
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolid-state nanopore quantification of discrete sequence motifs from DNA and RNA targets in human plasma†
Mohamed Amin
Elaguech
 a,
Komal
Sethi
a and
Adam R.
Hall
*ab
a,
Komal
Sethi
a and
Adam R.
Hall
*ab
aVirginia Tech-Wake Forest University School of Biomedical Engineering and Sciences, Wake Forest University School of Medicine, Winston-Salem, NC 27101, USA. E-mail: arhall@wakehealth.edu
bAtrium Health Wake Forest Baptist Comprehensive Cancer Center, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA
First published on 19th June 2025
Abstract
Fast and sensitive detection of target nucleic acid biomarker sequences in complex biofluids is essential for translational diagnostics. In this work, we report on the use of a solid-state nanopore assay to quantitate sequence motifs in human plasma. Extracted DNA or RNA is annealed to a biotinylated DNA oligonucleotide probe and then subjected to single-strand-specific enzymatic digestion to decompose off-target regions. The remaining duplex product is then bound to a protein tag that enables selective detection via resistive pulse sensing. We first demonstrate our approach on single-strand DNA and single-strand RNA spiked into human plasma and then extend the methodology to double-strand DNA, expanding the range of motifs that can be targeted. These advancements position our assay as a tool for the analysis of viral, bacterial, and human genetic markers.
Introduction
The detection of molecular biomarkers in patient biofluids provides an important pathway to rapid, frequent, and minimally- or non-invasive assessment across the spectrum of human health. While diverse molecules can serve this purpose, nucleic acids represent a particularly important class of biomarker because the rich information contained in their sequences can report on molecular origin, biological role, mutation status, and other features relevant to disease emergence or progression. For ex ample, the genomes of blood-borne pathogens like viruses and bacteria feature conserved sequence motifs1,2 that can be detected to identify their presence or quantitated to indicate infection level. Meanwhile, circulating cell-free DNA (cfDNA)3 has provided a means of probing tumor DNA to identify actionable mutations for therapy.4 Consequently, with appropriate analytical techniques, quantitative detection of specific sequences can provide tangible benefits to human health.A limited number of technologies are available for the task of assessing nucleic acid motifs. Next-generation sequencing (NGS) platforms like Oxford Nanopore5 and Pacific Biosciences6 have lowered the costs associated with reading nucleic acid sequences directly. However, these approaches are by nature untargeted and therefore require significant analyses to interpret. A more targeted approach is quantitative polymerase chain reaction (qPCR),7,8 wherein primer probes are designed to bind to specific sequences flanking the motif of interest, allowing it to be amplified. Incorporation of a reporter (typically fluorescent) provides real-time readout of the amplification process and enables quantitation relative to known standards. qPCR has become the gold-standard for sequence detection due to its relative ease of use and outstanding sensitivity as well as the selectivity afforded by the Watson–Crick base pairing at its core. However, some critical limitations to the approach still exist, including the lengthy cycling process, the need for expensive optics, and known biases associated with enzymatic amplification.9–11 For these reasons, there is still need for alternative technologies capable of selective and sensitive sequence detection at reduced cost and measurement time.
As a platform capable of electrical analysis at the single-molecule level,12 solid-state nanopores (SSNPs) are a major candidate technology for this purpose. In SSNP detection, a thin insulating membrane featuring a single, nanometer-scale through-hole13 is positioned as a barrier between two chambers of ionic solution and subjected to a voltage that drives ions through the opening to define an open-pore current. The subsequent addition of charged molecules to the appropriate chamber can impel their electrical transport through the pore, creating transient current interruptions (or ‘events’) that mark each translocation. Analysis of event characteristics can report on features of the threading molecules and has been used to probe diverse molecules like DNA,14–18 RNA,19–23 proteins,24–27 small molecules,28 and more29–32 with high sensitivity.
For the application of sequence motif assessment in particular, a significant limitation with SSNP detection is its lack of selectivity: because all passing molecules can in principle produce an event, it is challenging to identify one specific sequence among a mixture. While it is possible to detect a sequence-specific binding entity,33–35 the resulting signals can be subtle and difficult to interpret, inducing uncertainty. To address this, we introduced36 a selective SSNP assay that employs two constituents: a short, biotinylated duplex nucleic acid fragment (<250 bp) and a variant of the protein streptavidin mutated to feature only one active biotin-binding site (monovalent streptavidin, or MS37). When probed separately, these constituents yield insignificant event rates (Fig. 1a (i) and (ii)) because of their small size and rapid translocation speed coupled with the limited bandwidth of typical detection electronics. But when bound together, the larger nucleoprotein complex becomes detectable due to steric interactions with the pore walls36,38 and produces events (Fig. 1a(iii)), the rate of which can be used to determine concentration. Critically, we have shown that this contrasting behavior is only observed for duplex nucleic acids, with single-strand molecules being too compact to alter translocation dynamics38 (Fig. 1a(iv)). Consequently, by providing a single-strand biotinylated DNA probe designed to match a specific sequence in solution, a detectable duplex nucleoprotein complex can be formed only when the target is present, enabling its direct quantitation. The resulting approach utilizes the high selectivity of Watson–Crick base pairing like qPCR but avoids the need for amplification cycles due to the molecular sensitivity of the detection mechanism.
