented agreed with the consensus value (median result for all participating laboratories) for both food samples when quantifying CBD. The method also agreed with the consensus values for the controlled cannabinoids that could be reported, although there were only three consensus values out of a possible ten for the two food matrices due the lack of reported values. The lack of reported values is likely due to the cannabinoids being present at trace concentrations.46
Sample analysis
The validated methods were subsequently applied at both LGC and Kent Scientific Services (KSS) over two years to analyse commercially available samples to evaluate the products available on the market in the UK. Fig. 2 presents the CBD containing edible sample types analysed by the laboratories. Full data tables are provided in ESI 8 and Tables S1 and S2.† | ||
| Fig. 2 Categories of CBD containing food products tested at KSS and LGC. | ||
A total of 148 food products claiming to contain CBD, which could be bought online or on the high street in the UK between 2021 and 2023 were analysed. In 13 of the 148 samples (9%) CBD was below quantitation limits. The majority of products contained between 0.1–5% CBD.
91 (61%) of the 148 samples contained quantifiable levels of one or more controlled cannabinoids (CBN, Δ9-THC, Δ8-THC or THCV). However, as 9% of these products did not contain detectable levels of CBD, further data analysis was conducted based on the samples that did contain detectable levels of CBD to gain a better understanding of controlled cannabinoid prevalence in products containing CBD. Of the 135 (91%) samples that did have quantifiable levels of CBD, 66% of these had detectable amounts of one of more controlled cannabinoid with 64% of these samples containing Δ9-THC and/or Δ8-THC, 47% of these samples containing CBN, and 35% of these samples containing THCV. Additionally, 17% of these samples contained the non-controlled acid precursor THCA that could convert to THC. This is shown in Fig. 3. Some products labelled as ‘THC free’ did contain detectable levels of THC, this is shown in Table 5.
 | ||
| Fig. 3 Of the products that contained detectable levels of CBD (135) 66% contained detectable levels of one or more controlled cannabinoids as shown on the right. The additional graph on the left shows the percentage of each product which contained one of the controlled cannabinoids (blue) and THCA (green stripes). | ||
| Matrix | Calculated concentration of THC (μg g−1) | Label |
|---|---|---|
| Oil | 561 | THC <0.2% (2000 μg g−1) |
| Honey/jam/spread | 1.81 | THC free |
| Oil | 10.3 | THC free |
| Gummy sweet | 0.17 | THC free |
| Honey/jam/spread | 0.44 | THC free |
| Oil | 7.95 | THC <0.2% (2000 μg g−1) |
| Oil | 7.27 | THC free |
Of the 13 samples that did not contain detectable levels of CBD, two did contain quantifiable levels of controlled cannabinoids. One product contained all of the controlled cannabinoids as well as THCA. The other contained CBN and THCV.
Interestingly, a high content of CBN could indicate product deterioration, as CBN is the oxidized form of Δ9-THC.50 If the samples are being oxidised this could also explain why the expected concentration of CBD is occasionally not present, as CBD converts to its quinone formations.50 All the samples were within expiry dates upon analysis, thus potentially suggesting storage stability of commercial formulations requires more attention in the future.
It could be hypothesised that with higher concentrations of CBD in the products, there might be higher concentrations of other cannabinoids. From this data analysis we were able to plot CBD concentration against total controlled cannabinoid concentration in each product, this is shown in Fig. 4. Conversely, the figure demonstrates that there is no clear correlation observed between CBD concentration and controlled cannabinoid concentration. This shows a large variation in cannabinoid concentrations in commercial products, which is important to understand for consumer safety.
