
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlexible hand-made carbon electrode decorated with metronidazole imprinted polymer†
Dominik
Korol
 *,
Kostiantyn
Nikiforow
,
Paweł
Borowicz
,
Piyush S.
Sharma
and
Maciej
Cieplak
*
*,
Kostiantyn
Nikiforow
,
Paweł
Borowicz
,
Piyush S.
Sharma
and
Maciej
Cieplak
*
Institute of Physical Chemistry of the Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dkorol@ichf.edu.pl; mcieplak@ichf.edu.pl
First published on 28th April 2025
Abstract
Carbon paper was used as a cost-effective electrode material for flexible electrode fabrication. These electrodes were coated with polypyrrole film imprinted with metronidazole. SEM imaging indicated successful covering of the carbon paper fibers. Structure of the pre-polymerization complex was optimized via DFT simulations. This imprinted polymer-modified electrode responded linearly to the logarithm of metronidazole concentration in 0.2 to 200 nM range with the LOD of 0.4 nM in the DPV experiments in the presence of the Ru(NH3)6Cl3 redox probe. Selectivity of the fabricated sensor was appreciably high, and the apparent imprinting factor was equal to IF = 38. Such high selectivity and the imprinting factor confirmed successful imprinting in the polypyrrole matrix. The sensor was validated by metronidazole determination in honey samples. Moreover, robustness of the MIP-coated carbon paper electrode was proven. Only a slight loss of recorded current values was observed when the electrode was bent to approx. 45° and straightened multiple times.
1. Introduction
In industrial farming, several antimicrobial drugs, growth hormones, and other pharmaceuticals are commonly administered to farm animals to treat infections and, prophylactically, to prevent disease or spur growth. Over recent decades, public awareness of pharmaceutical toxicity in foods of animal origin has grown tremendously.1 Consumers continuously express their concern about the risks to health because of the presence of these substances in food products.2 Metronidazole (MTZ) is a broad-spectrum drug that has antibacterial and antiprotozoal activity. Residues of these substances may remain in food. Hence, they may pose a real menace to consumers.2 Therefore, the European Commission (EC) strictly regulates the use of veterinary drugs to feed farm animals.1 Maximum residue limits (MRLs) have been fixed for selected drugs, antibiotics, and hormones in animal products to protect consumers from potential health hazards regarding toxicity and allergic reactions. All these circumstances make the control of veterinary drug residues a necessary step in ensuring consumer protection. Animal foods must be controlled for the drug presence.3 Therefore, fast, inexpensive, and reliable procedures for determining these antibiotics in meat products are in demand.One of the appealing ways to execute these procedures is to use bio- or chemosensors. Antibiotic chemosensing in meat products is advantageous compared to classical or currently used screening methods, such as radioimmunoassays, ELISA, and HPLC.4 These methods are very accurate. However, they require expensive equipment and well-trained laboratory staff to operate it. These methods can be applied only in well-equipped laboratories. On the contrary, electrochemical sensors provide possibility of fast and inexpensive sample testing. Additional recognizing elements must be immobilized on electrode surfaces to provide a selective response. These elements may be of biological origin, i.e., antibodies, aptamers and enzymes, or chemical receptors can be applied. Molecularly imprinted polymers (MIPs) are examples of such chemically synthesized recognizing materials. Importantly, MIPs combine robustness, low production costs, and easy operation with sensitivity and selectivity similar to biological receptors.5 Therefore, MIP sensors found applications in numerous fields, including food safety analysis,6,7 clinical diagnosis,8 cancer biomarkers detection,9,10 forensic sciences,11 and environmental analysis8,12,13 as well as warfare agents detection.14 MIPs are being synthesized by polymerization of functional and cross-linking monomers in the presence of a molecular template.5 These template molecules are trapped in the polymer matrix. After their removal, imprinted molecular cavities emerge in the polymer. These cavities can selectively recognize target analytes due to perfect fitting in terms of size, shape, and pattern of interactions with target analyte molecules.
However, sensitivity and, subsequently, limits of detection (LOD) may be insufficient for trace analysis. Therefore, numerous ways to improve sensor performance were proposed. The easiest way to enhance the analytical signal of an electrochemical MIP sensor was to embed nanostructured materials, such as multi-walled carbon nanotubes (MWCNTs),15–18 graphene,19–22 quantum dots (QDs),16 or gold NPs22,23 in the MIP films. This approach offers only limited electrode surface development and signal enhancement despite the convenience. Electrodes of high specific surface area were applied to achieve a higher electrochemical signal. For example, deposition of various porous noble metal films on the surface of flat electrodes was reported.24–28 These methodologies resulted in the deposition of noble metals of significantly enhanced surface area. However, these procedures were laborious and expensive. Therefore, quite recently, George Whitesides proposed the application of inexpensive materials of high surface, such as paper, thread, or fabric, as a mold to prepare electrodes of the developed surface area.29 Towards that, a paper was soaked with noble metals salts (i.e., gold, silver, platinum, rhodium, palladium, and iridium) and burned off in a furnace. This process resulted in “paper-templated metals”, that is, metal–carbon composites of architecture that resembled paper or fabric. These composites consisted of 95% noble metals, their specific surface area was 20 times higher than for flat/continuous metal, and their conductivity was high enough to apply them as electrodes in electrochemical processes.29 Moreover, a method of enhancing electrode surfaces using paper was proposed.30 Chromatography paper was wax-printed with a designed pattern, sputtered with gold film, cut in proper shape, glued with adhesive in spray to carbon screen printed electrode (C-SPE) surface, and finally decorated with gold nanoparticles (Au NPs) via electrochemical reduction of HAuCl4. This method was very laborious. However, electrochemical signal enhancement was high enough to apply these electrodes for direct voltammetric determination of glucose in the samples, even if the glucose oxidation peak was not visible on unmodified C-SPEs.
Paper has also been widely used to support the fabrication of composite paper/MIP membranes. These membranes were applied for solid-phase extraction experiments,31 and drug delivery systems,32 as selective recognition elements in optical sensors.33–39 There is also one example of microfluidic thermal detection over MIP/paper chemosensors.40 Importantly, in all these sensors, the paper serves only as a solid support for the MIP synthesis in the form of a membrane, or it can also be applied as support for MIP film-based electrochemical sensors.41–46 However, different nanomaterials were employed to achieve appreciable sensitivity.38,44 For instance, a paper was decorated with Au and ZnO NPs.41 This material was conductive enough to electropolymerize pentachlorophenol-imprinted polypyrrole film on the top. Then, ascorbic acid was exploited as an efficient electron donor for scavenging photo-generated holes, facilitating the generation of stable photocurrent. This photocurrent decreased with increasing pentachlorophenol concentration. Moreover, paper modified with paraffin wax and carbon ink was applied as electrode material in the MIP film electrochemical sensor for bovine serum albumin (BSA) determination.42 BSA-MIP film was synthesized via free-radical polymerization, and after removal of the BSA template, the fabricated electrode was applied to impedimetric BSA determinations. However, neither selectivity performance nor imprinting factors were reported in either case. Therefore, it is difficult to determine if the imprinting process was successful.