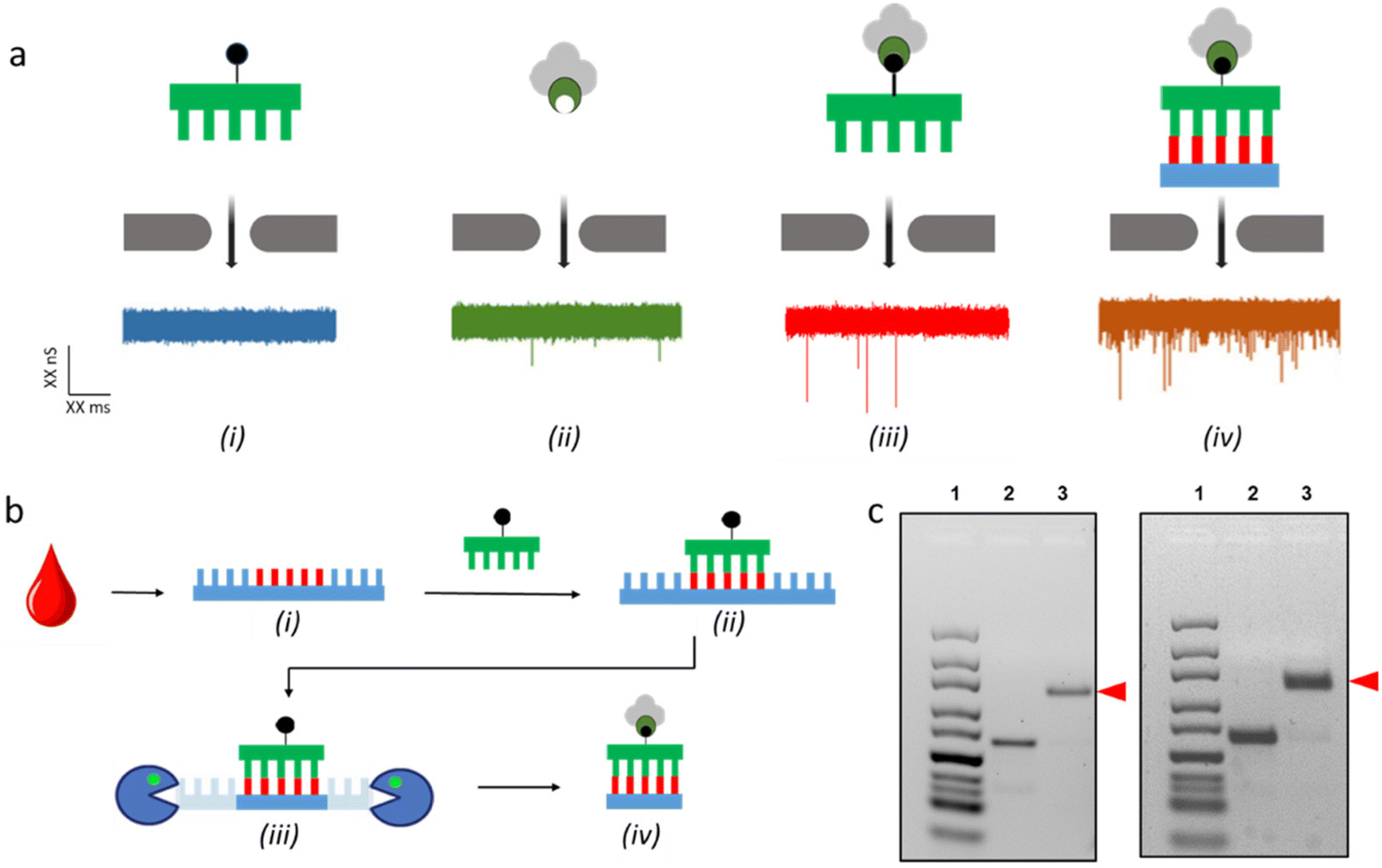 | ||
| Fig. 1 (a) Graphical representations (top) and example ionic current traces (bottom, 300 mV) for the translocation of (i) biotinylated probe, (ii) MS, (iii) probe + MS, and (iv) nucleoprotein complex. (b) Schematic representation of specific sequence isolation protocol from single-strand DNA and RNA targets showing (i) nucleic acids extraction from plasma, (ii) target annealing with the probe, and (iii) enzymatic digestion of single-strand regions to result in (iv) a duplex fragment capable of binding to MS. (c) Gel electrophoresis images of the product isolated from the DNA (left) and RNA (right) targets. In both images, lane 1 is a ladder, lane 2 is the duplex product, and lane 3 is the nucleoprotein complex resulting from MS binding (red arrow). Full gel in ESI Fig. S1.† | ||
We initially demonstrated selective SSNP sequence detection with microRNAs38 because they are intrinsically single-stranded and are of a size that is naturally within the dynamic range of the technique. However, considering the maximum size constraint, the types of nucleic acid sequences targetable with the assay are limited sharply. To expand the applicability of our approach, we recently reported on a variation of this approach that enables motifs within long, single-strand genomes to be quantitated.36 By annealing a short, biotinylated DNA probe to large target molecules and then digesting the remaining unannealed regions enzymatically, we showed that we could assay both single-strand DNA (M13mp18) and single-strand RNA (HIV-1B) genomes, broadening the scope of detection significantly.