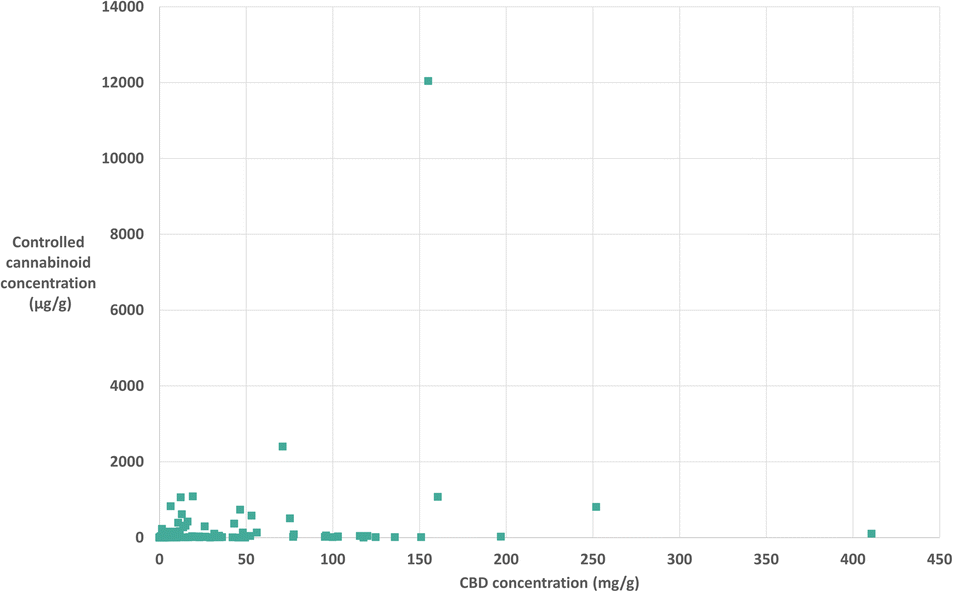 | ||
| Fig. 4 Lack of correlation between CBD concentration (mg g−1) and controlled cannabinoid concentration (μg g−1) in 135 samples. | ||
Discussion
Sample survey discussion
This was one of the largest comprehensive studies into CBD and trace level-controlled cannabinoid concentration in food products. Most of the commercially available CBD products contained controlled cannabinoids at trace levels despite some being labelled ‘THC free’. Moreover 9% of CBD products surveyed contained CBD < LOQ, with two of these containing controlled cannabinoids. These findings suggest the possibility of incompetent or fraudulent activity coupled with lack of, or incorrect quality control analysis (e.g. methods with unsuitable LOQs). There was no correlation between CBD concentration and controlled cannabinoid concentration demonstrating that consumers cannot assume that the lower CBD concentration the less likely there will be controlled cannabinoids in the product and furthermore, the concentrations seem to vary greatly between each product.As there is currently no defined limit of controlled cannabinoids in the UK and the CBD intake recommendation has recently changed, there needs to be an explicit set limit of CBD and controlled cannabinoid consumption defined soon to ensure the safety of the public and then testing laboratories will be able to adjust their methodology LOQs appropriately.
CBD product analysis discussion
Two validated LC-MS/MS methods were developed to quantify CBD and controlled cannabinoids (including non-controlled THCA) in CBD containing food matrices. The challenges met throughout the method development process can be discussed as pre-analytical challenges, sample preparation, matrix effects and analytical workflow and will be discussed below.It is also worth reiterating it is difficult to obtain a CBD standard that does not contain at least trace amounts of the controlled cannabinoids even when made synthetically, more information and a chromatogram of this is shared in ESI 9.† This limits the ability to measure both CBD and controlled cannabinoids in a single analytical method, as the trace levels of other cannabinoids found in the CBD standard could distort the concentration of the controlled cannabinoids.