The use of ready-made conductive materials as electrode materials allows to skip the stage related to sputtering or deposition of additional layers on their surface. It makes their preparation faster. Hence, cheaper equivalents of standard disk electrodes made of noble metals or glassy carbon are increasingly reported in the literature. One of the most readily available materials is pencil lead graphite, which is an inexpensive material with a high conductivity, offering cost as low as $1.30 per device.47 It can be used as a working and auxiliary electrode, but a significant problem limiting the use of pencil-drawn electrodes is high content of impurities (graphite constitutes only 27–60% of the composition of pencil lead) and high affinity for adsorption of solution components, making it difficult to obtain reproducible results in chemosensing. Nevertheless, electrode regeneration procedures, for example by oxidative cycling in NaOH, allow their practical use in the selective determination of p-nitrophenol.48
Higher purity of graphite can be found in sheets that can be conveniently cut to specific shapes and sizes, being adjustable to any electrochemical systems. Silva et al. comprehensively studied nine graphite sheets from different manufacturers and characterized by various thicknesses, placing particular emphasis on indicating the advantages of disposable electrodes.49 Analysis of the obtained voltammograms justifies the use of graphite sheets as working electrodes, indicating a linear dependence of the peak currents on the square root of the scan rate in the presence of ferri/ferrocyanide redox probe. However, the authors emphasize the importance of the quality of the material, since defects in the graphite structure, confirmed by Raman spectroscopy, cause significant limitations in the electrode conductivity. The morphology of such sheets was confirmed by SEM analysis. However, it indicated a flat character of the electrodes, without a greater development of the surface. Hence, it would make it difficult to conduct trace analyses, where a high ratio of the active surface to the specific surface of the electrode plays a crucial role. The surface development of graphite sheets was discussed by Marra et al. in their work on a sensor based on flexible graphite sheets for detecting ketamine in synthetic saliva samples.50 The graphite sheets were electrochemically activated with 0.5 M NaOH, leading to partial exfoliation of the surface. It resulted in developed surface morphology with additional edges, which improved the electrochemical properties of the material.
Electrode flexibility is an increasingly considered aspect when making hand-made sensors, as exemplified by the research conducted by Boček et al. on fully self-prepared inkjet-printed flexible electrodes for lactate sensors.51 Compact sizes of the electrodes, the possibility of preparing the electrode from scratch in the laboratory within one day and high flexibility facilitating the transport and storage of the finished sensor shed a new light on the design of chemosensors. However, the described procedure requires a laboratory inkjet printer, which increases the cost of single-use electrode preparation.
Although graphite is the leading material for disposable electrodes, studies can be found regarding gold leaf electrodes made from edible gold flakes52 as well as planar-disk and nanoband gold-leaf electrodes pressed with polyimide tape.53 The first procedure leads to obtaining electrodes characterized by excellent electrochemical response in the presence of ferrocenemethanol redox probe, however, it requires multi-step and long-term preparation. The study of Camargo et al.52 presents an innovative approach to hand-made preparation of the entire, three-electrode system, but the described procedure requires experience in manual skills. The latter work, led by Santos et al.,53 describes a much simpler method of obtaining electrodes based on gold leaves, with an interesting approach to obtaining nanoband electrodes in a sandwich approach, where the gold nanolayer is glued on both sides with a non-conductive Kapton foil, leading a submicron-thick electrodes. Moreover, such an electrode was characterized by high flexibility, due to the support of the Kapton foil. After subjecting the electrode to mechanical stress, no decrease in electrochemical properties was observed, which determines the possibility of using such a system in the construction of sensors operating directly on the skin. Despite the thickness of the electrodes being on the order of fractions of a micrometer, the use of gold in the construction of sensors is associated with higher production costs than in systems using carbon paper.
To sum up the examples of alternative materials for preparing electrodes, carbon paper is an excellent substrate for electrochemical sensors. The optimal price-to-quality ratio allows for the production of a much larger number of individual electrodes for the price of one gold electrode, and the readiness for use immediately after purchase allows for use even by people without extensive manual skills. The lower conductivity of graphite compared to gold does not limit the wide use of carbon paper for the construction of electrochemical sensors, confirmed by the numerous cited research works. The significant advantages of carbon paper over graphite sheets include the lack of the need for prior chemical or electrochemical treatment and a highly developed active surface area due to the network of alternately arranged thin graphite fibers. The excellent adhesion of the laminate to the carbon paper brings the flexibility of such laminated electrodes closer to that of substrates based on Kapton foil.
Until now, several examples of MIP sensors for MTZ have been reported (Tables S2 and S3, ESI data†). They mostly rely on direct electrochemical reduction of MTZ at highly negative potentials.54–58 This is because MTZ is electroactive at −0.65 V vs. SCE.59 Moreover, most of these sensors involved nanoparticles in achieving multi-step sensitivity and fabrication.56–58,60,61 One example of photoelectrochemical biosensor for selective MTZ determination was also reported.62 In that work, decoration of the electrode surface with coral-like g-C3N4 enabled detection of MTZ with LOD equal to 5 nM. Reported biosensor responded to MTZ concentration linearly in a semilogarithmic scale in 10 nM to 100 µM concentration range. Despite the high analytical parameters of this biosensor, its fabrication and operation were quite complicated. It highlights the need to design an inexpensive, sensitive, robust, and easy-to-use sensor for MTZ determination.
In this work, we propose to apply hand-made, inexpensive, flexible electrodes based on carbon paper to fabricate a selective electrochemical sensor for MTZ determination (Scheme 1). The carbon paper was modified with imprinted polypyrrole. SEM and IR confirmed carbon fiber coverage with MIP film. Such electrodes will be easy to transport and store. Moreover, they will not break during slight changes in storage conditions. Importantly, estimated price of materials used for one carbon electrode fabrication is less than 20¢. DPV experiments confirmed the fabricated sensor selectivity and sensitivity. Therefore, proposed MIP sensor was applied to MTZ determination in honey samples.
 | ||
| Scheme 1 (a) A flowchart showing steps of flexible carbon electrode preparation. (b) Optical camera photos of the hand-made flexible carbon electrode. (c) SEM image of MIP-coated carbon paper. (d) Schematic representation of Ru(NH3)6Cl3 DPV peak change in response to MTZ presence in the solution. For more details, see Fig. 2 and Fig. S9 (ESI data†). | ||
2. Experimental
2.1 Materials
Pyrrole (Py), metronidazole (MTZ), L-ascorbic acid, glyphosate, β-D(+)glucose, creatinine, vancomycin and penicillin G sodium salt were purchased from Merck (Sigma Aldrich), Germany. Hexaammineruthenium(III) chloride (Ru(NH3)6Cl3) was purchased from abcr, Germany, 3-(1H-pyrrol-1-yl)propanoic acid (Py-COOH) and 3-(1H-pyrrol-1-yl)propanamine (Py-NH2) were purchased from ChemBridge, USA. Potassium chloride (KCl), sodium carbonate (Na2CO3), sodium bicarbonate (NaHCO3), and potassium hexacyanoferrate(III) (K3Fe(CN)6) were supplied by Chempur. Potassium hexacyanoferrate(II) trihydrate (K4Fe(CN)6) was provided by POCh, Gliwice, Poland. Hydrochloric acid (HCl) was purchased from Stanlab Lublin, Poland, isopropanol from Linegal Chemicals, Poland, and compressed argon was supplied by Air Liquide, France. A glass micropipette column filled with aluminum oxide (Al2O3) for chromatographic adsorption analysis acc. to Brockman (II), Budapest, Hungary, was used for pyrrole purification. Honey used for real sample analysis was purchased on the open market from a Polish producer. All chemicals and solvents were of analytical grade. All aqueous solutions were prepared using distilled water.2.2 Instruments
All electrochemical measurements were performed on the BioLogic SP-150 potentiostat (France). The devised flexible electrode served as a working electrode (WE), platinum plate as a counter electrode (CE), and silver wire with an electrodeposited AgCl layer as a quasi-reference electrode. WE and CE were mounted parallelly in a special electrochemical holder fabricated at IPC PAS.63 A Carl Roth, Germany glass vial was used as the electrochemical cell, cut for measurements to a volume of approximately 3 mL. All curves presenting the results of electrochemical measurements were smoothed in the OriginLab 2020b software using the Savicky–Golay method with 10 points of the window. All experiments were carried out at room temperature in the laboratory, reaching an average of 25 °C.All CV experiments were performed with a scan rate of 50 mV s−1, if not otherwise stated. Pulse height, pulse width, step height, and step time in DPV experiments were 2.5 mV, 100.0 ms, 5.0 mV, and 500.0 ms, respectively. Frequencies during EIS experiments were scanned from 100 kHz to 100 mHz at −0.25 V vs. Ag quasi-reference electrode. Sinusoid amplitude was equal to 10 mV. All electrochemical experiments were performed in deaerated solutions.