In this report, we extend our SSNP sequence detection assay further by describing enhancements in two specific directions with translational relevance. First, to expand our protocols beyond in vitro measurements, we demonstrate sequence motif assessment in human biofluid by spiking DNA and RNA into human plasma and then performing quantitative detection for each. We find different sensitivities associated with the two types of nucleic acid, dictated primarily by differences in their respective efficiencies of recovery. Second, we advance our approach to enable the targeting of motifs internal to double-strand nucleic acids. By melting duplex DNA in the presence of probe molecules and then inhibiting large-scale re-annealing of the target genomes prior to digestion, we show that an internal motif from one target strand can be recovered and detected. Finally, we also establish the viability of this double-strand genome assessment in human plasma. Our results demonstrate the translational potential of our SSNP approach for detecting nucleic acid sequence biomarkers in human biofluids with sensitivity and specificity.
Materials and methods
Biomolecules and reagents
Biomolecules were obtained commercially, including M13mp18 single-strand (ss-) and double-strand (ds-) DNA (New England Biolabs, Ipswitch, MA, USA), Cas9 messenger RNA (mRNA) (TriLink Biotechnologies, San Diego, CA, USA), XbaI enzyme (New England BioLabs), and Mung Bean Nuclease (MBN) (New England Biolabs) along with 10× concentrated MBN buffer (New England Biolabs). MS was synthesized in-house following a protocol reported previously.37 Synthetic DNA oligonucleotide probes were purchased from Integrated DNA Technologies (Coralville, IA, USA). The sequence of the M13mp18 complementary probe was: CGA ACT AAC GGA ACA ACA TTA TTA CAG G![[T with combining low line]](https://www.rsc.org/images/entities/b_char_0054_0332.gif) A GAA AGA TTC ATC AGT TGA GAT TTA GGA ATA and the sequence of the Cas9 mRNA complementary probe was: TGC TCC GCT CCA TGA TGC CCA GCA GC CCTTCA CGC TCT TCA GCT TCT TGC TCT. In each sequence, represents the position of a biotinylated thymine. De-identified, pooled human plasma, phenol/chloroform/isoamyl alcohol (25
A GAA AGA TTC ATC AGT TGA GAT TTA GGA ATA and the sequence of the Cas9 mRNA complementary probe was: TGC TCC GCT CCA TGA TGC CCA GCA GC CCTTCA CGC TCT TCA GCT TCT TGC TCT. In each sequence, represents the position of a biotinylated thymine. De-identified, pooled human plasma, phenol/chloroform/isoamyl alcohol (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :24:1), and chloroform were each obtained commercially (Sigma Aldrich, St Louis, MO, USA).
:24:1), and chloroform were each obtained commercially (Sigma Aldrich, St Louis, MO, USA).
DNA/RNA spike-in and extraction from human plasma
40 μL of human plasma were mixed with 70 μL DNase-/RNase-free water before adding 30 μL ethylenediaminetetraacetic acid (EDTA) (Fischer Scientific, Hampton, NH, USA) to the mixture drop-wise to suppress native nuclease activity. After 2 hours incubation at room temperature, either M13mp18 (ssDNA or dsDNA) or Cas9 mRNA in water was added to a final concentration as indicated in the text and mixed by light vortexing. For extraction, the spiked plasma was mixed with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) (Acros Organics, Morris, NJ, USA) in a phase lock gel tube (Quantabio, Atlanta, GA, USA) and centrifuged at 17000g. The same process was repeated once with pure chloroform to remove remnant phenol. The resulting aqueous solution (containing DNA, RNA, and other soluble constituents) was collected, purified, and concentrated using an appropriate kit: DNA Clean & Concentrator kit (Zymo Research, Irvine, CA, USA) for DNA or RNA Clean & Concentrator kit (Zymo Research) for RNA. Final extracts were suspended in pure water and their concentrations were determined using a Qubit fluorometer (Fisher Scientific).
Sequence motif isolation from single-strand nucleic acid targets
Biotinylated DNA probe was mixed with the extracted nucleic acids at a molar ratio of 13:1 for the DNA and 40:1 for the RNA and then annealed for 5 minutes at 95 °C, followed by a gradual temperature decrease from 90 °C to 25 °C at a rate of 1 °C per minute. The need for a higher molar ratio of probe for RNA annealing was described in our previous work.39 For RNA in particular, 200 μM magnesium chloride (Sigma Aldrich) was included in the mixture to enhance the stability of the DNA/RNA heteroduplex. Following annealing, single-strand nucleic acids were digested by adding 40 U of MBN enzyme for every 1 μg of DNA or 180 U of MBN for every 1 μg of RNA, adding the appropriate volume of concentrated MBN buffer (final concentrations: 30 mM NaCl, 50 mM sodium acetate, 1 mM ZnSO4, pH 5), and incubated for 15 min at 30 °C. Following digestion, MBN activity was halted by mixing an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) in a phase lock gel tube, centrifuging at 17000g, then repeating once with pure chloroform to remove remnant phenol. The final products were purified using the kits as described above and then stored at 4 °C until further use.