At the time of method development, only four of the 12 controlled cannabinoids identified by the ACMD36 were available to purchase as standards – cannabinol (CBN), Δ 9-tetrahydrocannabinol (Δ9-THC), Δ 8-tetrahydrocannabinol (Δ8-THC) and tetrahydrocannabivarin (THCV). Since then some additional standards (Δ9-tetrahydrocannabinol-C4 (THCB), Δ9-tetrahydrocannabinol-C1, Cannabinol methyl ether-C5 (CBNM), Cannabinol-C4, Cannabinol-C3 (CBV), Cannabinol-C2, Cannabinol-C1) have become available (Cayman Chemicals, Michigan, USA), however all 12 remain to be readily available as standards.43
Isotopically labelled analogues to be used as internal standards were initially available for CBD, CBN, Δ9-THC and THCA. Additionally, labelled THCV is now also available (Merck, Gillingham, UK). Nonetheless, the labelled analogues are available at low concentrations. This meant that the labelled internal standards could not be added at the beginning of the sample preparation, as is best practice, but instead had to be used as a spike at the end. Use of an internal standard as a later addition allows for monitoring of instrument variability but not for monitoring recovery. Furthermore, there has been stability issues noted with some supplies of internal standards. There are currently no labelled internal standards for Δ8-THC the additional controlled cannabinoids identified by the ACMD (Δ9-tetrahydrocannabinol-C4 (THCB), Δ9-tetrahydrocannabinol-C1, Cannabinol methyl ether-C5 (CBNM), Cannabinol-C4, Cannabinol-C3 (CBV), Cannabinol-C2, Cannabinol-C1).36
Due to the lack of matrix reference materials, over-spiking experiments were undertaken to assess recovery of CBD in this method and as previously discussed, the low concentrations available made it difficult to over spike the controlled cannabinoids. If possible, either matrix materials or higher concentration-controlled cannabinoid solutions should be produced to make quality control samples or over-spiked samples to check recovery.
THCA is not controlled but readily converts to THC via a decarboxylation reaction, hence this additional analyte was included in the controlled cannabinoid method. As a result of our findings, supported by other research, the ACMD presented a series of recommendations to the government about the future of CBD product analysis including advising that THC and THCA content should be summed together. In addition, the ACMD recommended defined limits for controlled cannabinoids and more accurate and standardised testing and the UK government are considering these recommendations.43,51 The concentration of controlled cannabinoids allowed in samples should be defined explicitly, as there will most likely be some level of controlled cannabinoid in all CBD containing products as demonstrated through our analysis. When this is finalised, required limits of detection and method requirements will become clearer for testing laboratories.
The similar conversion can happen when cannabidiolic acid (CBDA) decarboxylates to CBD. CBDA was considered as an additional analyte to this method, however it did not meet the regulation criteria; it is not present on the labels to measure for label compliance, and it is not considered controlled for Home Office compliance. However, this additional cannabinoid and others to measure degradation of the product could be added to this method in the future to better understand why label concentrations often vary from measured concentrations.
Inhomogeneous distribution of CBD was found in gummy sweets. There was further work conducted by Kent Scientific Services and they found that occasionally more CBD was present on the outside surface of the gummy sweet than the inside. It was thus important to analyse the sweet as a whole and take multiple sweets to ensure a representative sample. Additionally, using a QuECHERS procedure rather than the dilution technique resulted in more analytically appropriate concentrations of CBD and controlled cannabinoids.
Additional to the food samples measured in this article, the method has broader applicability. For example, it has been used for both simple CBD oils, and more complex full spectrum CBD oils where higher concentrations of additional cannabinoids and terpenes are present. The method has also been used to analyse CBD isolates to check trace level cannabinoids; a chromatogram measuring a CBD isolate is shown in ESI 9.† Other subsequent matrices analysed using this methodology include cosmetics, moisturisers and e-liquids. Solid matrices and bath bombs require additional sample preparation as they need to be dissolved or cut into pieces first. This could lead to biased results if samples are not sufficiently homogeneous.
Some cannabinoids have the same precursor and product ions, for example CBD, Δ9-THC, Δ8-THC and other non-controlled cannabinoids such as cannabicyclol (CBL) and cannabichromene (CBC). Extended chromatography was needed for adequate separation of all analytes and occasionally, additional standards were required such as CBL, CBC and CBDV (cannabidivarin which has the same transitions as THCV) to identify additional peaks in the spectrum. Additionally, as discussed in the previous section, high concentrations of CBD can raise the background of the MRM channel and interfere with the sensitivity of the THC peaks, an example of this is shown in ESI 9.† To help resolve this, the CBD peak was eluted to waste instead of into the mass spectrometer when analysing for controlled cannabinoids, and there needed to be adequate separation between the CBD and other peaks of importance to achieve this.