Microscopic imaging was performed on the scanning electron microscope (SEM) FEI Nova NanoSEM 450 microscope, USA, with an energy-dispersive X-ray analyzer (EDX) Octane Elect Plus, USA. Fourier-transform infrared (FTIR) spectroscopy measurements were performed using the reflection mode with a Vertex 80 v spectrophotometer of Bruker using the deuterated triglycine sulfate detector controlled by OPUS v. 6.5 software. Spectra were recorded with a 2 cm−1 resolution. For each spectrum, 1024 scans were acquired. Measurements were performed under decreased (6 hPa) pressure. The photographs included in this publication were taken with Samsung Galaxy S22 mobile phone.
X-ray photoelectron spectroscopy (XPS) measurements were carried out on a PHI 5000 VersaProbe (ULVACPHI) scanning ESCA microprobe by using monochromatic Al Kα radiation (hν = 1486.6 eV). Casa XPS software then evaluated the XPS data. Background was subtracted using the Shirley method, and peaks were fitted with (Gaussian–Lorentzian)-shaped profiles. The binding energy (BE) scale was referenced to the C 1s peak (BE = 284.6 eV).
The electrodes were laminated with heat-shrinkable foil with a home ironing machine. Crystal Laminating Pouches 303 × 216 mm manufactured in China by ARGO S.A. (Poland) were applied for that purpose. The flexible TGP-H030 (30% wet proofing) carbon paper (TORAY Industries, Inc., Japan) was an electrode material. Au 100 nm-thick film-coated glass slides, with a 15 nm-thick underlayer of Ti were prepared by the Institute of Electronic Materials Technology (Warsaw, Poland) and served as an electrical contact.
Using the sessile drop method, the wetting properties of the used carbon paper and electrodeposited MIP film were characterized by their static water contact angle (WCA) with the manual optical tensiometer (Theta Lite, Attension). Hence, a 10 µL drop of distilled water was placed on the surface of the sample using a microsyringe. The contact angles within 30 s were monitored by 60 FPS (frames per second) cameras every 1 s, and the average of each measurement was calculated using its standard deviation.
DFT calculations with b3lyp functional and 3-21g* basis set were performed with Gaussian 16 software.64 Presence of solvent, namely water, was simulated using pcm model. Simulation results were visualized with Avogadro software.65
2.3 Preparation of the working electrode
The previously reported method was adapted to prepare working electrodes.66–69 Firstly, two sheets of laminating foil were pressed on one edge with a hot iron to join them thermally. Then, they were cut to about 18 × 10 mm. In one sheet, a hole of 6 mm in diameter (geometric area about 30 mm2) was punched with a corkborer. Carbon paper was cut to about 18 × 7 mm, and placed between the two sheets of laminating foil in such a way that it was exposed through the aforementioned hole. Then, a gold-coated glass slide serving as an electrical contact to the carbon paper was placed between the far side of the carbon paper and the foil sheets. Finally, everything was laminated using a hot iron, and the excess of foil was cut off. Furthermore, the exposed surface of the electrode was rinsed with isopropanol to remove any impurities and then dried.2.4 Imprinted and non-imprinted polymer films deposition
To deposit the MIP film, 2.5 mL of a solution containing 0.1 M pyrrole (17.34 µL), previously filtered on an Al2O3 column, 1 mM Py-COOH (0.348 mg), and 0.5 mM MTZ (0.214 mg), in 1.0 M aqueous solution of KCl (supporting electrolyte) was prepared. The solution was thoroughly stirred on a vortex and subjected to ultrasonication for 5 to 10 min until complete dissolution after each mixture component was added. Then, the solution was purged with argon for at least 10 min to remove oxygen.In the holder for electrochemical measurements, the hand-made carbon paper working electrode was mounted with the silver–silver chloride reference electrode and the platinum auxiliary electrode. This three-electrode system was immersed in the polymerization solution, and galvanostatic deposition was performed by passing a current of 200 µA for 400 s, corresponding to a charge of 80 mC (Fig. 1a and b). It corresponds to 830 nmol of oxidized monomers. On this basis it has been estimated that 54.5 µg of polymer was deposited on the electrode surface.
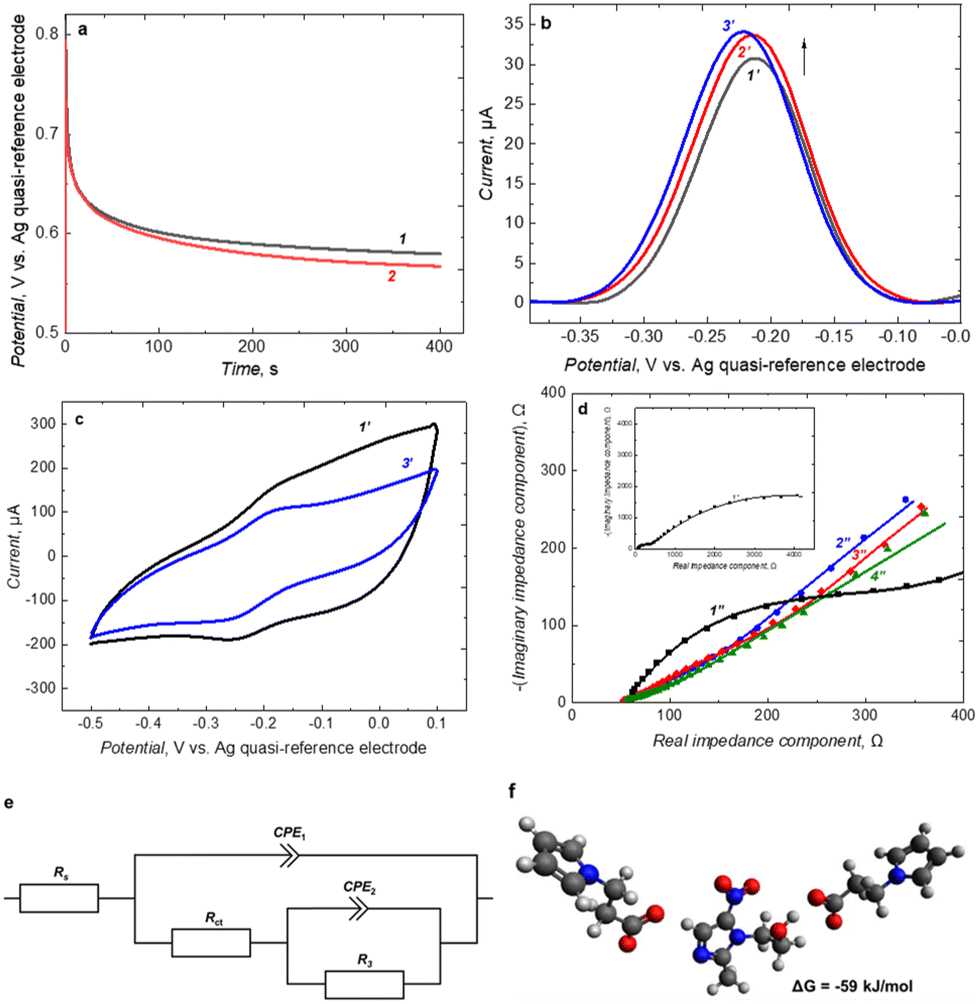 | ||
| Fig. 1 (a) Chronocoulometric plots recorded during galvanostatic deposition of (1) MIP and (2) NIP films. A current of 200 µA was passed for 400 s through the working electrode immersed in a solution of 0.1 M pyrrole, 1 mM Py-COOH, and 1.0 M KCl in water in the presence (1) or absence (2) of 0.5 mM MTZ. (b) DPV and (c) CV curves recorded in 5 mM Ru(NH3)6Cl3 solution in 0.1 M KCl(aq) on the MIP film-coated electrode (1′) after electrodeposition or after (2′) 30 min, and (3′) 60 min extraction in 0.1 M carbonate buffer of pH = 9.1. (d) EIS spectra and equivalent circuit fitting curves for (1″ squares) pristine carbon paper electrode, and carbon paper electrode coated with MIP film after (2″ circles) polymerization, (3″ diamonds) extraction in 0.1 M carbonate buffer of pH = 9.1 and (4″ triangles) in presence of 9.9 nM MTZ. (e) An equivalent circuit fitted to the EIS spectra. (f) DFT-simulated structure of the pre-polymerization complex formed between MTZ and two molecules of Py-COOH monomer. | ||
A template was removed from the MIP film by immersing the MIP-coated electrode for 60 minutes in 0.1 M carbonate buffer of pH = 9.1 under gentle stirring. Then, the residuals of the buffer were washed out with Milli-Q water for 30 minutes. As a control, non-imprinted polymer (NIP) film was deposited the same way as MIP but without adding MTZ (Fig. 1a and b).