Sequence motif isolation from dsDNA targets
Circular M13mp18 dsDNA was linearized by incubation with XbaI (2 U XbaI per 1 μg DNA) for 1 hour at 37 °C, followed by a heat inactivation step at 65 °C for 20 minutes. The linear dsDNA was mixed with the biotinylated DNA probe at 20× molar excess, incubated for 5 minutes at 95 °C to denature the dsDNA, and then reduced immediately to 75 °C before cooling first to 50 °C (at a rate of 1 °C per 4 min) and finally to 25 °C (at a rate of 1 °C min−1) to anneal probes. Next, single-strand nucleic acids (off target M13mp18 and unannealed probes) were digested by adding 50 U of MBN enzyme for every 1 μg of dsDNA and incubating for 15 min at 30 °C in 1× MBN buffer. Following digestion, the mixture was combined with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) in a phase lock gel tube and centrifuged at 17000g. The process was repeated once with pure chloroform as above. The final product was purified with the DNA Clean & Concentrator kit and stored at 4 °C until further use.
Gel electrophoresis
All gel analyses were conducted in 1× TBE buffer (445 mM Tris base, 444 mM Boric acid, and 10 mM EDTA pH 8) under an applied voltage of 180 V and in the presence of GelRed nucleic acid stain (Phenix Research Products, Candler, NC). Isolated 60 bp products were run on 3% agarose gel for 45 min, along with ultra-low range DNA ladder (Fischer Scientific). Gel images were acquired with an E-Gel imager (Thermo Fisher Scientific, Waltham, MA) and processed with the accompanying Gel Capture software.Nanopore measurements
4 × 4 mm silicon chips with a 20 nm thick, freestanding silicon nitride membrane were purchased commercially (Norcada, Edmonton, AB, Canada). In each membrane, a single aperture was produced using helium ion milling (Orion PLUS, Carl Zeiss, Peabody, MA) with a procedure described elsewhere.40 The resulting devices featured SSNPs with diameters between 8.5–12 nm as determined from the measured electrical resistance41 and assuming an effective thickness of 1/3 of the total membrane thickness.41 SSNP chips were stored in a 50% ethanol solution prior to use. In preparation for measurements, each chip was rinsed thoroughly with deionized water and ethanol, dried under filtered airflow, and then treated with air plasma (30 W, Harrick Plasma, Ithaca, NY, USA) for 60 seconds on each side to increase hydrophilicity and prevent bubble formation at the pore. The chip was placed immediately in a custom flow cell 3D printed in CE 221 resin (Carbon, Inc., Redwood City, CA, USA) and the chambers on either side of the chip were filled with optimized37 measurement buffer (0.75 M NaCl, 10 mM Tris, 1 mM EDTA, pH 8). The transmembrane voltage was applied through Ag/AgCl electrodes introduced to each chamber and connected via a patch-clamp amplifier (Axopatch 200B, Molecular Devices, San Jose, CA) for voltage application and ionic current measurement. Data were collected using custom LabView data acquisition software (National Instruments, Austin, TX). All presented measurements were conducted under an applied voltage of 300 mV and recorded at a rate of 200 kHz using a 100 kHz four-pole Bessel filter and subsequently analyzed using custom LabView software with which an additional 10 kHz low-pass filter was applied. Only events with amplitudes greater than 4.5× the standard deviation of the baseline current and with a duration >25 μs were considered. Data were recorded in discrete 3.2 s segments that were used to calculate the average rate as well as a standard deviation (error) for each measurement. All analyses reported here were performed in triplicate (on three independent SSNP devices) and at room temperature.Results and discussion
To establish the translational efficacy of SSNP sequence assessment, we first sought to demonstrate the quantitation of DNA and RNA motifs in human plasma. As a model, we employed a spike-in approach that allowed us to assay select sequences not found in humans, allowing us to avoid any possibility of interference by native sequences already present in the biofluid. For the DNA target, we selected the M13mp18 bacteriophage genome, a ssDNA with a length of 7249 nt. For RNA, we employed the 4522 nt Cas9 mRNA associated with Protein 9. While bare nucleic acids in vivo are prone to degradation by native nucleases, nucleic acid biomarker targets are typically shielded from such activity by barriers that include viral capsids and extracellular vesicles. In absence of these mechanisms of protection for our model molecules, we instead inhibited nucleases by chelating the Mg2+ cofactor required for their activity using EDTA42,43 prior to spike-in (see Materials and methods).With the integrity of the target sequences ensured, we prepared multiple aliquots (140 μL each) of human plasma with different amounts of either M13mp18 ssDNA or Cas9 mRNA spike-ins ranging from 0 to 12 μg. We then employed a protocol to yield biotinylated duplex product appropriate for SSNP analysis from each as shown schematically in Fig. 1b. Our approach began with a two-step process for removing other components of the plasma and isolating only DNA or RNA elements, respectively. In the first step, we performed a phenol–chloroform extraction44 to separate protein constituents to the organic phase and trap lipids at the organic–aqueous interface. This treatment preserved all nucleic acids in the aqueous phase along with other soluble molecules. In the second step, we subjected the remaining mixture to DNA- or RNA-specific extraction via a commercial kit. Here, we noted a difference in the extraction yields of the two kits (ESI Fig. S2†). Given that downstream SSNP analyses are performed on extracted materials, this disparity would manifest in a concomitant disparity in measurement sensitivity. However, this was partially offset by the high sample purity achieved as well as the volume reduction (∼6×) resulting from the kits, which scaled the relative concentrations of the targets and thus their detectability.