Furthermore, Δ8-THC and Δ9-THC co-elute and were challenging to baseline separate. Some columns allowed for good baseline separation of the two, at the cost of an increased LOD as the peaks were wider. Alternate columns and chromatography are included in ESI 7.† The final method called for a balance between these two factors with a good (but not baseline) separation between the two analytes and a low limit of detection. Many columns were trialled to achieve Δ8-THC and Δ9-THC separation as well as CBD separation from all other cannabinoids. Subsequently, a C8 column was chosen to achieve both goals. Both Δ8-THC and Δ9-THC were able to be integrated separately despite the lack of baseline separation and met calibration line and recovery acceptance criteria. Nevertheless, the method would benefit from future chromatographic improvements to allow for baseline separation of these isomeric compounds.
Some unknown interfering and co-eluting cannabinoids can also cause issues with chromatography during analysis and consequently measuring qualifier to quantifier ratios became very important in correct identification of peaks and to make sure the peak area was not biased. The interfering cannabinoids from the complex matrix contributes to the reason why the chromatographic method is lengthy, this is to ensure adequate separation.
In the future, more cannabinoids could be added to this method. Precursors such as CBDA and degradation products such as cannabidiol hydroxyquinone (CBD-HQ) and cannabielsoin (CBE) could help understand the label discrepancies and shelf life of these products. The additional controlled cannabinoids identified by the ACMD (Δ9-tetrahydrocannabinol-C4 (THCB), Δ9-tetrahydrocannabinol-C1, Cannabinol methyl ether-C5 (CBNM), Cannabinol-C4, Cannabinol-C3 (CBV), Cannabinol-C2, Cannabinol-C1) could be added in the future and any other key cannabinoids that may become popular in consumer products such as cannabigerol (CBG).
In countries where these four cannabinoids are not considered controlled or where cannabis is legal, this method would still be useful as it could be used to check label compliance and concentrations for consumer safety and the longer chromatography would be conductive to analysing additional cannabinoids that are country specific.
Conclusions
A sensitive LC-MS/MS method has been developed, validated and is now routinely used by testing laboratories for measuring CBD and controlled cannabinoids in a variety of consumer food products and matrices. For the first time a method of this kind was assessed through an interlaboratory comparison involving expert laboratories, demonstrating its performance to quantify CBD and controlled cannabinoids (including non-controlled THCA) in food samples.Multiple challenges encountered during method development are detailed here for the first time to aid practitioners in the CBD testing field. The production and use of natural and isotopically enriched standards for controlled cannabinoids and matrix reference materials is recommended to enable adequate quality control checks. Multiple MRM transitions are also recommended to ensure methods are accurate and free from interferences, matrix effects and bias.
Application of the method developed here to real market products demonstrated that not all CBD products contain CBD. The majority of the products monitored had controlled cannabinoids present at trace levels. The method quantified samples ranging from 100 ng g−1 to 500 mg g−1 CBD. The sensitivity of the method enabled detection of trace levels of controlled cannabinoids present in food samples from 5 ng g−1 to 15 mg g−1. Some product labels which say samples are ‘THC free’ may have used instrumentation with higher limits of detection, as 64% of CBD containing samples tested contained THC. It is important for consumers to be aware of what is present in their products and therefore accurate analysis is required. Additionally, two products that did not contain quantifiable levels of CBD did contain quantifiable levels of the controlled cannabinoids and the concentrations of cannabinoids seem to vary greatly between each product.
The methods presented here have a broader matrix applicability to test samples such as cosmetics and e-liquids as well as testing CBD isolates for trace level contamination. Although the method is very UK regulation focussed, it could be applicable in other countries for label compliance and cannabinoid monitoring.