2.5 Electrochemical determination of MTZ
For electrochemical MTZ determinations MIP- or NIP-coated electrodes were mounted in the electrochemical setup described above. Electrodes were immersed in 1 mM Ru(NH3)6Cl3 solution in 0.1 M KCl(aq). After each analyte injection, the solution was stirred with a magnetic stirrer for 0.5–1 min. Then, subsequently, CV and DPV curves were recorded. Each electrode was discarded after recording the calibration curve, and a new one was prepared.3. Results and discussion
3.1 Fabrication of flexible electrodes
Flexible (carbon paper)-based electrode is shown in Scheme 1a. Laminated carbon paper-based electrodes were prepared according to procedure 2.3 in the Experimental section. During lamination, the melted foil has sealed pores in the carbon paper. Therefore, gold had no direct contact with the solutions but provided an excellent electrical connection to carbon paper. Flexible hand-made carbon electrodes (Scheme 1b) were fabricated this way.3.2 Deposition of imprinted and non-imprinted polymer films
Strong interactions between template molecules and monomers are crucial for successfully introducing selective binding sites to MIP molecular cavities. For that purpose, we have considered two monomers: carboxylic or amine pyrrole derivatives, abbreviated as Py-COOH and Py-NH2, respectively.The electrochemical properties of both monomers were tested and compared with those of pristine Py (Fig. S3 and S4, ESI data†). It occurred that Py-COOH efficiently electropolymerizes under similar conditions as Py (Fig. S3, ESI data†). In contrast, Py-NH2 electropolymerization (Fig. S4a, ESI data†) resulted in the deposition of non-conducting film and blocking electrode surface (Fig. S5b, ESI data†). Therefore, Py-COOH was used in further experiments.
The MIP and NIP films were deposited under galvanostatic conditions. Chronocoulometric curves recorded during these electropolymerizations are shown in Fig. 1a. This process occurred to be more reproducible than electropolymerization under potentiodynamic conditions due to better potential stability over the measurement. Under these conditions, a thick enough layer was obtained to form a continuous film over the whole surface of carbon fibers. Moreover, the polymer film was not too thick, thus, pores in the carbon paper were still fully open. Therefore, neither redox probe ions nor MTZ could diffuse into the pores of the carbon paper. After the MIP film deposition, the gray carbon paper electrode turned deep black proving that the whole electrode surface was modified.
Template molecules should be extracted from the deposited polymer to obtain desired molecular cavities in the MIP structure. Both acidic (Fig. S5, ESI data†) and basic (Fig. 1b and c) extraction conditions were tested for that purpose. Apparently, extraction in carbonate buffer of pH = 9.1 appeared efficient. An increase in the DPV signal recorded for the MIP-coated electrode was observed after 30 minutes of extraction (Fig. 1b, curves 1′ and 2′). Only a slight increase was visible after another half an hour (Fig. 1b, curve 3′). Therefore, it has been decided that 60 minutes of extraction is sufficient to remove most of the MTZ template molecules from the MIP matrix.
Importantly, when the NIP film-coated electrode was subjected to a similar treatment with carbonate buffer, only a slight drop of DPV signal was observed (Fig. S6, ESI data†).
Moreover, the MIP-coated electrode was characterized using CV (Fig. 1c). After extraction, the capacitive current recorded for MIP-coated electrode was decreased, and the Ru(NH3)6Cl3 redox probe oxidation/reduction peaks were slightly more pronounced. Multicyclic CV experiments performed for MIP and NIP-coated electrodes proved that MIP film is electrochemically stable and does not deteriorate during electrochemical experiments (Fig. S7, ESI data†). Importantly, the current observed for the MIP film-coated electrode was about one order of magnitude higher than those observed for the uncoated carbon paper electrode (Fig. S8a, ESI data†). Moreover, peak-to-peak separation for redox probe is reduced after MIP film deposition. That is, on pristine carbon paper electrode Ru(NH3)6Cl3 redox probe peak-to-peak separation was equal to 190 mV (Fig. S7a, ESI data†). After MIP deposition and template extraction, it was equal to 85 mV (Fig. 1c, curve 3′). Reduction and oxidation of redox probe became more electrochemically reversible. Presumably, it is due to the higher conductivity of MIP film. As a result, we observed an enhancement of electrochemical response after MIP film deposition.
When selecting a functional monomer to MTZ ratio in pre-polymerization complex, a series of quantum mechanical calculations according to the DFT b3lyp functional with 3-21g* basis set were performed. Presence of solvent (water) was simulated using pcm model. Simulations confirmed the pre-polymerization complex formation of the Py-COOH monomer with the MTZ molecule with a stoichiometry of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 1f). The obtained Gibbs free energy value of complex formation reaction was equal to −59 kJ mol−1. It suggests that MTZ molecules form stable complexes with Py-COOH during polymerization, enabling simultaneous copolymerization of pyrrole with its carboxyl derivative and molecular imprinting of MTZ.
:1 (Fig. 1f). The obtained Gibbs free energy value of complex formation reaction was equal to −59 kJ mol−1. It suggests that MTZ molecules form stable complexes with Py-COOH during polymerization, enabling simultaneous copolymerization of pyrrole with its carboxyl derivative and molecular imprinting of MTZ.
Moreover, the MIP-coated paper electrode was characterized with EIS at each step of its fabrication (Fig. 1d). Two semicircles were visible in the presence of Ru(NH3)6Cl3 redox probe before MIP film deposition on carbon paper electrode (Fig. 1d, inset). The smaller one may be attributed to the charge transfer resistance (Rct) of the faradaic process on the electrode surface and the capacitance of the electrochemical double layer (CPE1). The second semicircle may originate from a diffusion of the redox probe through the porous structure of the electrode, which is described by parallel resistance (R3) and constant phase element (CPE2).70–74 The equivalent circuit model fitted to this plot is shown in Fig. 1e. After MIP film deposition, both semicircles were much less pronounced (Fig. 1d), but the recorded spectra were still well described by the same equivalent circuit. A higher conductivity of MIP-coated electrode may explain this behavior. Moreover, PPy MIP-coated electrode surface is more hydrophilic than pristine carbon paper since bare carbon paper consists of 30% wet-proof coating, according to its producer, and polypyrrole is known to have hydrophilic properties. This was supported by contact angle measurements, where 10 µL of water was dripped onto the surface of bare carbon paper and electrodeposited MIP film. In the first case, a contact angle of (147.97 ± 0.22)° was obtained, while after covering with a MIP layer, this angle decreased to (92.17 ± 0.31)° (Fig. S9, ESI data†). Therefore, penetration of redox probe solution into the pores of electrode material was easier. The changes in the EIS spectra caused by template extraction and subsequent MTZ binding were most pronounced in the region related to the diffusion of the redox probe.