After recovering and concentrating DNA or RNA from plasma (Fig. 1b(i)), the next step in our protocol was to bind a 60 nt biotinylated oligonucleotide probe to the target motifs in each sample (Fig. 1b(ii)). To ensure that all available motifs were coupled with a probe, we introduced the latter at an excess (see Materials and methods) and then brought the temperature to 95 °C down to 25 °C slowly (1 °C min−1) to promote annealing. Following this, we digested the remaining single-strand nucleic acids – including off-target native nucleic acids, unannealed probes, and the regions of the target nucleic acids flanking the annealed probe (Fig. 1b(iii)) – using MBN, an endonuclease that facilitates the breakdown of internal phosphodiester bonds with a strong preference for single-strands over duplexes.45,46 Because of the preponderance of native RNA in plasma,47 the total mass of RNA extracted from the negative (unspiked) specimen (1.65 ng μl−1) was significantly higher than the extracted DNA (0.5 ng μl−1) and consequently more MBN was required to digest the former. Finally, we performed a phenol–chloroform extraction to both inactivate MBN enzymes and remove them from the solution. The end products (Fig. 1b(iv)) of this treatment were 60 bp biotinylated dsDNA fragments for the M13mp18 target and 60 bp biotinylated DNA/RNA heteroduplex fragments for the Cas9 target, both suitable for the SSNP assay. We note that we designed our probes to feature their respective biotin moieties near their centers to avoid loss of the tag; despite the preference of MBN for single-strand nucleic acids, it is known to digest duplex molecules weakly through thermally-driven structural fluctuations (i.e., breathing) at molecular ends.39
Having assembled assay-compatible biotinylated constructs from specific motifs within the DNA and RNA targets, we next probed them with our assay. For this, the recovered duplexes from all plasma specimens were incubated with MS, added at a 5× molar ratio compared to the probe to ensure that all constructs were bound. The nucleoprotein complexes were confirmed by electromobility shift assay (EMSA; Fig. 1c) prior to being measured by SSNP analysis using optimized buffer conditions.37 For all samples, we observed a steady baseline current featuring events that could be easily distinguished (Fig. 2b and d). Analyses of event rates revealed linear trends for both M13mp18 and Cas9 targets as a function of spike-in concentration (Fig. 2a and c), similar to previous reports with the selective SSNP assay38,39,48,49 and confirmatory that its signals were proportional to the amount of target in the plasma specimens. The observed difference in slope for DNA (36 ± 4 fM−1 s−1) and RNA (150 ± 7 fM−1 s−1) appeared to stem solely from differences in the respective yields of extraction for DNA and RNA from plasma, given that all other experimental details were kept consistent. Indeed, plotting SSNP event rates against the recovered 60 bp construct concentrations (ESI Fig. S3†) yielded similar results for both molecules, demonstrating the regularity of the measurements themselves. While the consequence of the difference in slopes was a disparity in assay sensitivity, it also highlighted the extraction methods as primary targets for potential improvements to the overall approach. The negative controls (no spike-in) produced event rates close to zero (∼0.07 s−1), attributable mainly to unbound MS that was still added to the samples. These low values further signified the efficient removal of background molecules in our extraction protocol and contributed to the high sensitivity of our measurement for both DNA and RNA targets in plasma. Using the limit of detection (LoD) expression 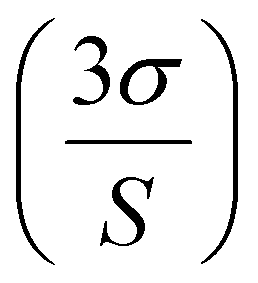 , where σ is the standard deviation of the negative control (noise floor) and S is the slope of the calibration curve, we determined that our measurements could resolve as little as 0.41 nM of M13mp18 ssDNA and 0.26 nM of Cas9 mRNA in human plasma with clear pathways to further improvements.
, where σ is the standard deviation of the negative control (noise floor) and S is the slope of the calibration curve, we determined that our measurements could resolve as little as 0.41 nM of M13mp18 ssDNA and 0.26 nM of Cas9 mRNA in human plasma with clear pathways to further improvements.
 | ||
| Fig. 2 (a) SSNP event rate as a function of ssDNA spike-in concentration (bottom axis) as well as the amount of DNA recovered from plasma (top axis). (b) Example current traces for measurements of (i) 0, (ii) 12.5, and (iii) 24.1 nM ssDNA spike-in. (c) SSNP event rate increase as a function of ssRNA spike-in concentration (bottom axis) and the amount of RNA recovered from plasma (upper axis). (d) Example current traces for measurements of (i) 0, (ii) 23.3, and (iii) 34 nM ssRNA spike-in. (a) and (c), Solid lines are linear fits to the data, and error bars represent the standard deviation of triplicate measurements on independent SSNP devices. All measurements were conducted at 300 mV applied voltage. Further analysis in ESI Fig. S4.† | ||
Following validation of single-strand sequence motif quantification in biofluid, we next sought to extend our approach to include targets in double-strand genomes. For this, we used as a model dsDNA the 7249 bp phage vector M13mp18 RF I: the circular, duplex form of the bacteriophage DNA used above. To demonstrate the process of motif extraction from this molecule, we initially performed the protocol in vitro. The restriction enzyme Xba I was first used to linearize the covalently-closed construct before being removed from solution by a clean-up kit (Fig. 3a(i)). Then, a 20× molar excess of biotinylated probe targeting a motif on one strand of the duplex DNA was added and the mixture was held at 95 °C for 5 minutes to fully denature the dsDNA (Fig. 3a(ii)). Then, the temperature was dropped to 75 °C cooled down to 50 °C, and finally brought to room temperature to promote annealing with the probe and prevent the strands from reannealing. However, we were not able to completely prevent melted strands from interacting, as faint bands were observed in the gel images (Fig. 3b, top), indicating minimal strand reannealing. Following probe annealing, the remaining steps were identical to those described above for single-strand targets, including enzymatic digestion (Fig. 3a(iii)), phenol–chloroform extraction, DNA Clean & Concentrator kit, and incubation with a 5× molar excess of MS (Fig. 3a(iv)). The efficacy of this process to isolate the target sequence was again confirmed by EMSA (Fig. 3b, top), indicating the upward shift that was characteristic of a 60 bp duplex product with an intact biotin moiety upon MS binding.