The method validation and sample analysis described in this work present intriguing sets of data which help gauge the ongoing narrative surrounding CBD analysis and CBD products and the research contributes to the global effort to keep unsafe products off the market.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Hambidge: investigation, methodology, validation, formal analysis, writing – original draft preparation, writing – review & editing. R. Nash: investigation, validation, writing – review & editing. S. Corless: investigation, methodology, writing – review & editing. P. Sanatcumar: investigation. P. Bowdery: investigation. J. Griffin: investigation. P. Sears: writing – review & editing. C. J. Hopley: conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described in this paper was funded by the UK government Department for Science, Innovation & Technology (DSIT). We thank Michael Walker and Ian Axford for scientific advice. We would like to acknowledge Abigail Cook for conducting some of the analysis. The authors would also like to thank Heidi Goenaga-Infante, Eva Illes-Toth and Christopher McElroy for their critical reading of the manuscript.References
- The Misuse of Drugs Regulations 2001; 2001, https://www.legislation.gov.uk/uksi/2001/3998/regulation/2/made (accessed 2023-01-27).
- Misuse of Drugs Act 1971, https://www.legislation.gov.uk/ukpga/1971/38/contents.
- WHO, WHO Expert Committee on Drug Dependence Critical Review, 2018, pp. 1–27 Search PubMed.
- M. J. Walker, D. T. Burns, I. Axford and G. P. Moss, Cannabinoids, a Tutorial Review Psychoactivity, Regulation, Common and IUPAC Nomenclature, Structures and Abbreviations in Relation to Cannabidiol (CBD) Products, J. Assoc. Public Anal., 2021, 1–28 Search PubMed.
- Drug licensing factsheets: cannabis, CBD and other cannabinoids - GOV.UK, https://www.gov.uk/government/publications/cannabis-cbd-and-other-cannabinoids-drug-licensing-factsheet/drug-licensing-factsheet-cannabis-cbd-and-other-cannabinoids (accessed 2023-07-05).
- F. Grotenhermen, E. Russo and A. W. Zuardi, Even High Doses of Oral Cannabidiol Do Not Cause THC-Like Effects in Humans: Comment on Merrick et al, Cannabis Cannabinoid Res., 2016, 1(1), 102–112, DOI:10.1089/Can.2015.0004; Cannabis Cannabinoid Res., 2017, 2 (1), 1–4, DOI:10.1089/can.2016.0036.
- J. Moltke and C. Hindocha, Reasons for Cannabidiol Use: A Cross-Sectional Study of CBD Users, Focusing on Self-Perceived Stress, Anxiety, and Sleep Problems, J. Cannabis Res., 2021, 3(1), 5, DOI:10.1186/s42238-021-00061-5.
- Food Standards Agency, Consumer Research Report on Cannabidiol (CBD) Extracts, 2020, No. October, pp. 1–26 Search PubMed.
- J. Li, R. Carvajal, L. Bruner and N. E. Kaminski, The Current Understanding of the Benefits, Safety, and Regulation of Cannabidiol in Consumer Products, Food Chem. Toxicol., 2021, 157, 112600, DOI:10.1016/j.fct.2021.112600.
- EMCDDA, Developments in the European Cannabis Market, EMCDDA., 2019, 1–19 Search PubMed.
- A. Hazekamp, The Trouble with CBD Oil, Med. Uses Cannabis Cannabinoids, 2018, 1(1), 65–72, DOI:10.1159/000489287.
- T. P. Freeman, C. Hindocha, S. F. Green and M. A. P. Bloomfield, Medicinal Use of Cannabis Based Products and Cannabinoids, BMJ Online, 2019, 365 DOI:10.1136/bmj.l1141.
- Food Standards Agency, Cannabidiol (CBD), https://www.food.gov.uk/safety-hygiene/cannabidiol-cbd (accessed 2023-08-02) Search PubMed.
- Food Standards Agency, CBD products linked to novel food applications Food Standards Agency, https://www.food.gov.uk/business-guidance/cbd-products-linked-to-novel-food-applications (accessed 2022-10-21).