The deposited MIP and NIP films were imaged using an SEM microscope to prove their successful deposition on the carbon paper electrode (Fig. S9, and S10, ESI data†). MIP (Fig. S9c, ESI data†), NIP (Fig. S9e, ESI data†), as well as pristine PPy (Fig. S9g, ESI data†), form lacy frost-like structures over carbon fibers (Fig. S9a and S9b, ESI data†). After extraction with carbonate buffer, MIP (Fig. S9d, ESI data†) and NIP (Fig. S9f, ESI data†) film morphology appeared more compact. SEM images of lower magnification (Fig. S10, ESI data†) and optical camera image (Fig. S9h, ESI data†) indicate that the whole surface of carbon fibers was coated with grainy polymer structures after deposition of MIP and NIP films. MIP film coating on these fibers was not affected by extraction in 0.1 M carbonate buffer of pH = 9.1 (Fig. S9, and S10, ESI data†).
The recorded ATR-IR and XPS spectra confirmed the successful deposition of MIP and NIP films on the carbon paper electrodes. IR spectra after polymerization showed a broad signal of aromatic C–H stretching (2950–2600 cm−1), and a set of overlapped signals between 1700 cm−1 and 1300 cm−1 emerged (Fig. S11, ESI data†). Also, XPS determined a higher content of nitrogen and a much lower content of fluorine compared to carbon paper electrodes, proving successful electropolymerization over carbon electrodes (Table S1, ESI data†). XPS signal originates only from the surface of the material (1–2nm, in depth). Therefore, the drop of fluorine content in XPS spectra suggests that carbon fibers of the carbon paper were coated with other material – MIP in this case.
3.3 Application of MIP- and NIP-coated electrodes for selective MTZ determination
The MIP-coated carbon paper electrode was applied for selective MTZ determination using DPV in the presence of Ru(NH3)6Cl3 redox probe (Fig. 2). The obvious choice of hexaammineruthenium chloride as the main redox probe was based on its charge which is opposite to the negatively charged polymer (due to the carboxyl group presence). Moreover, this redox probe is known to follow the outer-sphere mechanism, in which the electron transfer phenomenon occurs with no direct interaction with the electrode surface.75 Detail characterization of MIP film with outer- and inner-sphere redox probe is shown in Section 11 of ESI.†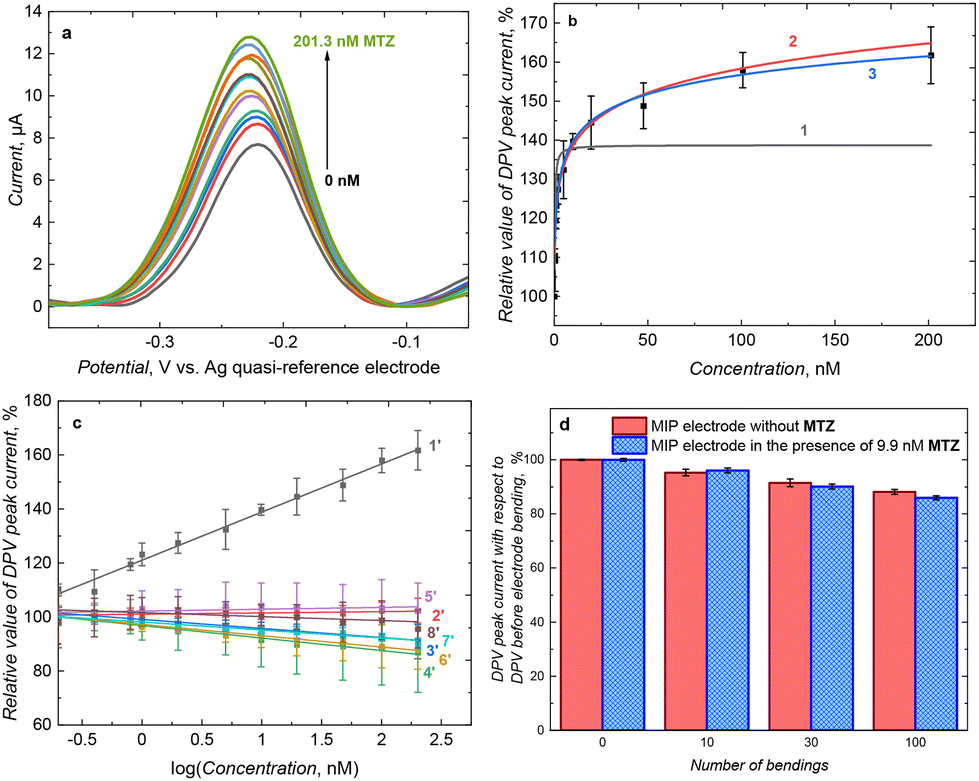 | ||
| Fig. 2 (a) DPV curves recorded in 1 mM Ru(NH3)6Cl3 and 0.1 M KCl(aq) solution on the MIP film-coated electrode for different MTZ concentrations. (b) Fitting of (1) Langmuir, (2) Freundlich, and (3) Langmuir–Freundlich isotherm to changes in DPV current with changes of MTZ concentration. (c) Constructed calibration plots for (1′ and 2′) MTZ, (3′) glyphosate, (4′) creatinine, (5′) L-ascorbic acid, (6′) glucose, (7′) penicillin G, and (8′) vancomycin on (1′ and 3′–8′) MIP or (2′) NIP film-coated carbon paper electrodes. (d) DPV signal measured in the absence and presence of the MTZ over MIP film-coated carbon paper electrode after multiple bending cycles by approx. 45° and straightening of the electrodes. | ||
We have exploited so-called “gate effect” in constructing the MIP based chemosensor.76,77 In theory, it is assumed that binding of analyte into MIP imprinted cavities affects the diffusion of the redox probe through the film to the electrode surface, resulting in changes in recorded current. Alternatively, we have proven in the past that in the case of conductive MIP films, changes in MIP film conductivity due to analyte binding can be the predominant factor influencing signal originating from redox probe.74,78,79 Herein, the diffusion of a positively charged redox probe, Ru(NH3)6Cl3, toward negatively charged MIP-modified electrode was enhanced. In this condition, subsequent increase in current during DPV measurements allowed for the quantitative determination of the analyte. In this case, DPV peak current increased with MTZ concentration increase from 0.2 to 200 nM (Fig. 2a). The response of the MIP film-coated carbon electrode was very well described by Langmuir–Freundlich isotherm (Fig. 2b and Table 1). The described system is characterized by a low value of homogeneity factor, namely, n = 0.144, indicating scarcely homogeneous distribution of molecular cavities in the polymer matrix.80 We may speculate that this is because of the highly developed surface of the carbon paper electrode, nevertheless, the performance of our fabricated chemosensor is satisfactory.
