
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the cellular lipid cascade of prostate cells explored using infrared microspectroscopy†
Thomas. A.
Gladwell
a,
Dougal
Ferguson
 ab,
Noel
Clarke
cd,
Michael D.
Brown
e and
Peter
Gardner
*ab
ab,
Noel
Clarke
cd,
Michael D.
Brown
e and
Peter
Gardner
*ab
aDepartment of Chemical Engineering, School of Engineering, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: peter.gardner@manchester.ac.uk
bPhoton Science Institute, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cDepartment of Surgery, The Christie Hospital NHS Foundation Trust, UK
dDepartment of Urology, Salford Royal Hospital, UK
eDivision of Cancer Sciences, University of Manchester, UK
First published on 1st May 2025
Abstract
Background: Although prostate cancer (PCa) is the most diagnosed cancer in men worldwide, there is geographical variance in both incidence and morbidity, with higher levels in developed “Western Diet” countries. In particular the high levels of the omega-6 polyunsaturated fatty acid, arachidonic acid (AA), in Western diets has been shown to promote aggressive PCa in vitro. However the exact mechanism through which AA induces the aggressive phenotype has not been fully characterised. Methods: In this study Fourier transform infrared (FTIR) imaging coupled with fluorescence microscopy (FM), is used to follow AA metabolism in PCa cell lines. This is achieved using partially deuterated AA, with a distinctive C–D stretch seen at 2251 cm−1 providing molecular specificity, coupled with Nile Red Fluorescence imaging. Results: We show that, invasive cell lines PC-3, LNCaP C4-2B and DU145 readily uptake and metabolise AA, producing prostaglandins via the COX-2 pathway. Inhibition of the COX-2 pathway with either NS938 or the omega-3 polyunsaturated fatty acid Docosahexaenoic acid (DHA), reduces the invasive stimulus of AA and blocks its uptake. Conclusion: This demonstrates that FTIR imaging can be utilised to follow metabolomics processes within a PCa model and provide an insight to the molecular pathways underlying the cancer metabolome. Additionally, these works provide key insights into the rapid uptake of AA within certain invasive cell lines of prostate cancer, suggesting that AA exposure initiates early cellular responses prior to the uptake and processing of lipids within the cells.
Introduction
Infrared Prostate cancer (PCa) is a global medical burden with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 414259 men diagnosed and 375304 deaths recorded world-wide in 2020.1 The main cause of PCa related morbidity and mortality is due to PCa predilection to metastasis to areas of red bone marrow activity in the bony skeleton.2,3 Although the mechanism has still to be fully characterised, in vitro modelling has demonstrated that bone marrow adipocytes (BM-ad), or the fats within, play a critical role in this process.4 Epidemiological evidence, which shows geographical variations in both PCa incidence and mortality, with a 3-fold difference in incidence between developed and developing countries.1,2,5 Studies6–8 have shown that dietary fats may play an important role in the aetiology of PCa, although the extent is currently unknown.6
414259 men diagnosed and 375304 deaths recorded world-wide in 2020.1 The main cause of PCa related morbidity and mortality is due to PCa predilection to metastasis to areas of red bone marrow activity in the bony skeleton.2,3 Although the mechanism has still to be fully characterised, in vitro modelling has demonstrated that bone marrow adipocytes (BM-ad), or the fats within, play a critical role in this process.4 Epidemiological evidence, which shows geographical variations in both PCa incidence and mortality, with a 3-fold difference in incidence between developed and developing countries.1,2,5 Studies6–8 have shown that dietary fats may play an important role in the aetiology of PCa, although the extent is currently unknown.6
Of particular interest is the role of omega 3(ω-3) and 6 (ω-6) polyunsaturated fatty acid (PUFA) in PCa progression. Sumida et al. (1965)9 and Denizot et al. (1998, 1999)10,11 characterised the lipid rich bone marrow environment and showed that both the ω-6 PUFAs arachidonic acid and linoleic acid were present, comprising 2.5–9.5% and 4.1–15.3% of the total lipid present respectively. There has also been a major shift in the dietary balance of ω-6:ω-3 PUFAs in developed countries, through industrialisation and changing diets, with parts the USA consuming a ω-6:ω-3 PUFA dietary ratio of 20:112 as compared with 4:1 in Japan. Both of these are above the perceived optimal ratio of 2.3:113 and suggests a potential role of the high fat/ω-6-PUFA Westernised diet in driving PCa progression.12,14
The association between ω-6 PUFAs, in particular arachidonic acid (AA), have been strengthened by in vitro studies that show ω-6 PUFAs such as linoleic and AA can promote PCa cell proliferation15,16 and stimulate transendothelial invasion across a bone marrow endothelial barrier enabling extravasation into the bone marrow stroma (BMS).17 Once within the BMS, PCa cells migrate towards and interact with AA loaded BM-ad leading to a reduction of BM-ad size and PCa uptake of AA.4 Although the specific mechanisms surrounding these studies remain unclear, AA was seen to play a key role in driving the metastatic process.4,17,18
Arachidonic acid is metabolised by cyclooxygenases (COX1/2) and lipooxygenases (LOX) to generate prostaglandins, prostacyclins and thromboxanes, and leukotrienes and hydroxyeicosatetraenoic acids respectively.19 Arachidonic acid induced PCa invasion is dependent on the production of PGE2 by COX-2.20 Inhibition of PGE2 production by either the specific COX-2 inhibitor NS398 or by an ω-3 PUFA (eicosapentaenoic acid (EPA) or docosahexaenoic acid (DHA)) significantly reduced PCa invasion in vitro.21–24
Here we seek to track the cellular uptake and metabolism of AA using Fourier transformed infrared (FTIR) imaging in a PCa model. FTIR imaging is a powerful vibrational spectroscopic technique which enables different functional groups, relating to cellular proteins, lipids, carbohydrates, sugars and DNA to be easily identified and characterized.25,26 In recent years it has been applied to cellular systems for the study of different cell lines,27 stem cell characterisation,28,29 drug-cell interactions30–34 but has rarely been used to shed light on a specific metabolic pathway.