- Food Standards Agency, Novel Foods Authorisation Requirements and What you Need to Submit as Part of a Novel Food Application, https://www.food.gov.uk/business-guidance/regulated-products/novel-foods-guidance (accessed 2023-07-05).
- C. R. Nyland and D. C. Moyer, Regulating for Safety: Cannabidiol Dose in Food: A Review, J. Food Prot., 2022, 85(9), 1355–1369, DOI:10.4315/JFP-21-374.
- D. Caprioglio, H. I. M. Amin, O. Taglialatela-Scafati, E. Muñoz and G. Appendino, Minor Phytocannabinoids: A Misleading Name but a Promising Opportunity for Biomedical Research, Biomolecules, 2022, 12(8), 1084, DOI:10.3390/biom12081084.
- D. G. Evans, Medical Fraud, Mislabeling, Contamination: All Common in CBD Products, Mo. Med, 2020, 117(5), 394–399 Search PubMed.
- B. Haynes, T. Khan, R. Willis, C. Alexander-White and A. Boobis, Joint Position Paper from the Advisory Committee on Novel Foods and Processes (ACNFP) & Committee on Toxicity (COT) on Establishing a Provisional Acceptable Daily Intake (ADI) for Pure Form (≥98%) Cannabidiol (CBD) in Foods, Based on New Evidence, 2023, No. March 2021, DOI:10.46756/sci.fsa.zcg392.
- J. P. Liebling; N. J. Clarkson; B. W. Gibbs; A. S. Yates; S. E. O'SullivanAn Analysis of Over-the-Counter Cannabidiol Products in the United Kingdom. https://home.liebertpub.com/can 2022, vol. 7 (2), pp. 207–213, DOI:10.1089/CAN.2019.0078.
- D. A. Johnson, M. Hogan, R. Marriot, L. M. Heaney, S. J. Bailey, T. Clifford and L. J. James, A Comparison of Advertised versus Actual Cannabidiol (CBD) Content of Oils , Aqueous Tinctures , e - Liquids and Drinks Purchased in the UK, J. Cannabis Res., 2023, 5, 28, DOI:10.1186/s42238-023-00183-y.
- B. J. Gurley, T. P. Murphy, W. Gul, L. A. Walker and M. ElSohly, Content versus Label Claims in Cannabidiol (CBD)-Containing Products Obtained from Commercial Outlets in the State of Mississippi, J. Diet. Suppl., 2020, 17(5), 599–607, DOI:10.1080/19390211.2020.1766634.
- E. Johnson, M. Kilgore and S. Babalonis, Label Accuracy of Unregulated Cannabidiol (CBD) Products: Measured Concentration vs. Label Claim, J. Cannabis Res., 2022, 4(1), 1–7, DOI:10.1186/s42238-022-00140-1.
- E. Johnson, M. Kilgore and S. Babalonis, Cannabidiol (CBD) Product Contamination: Quantitative Analysis of Δ9-Tetrahydrocannabinol (Δ9-THC) Concentrations Found in Commercially Available CBD Products, Drug Alcohol Depend., 2022, 237, 109522, DOI:10.1016/j.drugalcdep.2022.109522.
- C. Duchateau, M. Canfyn, B. Desmedt, J.-M. Kauffmann, C. Stévigny, K. De Braekeleer and E. Deconinck, CBD Oils on the Belgian Market: A Validated MRM GC-MS/MS Method for Routine Quality Control Using QuEChERS Sample Clean Up, J. Pharm. Biomed. Anal., 2021, 205, 114344, DOI:10.1016/j.jpba.2021.114344.
- I. Di Marco Pisciottano, G. Guadagnuolo, V. Soprano, M. Esposito and P. Gallo, A Survey of Δ9-THC and Relevant Cannabinoids in Products from the Italian Market: A Study by LC–MS/MS of Food, Beverages and Feed, Food Chem., 2021, 346, 128898, DOI:10.1016/j.foodchem.2020.128898.