| Isotherm | Isotherm equation | Fitted parameters | R 2 | ||
|---|---|---|---|---|---|
| ΔIDPV,max, % | K, nM−1 | n | |||
| a K L – Langmuir constant. b K F – Freundlich constant. c K LF – Langmuir–Freundlich constant. d Adsorption intensity. e Homogeneity factor. | |||||
| Langmuir |

|
138.7 (±3.8) | 16.81a (±4.14) | — | 0.7139 |
| Freundlich | ΔIDPV = KFcMTZn | — | 121.43b (±0.44) | 0.058d (±0.002) | 0.9932 |
| Langmuir–Freundlich |
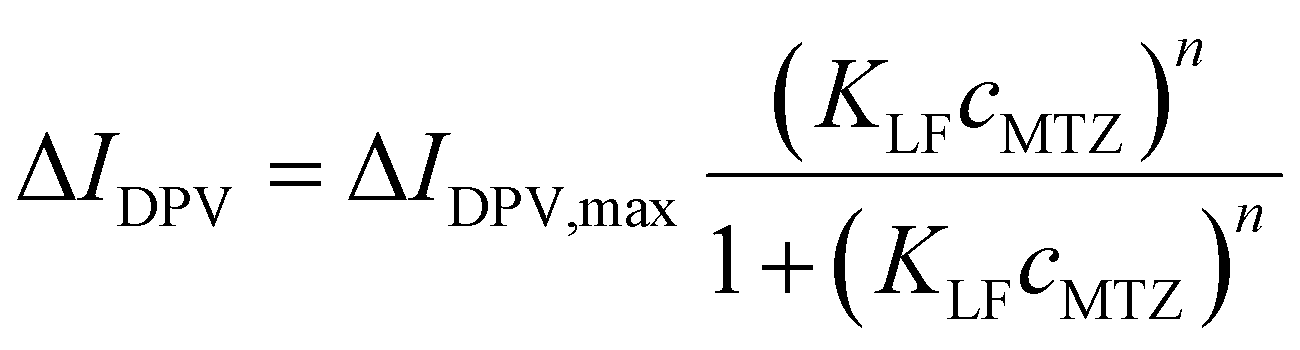
|
226.9 (±39.2) | 2.62c (±6.07) | 0.144e (±0.038) | 0.9930 |
From a practical point of view, it is more convenient to use linear fitting. The linear fitting enables easier comparison of the analytical parameters of the sensor. For that purpose, a linear calibration curve to the logarithm of MTZ concentration was prepared (Fig. 2c). The calibration plots followed the linear equation (eqn (1))
 | (1) |
 | (2) |
cMTZ [log(nM)] for MIP and NIP, respectively. The correlation coefficient and the limit of detection at S/N = 3 were R2 = 0.9882 and LOD = 0.4 nM, respectively. The apparent imprinting factor estimated by slopes of calibration curves constructed for MIP and NIP films-coated electrodes was equal to IF = 38.
The main purpose of decorating electrodes with MIP films is to introduce selective recognition. Therefore, the MIP film-coated electrode response toward chosen interfering compounds, including glyphosate, creatinine, L-ascorbic acid, glucose, penicillin G, and vancomycin, was tested (Fig. 2c). Only in the case of glucose it was possible to calculate the selectivity factor, which was equal to 26.1. In the case of other interfering compounds, sensor responses were negligible, or changes were in the opposite direction compared to MTZ. However, absolute values of these changes were much lower than sensors response to MTZ and it may be concluded that MIP sensor selectivity was high.
Elasticity constitutes the main advantage of carbon-based electrodes, since it may be a crucial factor in designing wearable sensors.81–83 The MIP-coated laminated carbon paper electrode was bent to around 45° and straightened multiple times to estimate its robustness. After 100 such events, currents recorded in both solutions of the pristine redox probe and in the presence of 9.9 nM MTZ dropped only by 12–14% (Fig. 2d). It proved that the MIP film coating over the carbon paper electrode was stable and did not break or damage the electrode by bending it. Only the conductivity of the electrode material (carbon paper) dropped slightly. The flexibility and robustness of the fabricated electrodes may be beneficial in future applications, especially for in-field measurements using portable sensing devices.81–84
3.4 Real sample analysis
The MIP film-coated carbon-based electrodes were applied for MTZ determination in spiked honey samples (Fig. 3). For that purpose, honey sample was diluted 1000 times with a solution of 1 mM Ru(NH3)6Cl3 in 0.1 M KCl(aq). Then, it was spiked with known amounts of MTZ. Following the studies done by de Silva et al.,85 sugars are by far the largest group of compounds in the matrix, constituting 75% of monosaccharides, mainly fructose (38.5%) and glucose (31%). It is worth mentioning, proteins (3.3% in honey) known by strong adsorption on the electrode, which often hinders parallel determination of target analytes, 0.57% of organic acids lowering pH relative to the sample without matrix, as well as redox-active vitamins, which can interfere with the reactions of the redox probe in the observed ‘gate effect’ phenomenon. | ||
| Fig. 3 Comparison of constructed calibration plots for MTZ in the same concentration range prepared in 1 mM Ru(NH3)6Cl3 solution in (1) 0.1 M KCl(aq) and (2) honey diluted 1000-times with 1 mM Ru(NH3)6Cl3 solution in 0.1 M KCl(aq) in (a) a normal and (b) semi-log scale. | ||
As expected, some matrix effect was observed, and slightly smaller changes in current were observed in a honey sample than in pristine KCl solution. When obtained data were visualized in a semi-log scale, it became visible that response in the real sample was only slightly lower, but the slopes of both calibration curves were similar. That is, it is equal to 15.63(±0.73)[%/log(nM)] in honey in comparison to 17.82(±0.65)[%/log(nM)] reported for pristine KCl solution. Therefore, equation (eqn (3)) fitted for sensor response had form of
 | (3) |
Then, we performed as separate experiment on freshly prepared electrode determination of MTZ in spiked honey samples using eqn (3) as a calibration plot (Table 2). Obtained recoveries were on acceptable level. It proves that matrix effect can be minimized by calibrating our sensor in proper matrix before performing measurements.
| No. | Known concentration of spiked sample, nM | DPV current response of the sensor, % | Calculated MTZ concentration, nM | Recovery, % |
|---|---|---|---|---|
| 1 | 0.4 | 104.3 | 0.40 | 99.0 |
| 2 | 1.0 | 109.4 | 0.83 | 83.4 |
| 3 | 2.0 | 115.6 | 2.07 | 103.7 |
| 4 | 5.0 | 121.2 | 4.76 | 95.2 |
| 5 | 9.9 | 126.2 | 9.85 | 99.5 |
4. Conclusions
Carbon paper was applied as an electrode material for flexible electrode fabrication. Furthermore, this electrode was coated with polypyrrole film imprinted with MTZ. The observed current dropped only by 12–14%. The modified electrode responded in DPV experiments with respect to MTZ in a concentration range of 0.2 nM up to 200 nM. Changes in DPV current were fitted to the Langmuir–Freundlich isotherm. Moreover, these changes were well described by a linear equation with respect to the logarithm of MTZ concentration. Electrode durability was tested, and even after bending 100 times, response of the electrode was only slightly affected. A fabricated chemosensor was applied for MTZ determination in spiked honey samples for the sensor validation. The matrix effect was substantial, however, sensor response to MTZ in honey was still linear on logarithm scale. Therefore, the sensor will require separate calibration for samples of different types. These flexible electrodes in field applications will be more convenient than traditional costly disk electrodes. Screen-printed electrodes have already become popular because of similar features; however, they lack flexibility. Importantly, the estimated price of materials for one carbon electrode fabrication is less than ∼20¢. These are essential parameters if we target mass production.Author contributions
All authors have contributed to the research and they have reviewed text of manuscript.Data availability
Data may be available on demand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank The National Science Centre (NCN) of Poland financially supported our research through the SONATA 14 Grant No. 2018/31/D/ST5/02890 to MC. We thank Kamila Łępicka and Monika Mierzejewska for fruitful discussions and suggestions.References
- N. Gilbert, Nature, 2012, 481, 125 CrossRef CAS PubMed.
- R. Gehring, S. R. Haskell, M. A. Payne, A. L. Craigmill, A. I. Webb and J. E. Riviere, J. Am. Vet. Med. Assoc., 2005, 227, 63–66 CAS.
- V. F. Samanidou and E. N. Evaggelopoulou, J. Sep. Sci., 2007, 30, 2549–2569 CrossRef CAS PubMed.