 | ||
| Fig. 3 (a) Schematic representation of target motif isolation from dsDNA, showing (i) a duplex containing a target sequence is (ii) melted and reannealed in the presence of a biotinylated probe and then (iii) single-strand regions are digested to yield (iv) a duplex fragment capable of MS binding. (b) Gel electrophoresis images of the product isolated from the dsDNA target in water (upper gel) and spiked into plasma (lower gel). In both images, lane 1 is a ladder, lane 2 is the duplex product, and lane 3 is the nucleoprotein complex resulting from MS binding (red arrow). Full gels in ESI Fig. S5.† | ||
We next demonstrated that dsDNA motif extraction and detection could also be performed in biofluid. For this, we prepared a 140 μL human plasma solution spiked with 2.5 μg (3.8 nM) of M13mp18 RF I. As in our prior measurements, EDTA was used to chelate Mg2+ in advance of adding the spike-in to inhibit nuclease activity and protect nucleic acids from degradation. Total DNA was then isolated from the plasma using a commercial kit, the efficiency of which was the main limit on assay sensitivity as described above. Using the isolated dsDNA, subsequent protocol steps were identical to those employed in vitro but with an additional cleaning step with pure chloroform following each phenol–chloroform extraction. The successful retrieval of the final construct was verified by EMSA (Fig. 3b, bottom), showing results identical to the in vitro process. Finally, we performed SSNP assessments. Fig. 4 (red) shows the event rate of the 60 bp plasma extract complexed with MS as a function of applied voltage, showing the linear dependence reported previously.39 The measured rates for the complex were significantly higher than both the biotinylated 60 bp DNA (Fig. 4, black) and the MS (Fig. 4, blue) alone at the same concentration across the entire investigated voltage range; for example, at 300 mV the spiked measurement was ∼3-fold higher than the negative control (Fig. 4, inset). These results demonstrate the selectivity of the assay for dsDNA sequence targets.
 | ||
| Fig. 4 (a) Voltage-dependent translocation event rates for isolated nucleoprotein complex (red), biotinylated 60 bp alone (black), and MS alone (blue). All error bars represent the standard deviation of triplicate measurements on independent SSNP devices. Solid lines are linear fits to the data. Inset: comparison of event rates for the nucleoprotein complex isolated from plasma and the negative control measured at an applied voltage of 300 mV. Values differ with a significance of p < 0.001 as determined by Student's t-test. | ||
Conclusion
In this study, we presented a method for isolating specific sequence motifs from single- and double-strand nucleic acid targets in complex biofluid and detecting them using a selective SSNP assay. Large DNA and RNA targets spiked into human plasma were isolated and then annealed to a synthetic biotinylated DNA probe with complementarity to a specific internal sequence. Once annealed, the unpaired regions of the genomic template were digested with a single-strand specific endonuclease, creating duplex constructs that match the length of the probe. The biotin moiety in these duplexes allowed them to bind to a MS protein, facilitating their analysis by a selective SSNP assay. In this work, we first investigated the performance of our assay using two different single-strand nucleic acid targets spiked into human plasma: M13mp18 ssDNA and Cas9 mRNA. Here, we demonstrated that our assay could retrieve a consistent amount of the specific sequence across the investigated concentration range of the nucleic acid templates. Our SSNP assay exhibited good selectivity and sensitivity, yielding a LoD of 0.41 and 0.26 nM for ssDNA and RNA, respectively. After assessing assay performance on single-strand targets in plasma, we extended our assay by adapting our protocols to accommodate a dsDNA target: M13mp18RF I. Similar to our findings for single-strands, the dsDNA assay exhibited good selectivity and sensitivity.While these outcomes demonstrate the translational potential of our SSNP assay, there are aspects of our approach that will provide key improvements in the future. For example, since the isolation of nucleic acid templates involves multiple purification and extraction steps, optimization of this process is likely to enhance the detection limit of the assay. In addition, we have shown39 that using multiple distinct probes designed to bind to different regions of the target nucleic acids simultaneously can increase the SSNP event rate concomitantly, yielding greater sensitivity. Overall, our results have demonstrated the isolation and quantification of sequence motifs within large single- and double-strand DNA and RNA targets from human biofluids. These findings enhance the versatility of the selective SSNP assay and its associated protocols and enable a range of new applications for the platform, including pathogen screening of viral genomes and mutation analysis for personalized medicine in human biofluids.