- FSAI, Survey: Regulatory Issues with Hemp-Based Food and Food Supplements on the Irish Market, 2020, No. February, pp. 1–13 Search PubMed.
- Q. Meng, B. Buchanan, J. Zuccolo, M. M. Poulin, J. Gabriele and D. C. Baranowski, A Reliable and Validated LC-MS/MS Method for the Simultaneous Quantification of 4 Cannabinoids in 40 Consumer Products, PLoS One, 2018, 13(5), 1–16, DOI:10.1371/journal.pone.0196396.
- Shimadzu, The Determination of CBD and General Cannabinoid Content in Hemp Oils Using HPLC with UV Detection, Shimadzu, 2018, 1–10 Search PubMed.
- R. Pavlovic, G. Nenna, L. Calvi, S. Panseri, G. Borgonovo, L. Giupponi, G. Cannazza and A. Giorgi, Quality Traits of “Cannabidiol Oils”: Cannabinoids Content, Terpene Fingerprint and Oxidation Stability of European Commercially Available Preparations, Mol, 2018, 23(5), 1230, DOI:10.3390/MOLECULES23051230.
- E. Jang, H. Kim, S. Jang, J. Lee, S. Baeck, S. In, E. Kim, Y. Kim and E. Han, Concentrations of THC, CBD, and CBN in Commercial Hemp Seeds and Hempseed Oil Sold in Korea, Forensic Sci. Int., 2020, 306, 110064, DOI:10.1016/j.forsciint.2019.110064.
- M. O. Bonn-Miller, M. J. E. Loflin, B. F. Thomas, J. P. Marcu, T. Hyke and R. Vandrey, Labeling Accuracy of Cannabidiol Extracts Sold Online, JAMA, J. Am. Med. Assoc., 2017, 318(17), 1708–1709, DOI:10.1001/jama.2017.11909.
- M. C. Christodoulou, A. Christou, I. J. Stavrou and C. P. Kapnissi-Christodoulou, Evaluation of Different Extraction Procedures for the Quantification of Seven Cannabinoids in Cannabis-Based Edibles by the Use of LC-MS, J. Food Compos. Anal., 2023,(September 2022), 115, DOI:10.1016/j.jfca.2022.104915.
- G. A. Dubrow, R. S. Pawar, C. Srigley, J. Fong Sam, C. Talavera, C. H. Parker and G. O. Noonan, A Survey of Cannabinoids and Toxic Elements in Hemp-Derived Products from the United States Marketplace, J. Food Compos. Anal., 2021, 97, 103800, DOI:10.1016/j.jfca.2020.103800.
- Food. 2018No. 154 the Novel Foods (England) Regulations 2018, England Search PubMed.
- S. N. Mp ACMD Advisory Council on the Misuse of Drugs Report - Phytocannabinoids, 2016.
- N. Christinat, M. C. Savoy and P. Mottier, Development, Validation and Application of a LC-MS/MS Method for Quantification of 15 Cannabinoids in Food, Food Chem., 2020, 318, 126469, DOI:10.1016/j.foodchem.2020.126469.
- A. Laura, Ciolino, L. Tracy and A. M. T. Ranieri, Commercial Cannabis Consumer Products Part 1: GC–MS Qualitative Analysis of Cannabis Cannabinoids, Forensic Sci. Int., 2018, 289, 429–437 CrossRef PubMed.
- L. Vaclavik, F. Benes, M. Fenclova, J. Hricko, A. Krmela, V. Svobodova, J. Hajslova and K. Mastovska, Quantitation of Cannabinoids in Cannabis Dried Plant Materials, Concentrates, and Oils Using Liquid Chromatography–Diode Array Detection Technique with Optional Mass Spectrometric Detection: Single-Laboratory Validation Study, First Action 2018.11, J. AOAC Int., 2019, 102(6), 1822–1833, DOI:10.1093/jaoac/102.6.1822.