- C. Chafer-Pericas, A. Maquieira and R. Puchades, TrAC, Trends Anal. Chem., 2010, 29, 1038–1049 CrossRef CAS.
- M. Cieplak and W. Kutner, Trends Biotechnol., 2016, 34, 922–941 CrossRef CAS PubMed.
- V. Ayerdurai, P. Lach, A. Lis-Cieplak, M. Cieplak, W. Kutner and P. S. Sharma, Crit. Rev. Food Sci., 2024, 64, 3407–3440 CrossRef CAS PubMed.
- V. Ayerdurai, M. Cieplak and W. Kutner, TrAC, Trends Anal. Chem., 2023, 158, 116830 CrossRef CAS.
- Y. L. Mustafa, A. Keirouz and H. S. Leese, J. Mater. Chem. B, 2022, 10, 7418–7449 RSC.
- G. Selvolini and G. Marrazza, Sensors, 2017, 17, 718 CrossRef PubMed.
- S. Bhakta and P. Mishra, Sens. Actuators, Rep., 2021, 3, 100061 CrossRef.
- E. Yılmaz, B. Garipcan, H. Patra and L. Uzun, Sensors, 2017, 17, 691 CrossRef PubMed.
- P. Rebelo, E. Costa-Rama, I. Seguro, J. G. Pacheco, H. P. A. Nouws, M. N. D. S. Cordeiro and C. Delerue-Matos, Biosens. Bioelectron., 2021, 172, 112719 CrossRef CAS PubMed.
- B. Keitel, A. D. Batista, B. Mizaikoff and B. Fresco-Cala, in Encyclopedia of Sensors and Biosensors, 2023, pp. 851–867, DOI:10.1016/b978-0-12-822548-6.00158-8.
- M. Grabka, Z. Witkiewicz, K. Jasek and K. Piwowarski, Sensors, 2022, 22, 5607 CrossRef CAS PubMed.
- D. Kumar and B. B. Prasad, Sens. Actuators, B, 2012, 171, 1141–1150 CrossRef.
- B. B. Prasad, A. Prasad and M. P. Tiwari, Talanta, 2013, 109, 52–60 CrossRef CAS PubMed.
- S. Patra, E. Roy, R. Madhuri and P. K. Sharma, Biosens. Bioelectron., 2015, 66, 1–10 CrossRef CAS PubMed.
- H. J. Chen, Z. H. Zhang, L. J. Luo and S. Z. Yao, Sens. Actuators, B, 2012, 163, 76–83 CrossRef CAS.
- A. Fatoni, A. Numnuam, P. Kanatharana, W. Limbut and P. Thavarungkul, Analyst, 2014, 139, 6160–6167 RSC.
- B. V. M. Silva, B. A. G. Rodriguez, G. F. Sales, M. D. T. Sotomayor and R. F. Dutra, Biosens. Bioelectron., 2016, 77, 978–985 CrossRef CAS PubMed.
- J. F. Xia, X. Y. Cao, Z. H. Wang, M. Yang, F. F. Zhang, B. Lu, F. Li, L. Xia, Y. H. Li and Y. Z. Xia, Sens. Actuators, B, 2016, 225, 305–311 CrossRef CAS.
- X. D. Wang, J. Dong, H. M. Ming and S. Y. Ai, Analyst, 2013, 138, 1219–1225 RSC.
- L. Li, L. M. Fan, Y. L. Dai and X. W. Kan, Microchim. Acta, 2015, 182, 2477–2483 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS PubMed.
- Y. G. Sun, B. Mayers and Y. N. Xia, Adv. Mater., 2003, 15, 641–646 CrossRef CAS.
- H. Y. Son, J. H. Ryu, H. Lee and Y. S. Nam, ACS Appl. Mater. Interfaces, 2013, 5, 6381–6390 CrossRef CAS PubMed.
- M. Heim, S. Reculusa, S. Ravaine and A. Kuhn, Adv. Funct. Mater., 2012, 22, 538–545 CrossRef CAS.
- H. Xu and W. A. Goedel, Small, 2005, 1, 808–812 CrossRef CAS PubMed.
- D. C. Christodouleas, F. C. Simeone, A. Tayi, S. Targ, J. C. Weaver, K. Jayaram, M. T. Fernandez-Abedul and G. M. Whitesides, Adv. Mater. Technol., 2017, 2, 1600229 CrossRef.
- E. Nunez-Bajo, M. Blanco-Lopez, A. Costa-Garcia and M. T. Fernandez-Abedul, Talanta, 2018, 178, 160–165 CrossRef CAS PubMed.
- M. Yoshikawa and K. Tharpa, in Handbook of Polymer Nanocomposites. Processing, Performance and Application, Springer-Verlag, Berlin Heidelberg, 2015, vol. c, ch. 20, p. 396 Search PubMed.
- C. Jantarat, N. Tangthong, S. Songkro, G. P. Martin and R. Suedee, Int. J. Pharm., 2008, 349, 212–225 CrossRef CAS PubMed.
- W. Liu, Y. M. Guo, J. Luo, J. Kou, H. Y. Zheng, B. X. Li and Z. J. Zhang, Spectrochim. Acta, Part A, 2015, 141, 51–57 CrossRef CAS PubMed.
- M. Fang, L. Zhou, H. Zhang, L. Liu and Z. Y. Gong, Food Chem., 2019, 274, 156–161 CrossRef CAS PubMed.
- C. Zhang, H. Y. Cui, Y. F. Han, F. F. Yu and X. M. Shi, Food Chem., 2018, 240, 893–897 CrossRef CAS PubMed.
- Q. Kong, Y. Wang, L. Zhang, S. Ge and J. Yu, Sens. Actuators, B, 2017, 243, 130–136 CrossRef CAS.
- J. Qi, B. Li, X. Wang, L. Fu, L. Luo and L. Chen, Anal. Chem., 2018, 90, 11827–11834 CrossRef CAS PubMed.
- W. Alahmad, A. Cetinkaya, S. I. Kaya, P. Varanusupakul and S. A. Ozkan, TrAC, Trends Anal. Chem., 2024, 170, 117475 CrossRef CAS.
- N. Duan, X. Chen, X. Lin, D. Ying, Z. Wang, W. Yuan and S. Wu, Sens. Actuators, B, 2023, 384, 133665 CrossRef CAS.
- S. Casadio, J. W. Lowdon, K. Betlem, J. T. Ueta, C. W. Foster, T. J. Cleij, B. van Grinsven, O. B. Sutcliffe, C. E. Banks and M. Peeters, Chem. Eng. J., 2017, 315, 459–468 CrossRef CAS.
- G. Q. Sun, P. P. Wang, S. G. Ge, L. Ge, J. H. Yu and M. Yan, Biosens. Bioelectron., 2014, 56, 97–103 CrossRef CAS PubMed.
- N. S. Ferreira, A. P. T. Moreira, M. H. M. de Sa and M. G. F. Sales, Sens. Actuators, B, 2017, 243, 1127–1136 CrossRef CAS.
- M. Amatatongchai, J. Sitanurak, W. Sroysee, S. Sodanat, S. Chairam, P. Jarujamrus, D. Nacapricha and P. A. Lieberzeit, Anal. Chim. Acta, 2019, 1077, 255–265 CrossRef CAS PubMed.
- K. Somnet, S. Chimjarn, S. Wanram, P. Jarujamrus, D. Nacapricha, P. A. Lieberzeit and M. Amatatongchai, Talanta, 2024, 269, 125512 CrossRef CAS PubMed.
- P. Rebelo, I. Martins, J. G. Pacheco, R. Banegas, E. Costa-Rama, M. M. Moreira, H. P. A. Nouws and C. Delerue-Matos, Microchem. J., 2024, 205, 111410 CrossRef CAS.
- L. Fan, Q. Hao and X. Kan, Sens. Actuators, B, 2018, 256, 520–527 CrossRef CAS.