Author contributions
M. A. E. contributed to experimental design, performed measurements, analyzed the data, and wrote the manuscript. K. S. performed initial measurements and contributed to experimental design. A. R. H. oversaw the project, contributed to experimental design, and wrote the manuscript. All authors contributed to the editing and review of the manuscript.Conflicts of interest
A. R. H. is listed as an inventor on a patent covering the SSNP assay. The other authors declare no conflicts.Data availability
The data underlying this study's findings are available from the corresponding author upon reasonable request.Acknowledgements
This project was supported by NIH award R33CA246448 and by the Wake Forest Technology Development Program through a Catalyst Phase II award, both to A. R. H.References
- G. A. Marsh, R. Rabadán, A. J. Levine and P. Palese, J. Virol., 2008, 82, 2295–2304 CrossRef CAS PubMed.
- S. H. Portakal, B. Kanat, M. Sayan, B. Berber and O. Doluca, J. Virol. Methods, 2021, 293, 114146 CrossRef CAS PubMed.
- M. Cisneros-Villanueva, L. Hidalgo-Pérez, M. Rios-Romero, A. Cedro-Tanda, C. A. Ruiz-Villavicencio, K. Page, R. Hastings, D. Fernandez-Garcia, R. Allsopp, M. A. Fonseca-Montaño, S. Jimenez-Morales, V. Padilla-Palma, J. A. Shaw and A. Hidalgo-Miranda, Br. J. Cancer, 2022, 126, 391–400 CrossRef CAS PubMed.
- S. Liebs, U. Keilholz, I. Kehler, C. Schweiger, J. Haybäck and A. Nonnenmacher, Cancer Med., 2019, 8, 3761–3769 CrossRef CAS PubMed.
- P. Zheng, C. Zhou, Y. Ding, B. Liu, L. Lu, F. Zhu and S. Duan, MedComm, 2023, 4, e316 CrossRef CAS PubMed.
- P. Cuber, D. Chooneea, C. Geeves, S. Salatino, T. J. Creedy, C. Griffin, L. Sivess, I. Barnes, B. Price and R. Misra, Ecol. Genet. Genomics, 2023, 28, 100181 CAS.
- M. N. Emaus, C. Zhu and J. L. Anderson, Anal. Chim. Acta, 2020, 1094, 1–10 CrossRef CAS PubMed.
- A. Marengo, M. N. Emaus, C. M. Bertea, C. Bicchi, P. Rubiolo, C. Cagliero and J. L. Anderson, Anal. Bioanal. Chem., 2019, 411, 6583–6590 CrossRef CAS PubMed.
- S. Yang and R. E. Rothman, Lancet Infect. Dis., 2004, 4, 337–348 CrossRef CAS PubMed.
- J. M. Kebschull and A. M. Zador, Nucleic Acids Res., 2015, gkv717 CrossRef PubMed.
- P. Ménová, V. Raindlová and M. Hocek, Bioconjugate Chem., 2013, 24, 1081–1093 CrossRef PubMed.
- Y. He and M. Tsutsui, NPG Asia Mater., 2021, 48, 1–26 Search PubMed.
- K. Briggs, H. Kwok and V. Tabard-Cossa, Small, 2014, 10, 2077–2086 CrossRef CAS PubMed.
- J. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, Nat. Mater., 2003, 2, 611–615 CrossRef CAS PubMed.
- A. J. Storm, C. Storm, J. Chen, H. Zandbergen, J.-F. Joanny and C. Dekker, Nano Lett., 2005, 5, 1193–1197 CrossRef CAS PubMed.
- A. T. Carlsen, O. K. Zahid, J. Ruzicka, E. W. Taylor and A. R. Hall, ACS Nano, 2014, 8, 4754–4760 CrossRef CAS PubMed.
- S. King, K. Briggs, R. Slinger and V. Tabard-Cossa, ACS Sens., 2022, 7, 207–214 CrossRef CAS PubMed.
- M. A. Elaguech, Y. Yin, Y. Wang, B. Shao, C. Tlili and D. Wang, Sens. Diagn., 2023, 2, 1612–1622 RSC.
- G. M. Skinner, M. Van Den Hout, O. Broekmans, C. Dekker and N. H. Dekker, Nano Lett., 2009, 9, 2953–2960 CrossRef CAS PubMed.
- M. Van Den Hout, G. M. Skinner, S. Klijnhout, V. Krudde and N. H. Dekker, Small, 2011, 7, 2217–2224 CrossRef CAS PubMed.
- C. Chau, F. Marcuccio, D. Soulias, M. A. Edwards, A. Tuplin, S. E. Radford, E. Hewitt and P. Actis, ACS Nano, 2022, 16, 20075–20085 CrossRef CAS PubMed.
- R. Ren, S. Cai, X. Fang, X. Wang, Z. Zhang, M. Damiani, C. Hudlerova, A. Rosa, J. Hope, N. J. Cook, P. Gorelkin, A. Erofeev, P. Novak, A. Badhan, M. Crone, P. Freemont, G. P. Taylor, L. Tang, C. Edwards, A. Shevchuk, P. Cherepanov, Z. Luo, W. Tan, Y. Korchev, A. P. Ivanov and J. B. Edel, Nat. Commun., 2023, 14, 7362 CrossRef CAS PubMed.