- L. Nahar, A. Onder and S. D. Sarker, A Review on the Recent Advances in HPLC, UHPLC and UPLC Analyses of Naturally Occurring Cannabinoids (2010–2019), Phytochem. Anal., 2020, 31(4), 413–457, DOI:10.1002/pca.2906.
- S. Zivovinovic, R. Alder, M. D. Allenspach and C. Steuer, Determination of Cannabinoids in Cannabis Sativa L. Samples for Recreational, Medical, and Forensic Purposes by Reversed-Phase Liquid Chromatography-Ultraviolet Detection, J. Anal. Sci. Technol., 2018, 9(1), 0–10, DOI:10.1186/s40543-018-0159-8.
- R. Deidda, H. T. Avohou, R. Baronti, P. L. Davolio, B. Pasquini, M. Del Bubba, C. Hubert, P. Hubert, S. Orlandini and S. Furlanetto, Analytical Quality by Design: Development and Control Strategy for a LC Method to Evaluate the Cannabinoids Content in Cannabis Olive Oil Extracts, J. Pharm. Biomed. Anal., 2019, 166, 326–335, DOI:10.1016/j.jpba.2019.01.032.
- A. Sullivan, ACMD Advisory Council on the Misuse of Drugs Consumer Cannabidiol (CBD) Products, accessed 10 October, 2024, https://www.gov.uk/government/publications/acmd-advice-on-consumer-cannabidiol-cbd-products/consumer-cannabidiol-cbd-products-report-accessible-version.
- A. L. Capriotti, G. Cannazza, M. Catani, C. Cavaliere, A. Cavazzini, A. Cerrato, C. Citti, S. Felletti, C. M. Montone, S. Piovesana and A. Laganà, Recent Applications of Mass Spectrometry for the Characterization of Cannabis and Hemp Phytocannabinoids: From Targeted to Untargeted Analysis, J. Chromatogr. A, 2021, 1655, 462492, DOI:10.1016/j.chroma.2021.462492.
- M. Walker; I. AxfordGovernment Chemist Guidance Analytical Limits for Controlled Cannabinoids in Specified Products Containing Cannabidiol (CBD). 2021, No. January Search PubMed.
- T. Hambidge, Government Chemist CBD Food and Cosmetic Ring Trial Final Report Cannabidiol and Controlled Cannabinoids, accessed 10 October, 2024, https://www.gov.uk/government/news/cbd-and-controlled-cannabinnoids-results-from-a-ring-trial.
- C. Deckers; J.-F. RoyAgilent Application Note: Quantification of THC and CBD in Gummies and Hard Candies. Decmeber 19 2022 Search PubMed.
- G. Mandolino, G. Roda, D. Stewart, C. Citti, G. Cannazza, P. Linciano, S. Panseri, F. Vezzalini, F. Forni and M. A. Vandelli, Cannabinoid Profiling of Hemp Seed Oil by Liquid Chromatography Coupled to High-Resolution Mass Spectrometry, Front. Plant Sci., 2019, 10, 120, DOI:10.3389/fpls.2019.00120.
- B. Magnusson and U. Örnemark, Eurachem Guide: the Fitness for Purpose of Analytical Methods - a Laboratory Guide to Method Validation and Related Topics, 2nd edn; Eurachem, 2014 Search PubMed.
- W. Jaidee, I. Siridechakorn, S. Nessopa, V. Wisuitiprot, N. Chaiwangrach, K. Ingkaninan and N. Waranuch, Kinetics of CBD, Δ9 -THC Degradation and Cannabinol Formation in Cannabis Resin at Various Temperature and PH Conditions, Cannabis Cannabinoid Res., 2022, 7(4), 537–547, DOI:10.1089/can.2021.0004.
- M. P. Rt Hon Chris Philp, Government response to the ACMD ’ s advice on consumer CBD products (accessible version), https://www.gov.uk/government/publications/acmd-advice-on-consumer-cannabidiol-cbd-products/government-response-to-the-acmds-advice-on-consumer-cbd-products-accessible-version (accessed 2023-12-21).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ay01946f |
| This journal is © The Royal Society of Chemistry 2025 |