- R. Torre, M. Cerrato-Alvarez, H. P. A. Nouws, C. Delerue-Matos, M. T. Fernández-Abedul and E. Costa-Rama, Sens. Actuators, B, 2025, 423, 136705 CrossRef CAS.
- E. Witkowska Nery, M. Kundys-Siedlecka, Y. Furuya and M. Jönsson-Niedziółka, Sensors, 2018, 18, 4037 CrossRef PubMed.
- L. A. J. Silva, J. S. Stefano, R. M. Cardoso, N. S. Prado, P. H. T. Soares, E. Nossol, R. A. A. Munoz, L. Angnes and E. M. Richter, J. Electroanal. Chem., 2019, 833, 560–567 CrossRef CAS.
- M. C. Marra, T. C. Oliveira, R. G. Rocha, G. P. Siqueira, S. C. Chaves, E. M. Richter, R. V. Gelamo and R. A. A. Muñoz, Electrochim. Acta, 2024, 508, 145219 CrossRef CAS.
- Ž. Boček, M. Zubak and P. Kassal, Biosensors, 2025, 15, 28 CrossRef PubMed.
- J. R. Camargo, S. Cleto, A. Neumann, D. C. Azzi, R. D. Crapnell, C. E. Banks and B. C. Janegitz, Electrochim. Acta, 2024, 478, 143825 CrossRef CAS.
- M. S. F. Santos, W. A. Ameku, I. G. R. Gutz and T. R. L. C. Paixão, Talanta, 2018, 179, 507–511 CrossRef CAS PubMed.
- M. B. Gholivand and M. Torkashvand, Talanta, 2011, 84, 905–912 CrossRef CAS PubMed.
- J. Liu, H. Tang, B. Zhang, X. Deng, F. Zhao, P. Zuo, B.-C. Ye and Y. Li, Anal. Bioanal. Chem., 2016, 408, 4287–4295 CrossRef CAS PubMed.
- J. Wang, W. Du, X. Huang, J. Hu, W. Xia, D. Jin, Y. Shu, Q. Xu and X. Hu, Anal. Methods, 2018, 10, 4985–4994 RSC.
- A. A. Ensafi, P. Nasr-Esfahani and B. Rezaei, Sens. Actuators, B, 2018, 270, 192–199 CrossRef CAS.
- Y. Wang, L. Yao, X. Liu, J. Cheng, W. Liu, T. Liu, M. Sun, L. Zhao, F. Ding, Z. Lu, P. Zou, X. Wang, Q. Zhao and H. Rao, Biosens. Bioelectron., 2019, 142, 111483 CrossRef CAS PubMed.
- K. Plotnikova, L. Dubenska, P. Rydchuk, S. Pysarevska, M. Rydchuk, S. Ivakh, D. Yanovych, Z. Zasadna and S. Plotycya, J. Food Meas. Charact., 2022, 16, 891–900 CrossRef.
- Z. Zhang, Y. Liu, P. Huang, F.-Y. Wu and L. Ma, Talanta, 2021, 232, 122411 CrossRef CAS PubMed.
- Y. Li, Y. Liu, Y. Yang, F. Yu, J. Liu, H. Song, J. Liu, H. Tang, B.-C. Ye and Z. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 15474–15480 CrossRef CAS PubMed.
- X. Li, Y. J. Yuan, X. M. Pan, L. Z. Zhang and J. M. Gong, Biosens. Bioelectron., 2019, 123, 7–13 CrossRef CAS PubMed.
- M. Dabrowski, M. Cieplak, P. S. Sharma, P. Borowicz, K. Noworyta, W. Lisowski, F. D'Souza, A. Kuhn and W. Kutner, Biosens. Bioelectron., 2017, 94, 155–161 CrossRef CAS PubMed.
- G. W. Trucks, M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- K. Lepicka, P. Pieta, R. Gupta, M. Dabrowski and W. Kutner, Electrochim. Acta, 2018, 268, 111–120 CrossRef CAS.
- M. Mierzejewska, K. Łępicka, J. Kalecki, W. Lisowski and P. S. Sharma, ACS Appl. Mater. Interfaces, 2022, 14, 33768–33786 CrossRef CAS PubMed.
- K. Łępicka, P. Pięta, R. Rybakiewicz, M. Zagórska, W. Kutner and M. Majewska, PL 234733, 2018.
- K. Łępicka, P. Pięta, P. Borowicz, A. Sukhenko, L. Stobiński and W. Kutner, PL 232126, 2016.
- P. Leuaa, D. Priyadarshani, A. K. Tripathi and M. Neergat, J. Phys. Chem. C, 2019, 123, 21440–21447 CrossRef CAS.
- S. Omanovic and S. G. Roscoe, Langmuir, 1999, 15, 8315–8321 CrossRef CAS.
- S. Omanovic and S. G. Roscoe, J. Colloid Interface Sci., 2000, 227, 452–460 CrossRef CAS PubMed.
- J. Bisquert, G. Garcia-Belmonte, F. Fabregat-Santiago and P. R. Bueno, J. Electroanal. Chem., 1999, 475, 152–163 CrossRef CAS.
- V. Ayerdurai, M. Cieplak, K. R. Noworyta, M. Gajda, A. Ziminska, M. Sosnowska, J. Piechowska, P. Borowicz, W. Lisowski, S. Shao, F. D'Souza and W. Kutner, Bioelectrochemistry, 2021, 138, 107695 CrossRef CAS PubMed.
- N. Kurapati, P. Pathirathna, C. J. Ziegler and S. Amemiya, ChemElectroChem, 2019, 6, 5651–5660 CrossRef CAS.
- P. S. Sharma, A. Garcia-Cruz, M. Cieplak, K. R. Noworyta and W. Kutner, Curr. Opin. Electrochem., 2019, 16, 50–56 CrossRef CAS.
- Y. Yoshimi, A. Narimatsu, K. Nakayama, S. Sekine, K. Hattori and K. Sakai, J. Artif. Organs, 2009, 12, 264–270 CrossRef CAS PubMed.
- P. Lach, M. Cieplak, M. Majewska, K. R. Noworyta, P. S. Sharma and W. Kutner, Anal. Chem., 2019, 91, 7546–7553 CrossRef CAS PubMed.
- P. Lach, M. Cieplak, K. R. Noworyta, P. Pieta, W. Lisowski, J. Kalecki, R. Chitta, F. D'Souza, W. Kutner and P. S. Sharma, Sens. Actuators, B, 2021, 344, 130276 CrossRef CAS.
- V. Ayerdurai, A. Garcia-Cruz, J. Piechowska, M. Cieplak, P. Borowicz, K. R. Noworyta, G. Spolnik, W. Danikiewicz, W. Lisowski, A. Pietrzyk-Le, F. D'Souza, W. Kutner and P. S. Sharma, J. Agric. Food Chem., 2021, 69, 14689–14698 CrossRef CAS PubMed.
- W. Gao, H. Ota, D. Kiriya, K. Takei and A. Javey, Acc. Chem. Res., 2019, 52, 523–533 CrossRef CAS PubMed.
- Y. Liu, M. Pharr and G. A. Salvatore, ACS Nano, 2017, 11, 9614–9635 CrossRef CAS PubMed.
- T. Q. Trung and N.-E. Lee, Adv. Mater., 2016, 28, 4338–4372 CrossRef CAS PubMed.
- T. Delipinar, A. Shafique, M. S. Gohar and M. K. Yapici, ACS Omega, 2021, 6, 8744–8753 CrossRef CAS PubMed.
- P. M. da Silva, C. Gauche, L. V. Gonzaga, A. C. O. Costa and R. Fett, Food Chem., 2016, 196, 309–323 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00146c |
| This journal is © The Royal Society of Chemistry 2025 |