- G. Patiño-Guillén, J. Pešović, M. Panić, D. Savić-Pavićević, F. Bošković and U. F. Keyser, Nat. Commun., 2024, 15, 1699 CrossRef PubMed.
- C. Plesa, S. W. Kowalczyk, R. Zinsmeester, A. Y. Grosberg, Y. Rabin and C. Dekker, Nano Lett., 2013, 13, 658–663 CrossRef CAS PubMed.
- J. Larkin, R. Y. Henley, M. Muthukumar, J. K. Rosenstein and M. Wanunu, Biophys. J., 2014, 106, 696–704 CrossRef CAS PubMed.
- D. B. Roy and A. R. Hall, Analyst, 2017, 142, 1676–1681 RSC.
- K. Chuah, Y. Wu, S. R. C. Vivekchand, K. Gaus, P. J. Reece, A. P. Micolich and J. J. Gooding, Nat. Commun., 2019, 10, 1–9 CrossRef CAS PubMed.
- M. A. Elaguech, M. Bahri, K. Djebbi, D. Zhou, B. Shi, L. Liang, N. Komarova, A. Kuznetsov, C. Tlili and D. Wang, Food Chem., 2022, 389, 133051–133051 CrossRef CAS PubMed.
- K. Lee, K. Park, H. Kim, J. Yu, H. Chae, H. Kim and K. Kim, Adv. Mater., 2018, 30, 1704680 CrossRef PubMed.
- F. Rivas, O. K. Zahid, H. L. Reesink, B. T. Peal, A. J. Nixon, P. L. DeAngelis, A. Skardal, E. Rahbar and A. R. Hall, Nat. Commun., 2018, 9, 1037 CrossRef PubMed.
- L. Xue, H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov and J. B. Edel, Nat. Rev. Mater., 2020, 5, 931–951 CrossRef CAS.
- C. C. C. Chau, N. E. Weckman, E. E. Thomson and P. Actis, ACS Nano, 2025, 19, 3839–3851 CrossRef CAS PubMed.
- W. Yang, L. Restrepo-Pérez, M. Bengtson, S. J. Heerema, A. Birnie, J. Van Der Torre and C. Dekker, Nano Lett., 2018, 18, 6469–6474 CrossRef CAS PubMed.
- N. E. Weckman, N. Ermann, R. Gutierrez, K. Chen, J. Graham, R. Tivony, A. Heron and U. F. Keyser, ACS Sens., 2019, 4, 2065–2072 CrossRef CAS PubMed.
- C. M. Platnich, M. K. Earle and U. F. Keyser, J. Am. Chem. Soc., 2024, 146, 12919–12924 CrossRef CAS PubMed.
- A. T. Carlsen, O. K. Zahid, J. A. Ruzicka, E. W. Taylor and A. R. Hall, Nano Lett., 2014, 14, 5488–5492 CrossRef CAS PubMed.
- S. Abu Jalboush, I. D. Wadsworth, K. Sethi, L. C. Rogers, T. Hollis and A. R. Hall, ACS Sens., 2024, 9, 1602–1610 CrossRef CAS PubMed.
- I. D. Wadsworth and A. R. Hall, Nano Res., 2022, 15, 9936–9942 CrossRef CAS.
- K. Sethi, G. P. Dailey, O. K. Zahid, E. W. Taylor, J. A. Ruzicka and A. R. Hall, ACS Nano, 2021, 15, 8474–8483 CrossRef CAS PubMed.
- J. Yang, D. C. Ferranti, L. A. Stern, C. A. Sanford, J. Huang, Z. Ren, L.-C. Qin and A. R. Hall, Nanotechnology, 2011, 22, 285310 CrossRef PubMed.
- M. Wanunu, T. Dadosh, V. Ray, J. Jin, L. McReynolds and M. Drndić, Nat. Nanotechnol., 2010, 5, 807–814 CrossRef CAS PubMed.
- T. Allers, Nucleic Acids Res., 2000, 28, 6e–66 CrossRef PubMed.
- B. B. Panda, A. S. Meher and R. K. Hazra, J. Parasit. Dis., 2019, 43, 337–342 CrossRef PubMed.
- Ş. Tüzmen, Y. Baskın, A. F. Nursal, S. Eraslan, Y. Esemen, G. Çalıbaşı, A. B. Demir, D. Abbasoğlu and C. Hızel, in Omics Technologies and Bio-Engineering, Elsevier, 2018, pp. 247–315 Search PubMed.
- N. A. Desai and V. Shankar, FEMS Microbiol. Rev., 2003, 26, 457–491 CrossRef CAS PubMed.
- W. Ardelt and M. Laskowski, Biochem. Biophys. Res. Commun., 1971, 44, 1205–1211 CrossRef CAS PubMed.
- A. V. Savelyeva, E. V. Kuligina, D. N. Bariakin, V. V. Kozlov, E. I. Ryabchikova, V. A. Richter and D. V. Semenov, BioMed Res. Int., 2017, 2017, 1–10 CrossRef PubMed.
- O. K. Zahid, B. S. Zhao, C. He and A. R. Hall, Sci. Rep., 2016, 6, 29565 CrossRef PubMed.
- O. K. Zahid, F. Wang, J. A. Ruzicka, E. W. Taylor and A. R. Hall, Nano Lett., 2016, 16, 2033–2039 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00373c |
| This journal is © The Royal Society of Chemistry 2025 |