Materials and methods
Materials
All cell culture general reagents inhibitors and stains were purchased from Sigma-Aldrich (Poole, UK). Arachidonic acid (AA) and deuterated arachidonic acid (d8-AA) were purchased from MP Biomedicals (London, UK) was and were made up in methyl-β-cyclodextrin (Sigma-Aldrich (Poole, UK)) producing a 10 mg ml−1 emulsion. Foetal calf serum was provided by PAA Laboratories (Yoevil, UK). Calcium fluoride (CaF2) substrates were supplied by Crystran Limited (Poole, UK).Cell line culture
All cell lines were verified by the Cancer Research UK Manchester Institute tissue typing service and cultured as previously described.35 All cell lines were cultured onto CaF2 substrates, with the disks sterilised with 90% w/w ethanol for 1 hour and air dried before use. All cells were serum starved in RPMI 1640 for 24 hours before use and were free from antibiotics.Cell treatment with arachidonic acid
All AA and d8-AA treatments were made up to a 20 μM concentration in RPMI 1640. The cell lines were cultured on CaF2 substrates. All cells were washed in PBS after treatment and formalin fixed 4% for 20 minutes. Cultured substrates were then dipped in double distilled water following the protocol of Gazi et al.36 and air dried.COX-2 inhibition
Cell lines were exposed to 10 μM NS-398 (N-[2-(cyclohexyloxy)-4-nitrophenyl] methanesulfonamide) in RPMI 1640 and cultured at 37 °C in 5% CO2 for 30 minutes prior to treatment.Fluorescent microscopy
All cells were cultured in Hank's Buffer Salt Solution (HBSS) without phenol red prior to staining with 5 μM Nile-red 5 minutes before fatty acid treatment. Cultures were washed in PBS and fixed in 4% formalin for 20 minutes, with a final wash in DD water and air drying. Cultures were imaged on a Nikon Eclipse 90i Fluorescence microscope.Fourier transform infrared microscopy
All measurements were taken in transmission mode and hyperspectral chemical images were obtained using an Agilent 670 FTIR spectrometer coupled with an Agilent-620 IR microscope. The system was equipped with a 128 × 128 liquid-nitrogen-cooled mercury cadmium telluride focal plane array detector, using 256 background scans with 128 sample scans at 4 cm−1 resolution over a range 950 cm−1 to 3800 cm−1. A 15× magnification objective was used giving a pixel size of 5.5 μm × 5.5 μm.Data pre-processing and statistics
A cell finder algorithm was used identifying high protein threshold regions discarding the spectra with no obvious biological spectral characteristics.29 All spectra were processed using an RMieS correction algorithm,37–39 removing spectral distortions relating to Mie scattering. All spectra were de-noised using 8 Principle Components (PC) and vector normalised. All FTIR spectra were converted to the first derivative, allowing PCA to identify subtle biochemical features required to track lipid metabolism. For First derivative analysis 7 smoothing points were used throughout.All principle component analysis (PCA) scores were converted into median PCA positional plots, where the median of each time point of a given PC was plotted as a function of time. The relative median positional values were calculated setting the position of t = 0 min to zero. Seeded PCA was used following the method outlined by Keating,40 and recently used by Gardner et al. termed “Guided PCA”.41 This process involves the insertion of a user defined spectrum (typically the spectral bands of the target molecule) which can be deliberately scaled by a large weighting. This is then be interpreted by the PCA model as being the main contribution of dataset variance, and subsequently becomes the most important principal component loadings. This forces the loading plot for PCA to be reflective of the pure molecules required for tracking, allowing chemical variance to be seen specific to a given molecules. PCA was carried out over the spectral range 1720 cm−1 to 1750 cm−1 to investigate the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch vibration assigned to fatty acid molecules, specifically AA.
O stretch vibration assigned to fatty acid molecules, specifically AA.
Results
PCA analysis of AA uptake and COX-2 suppression
The PCA scores plot of invasive PC-3 cells treated with AA over a 60-minute time-course can be seen in Fig. 1a–d, scatter plots for the invasive LNCaP C4-2B, DU-145, LNCaP C4-2 and non-invasive PNT2 and LNCaP cell lines are show in the ESI (ESI: Fig. 2–6†). As early as t = 5 min there is separation within the plot from the initial t = 0 min. Maximum separation occurs at t = 15 min, as indicated by the most biochemical variance from the initial time plot. From t = 30 min to t = 60 min the PC-3 cells display progressively less separation returning to a state chemically similar to that at t = 0 min. This PCA score can be represented as a median positional plot, which describes the movement and position of the clusters inside the PCA score space. The median value of each time point cluster for PC 1 was taken and plotted as a function of time. The PCA median plots for both invasive and non-invasive cell lines treated with AA can be seen in Fig. 2, where the COX-2 active and inhibited curves appear in blue and red respectively.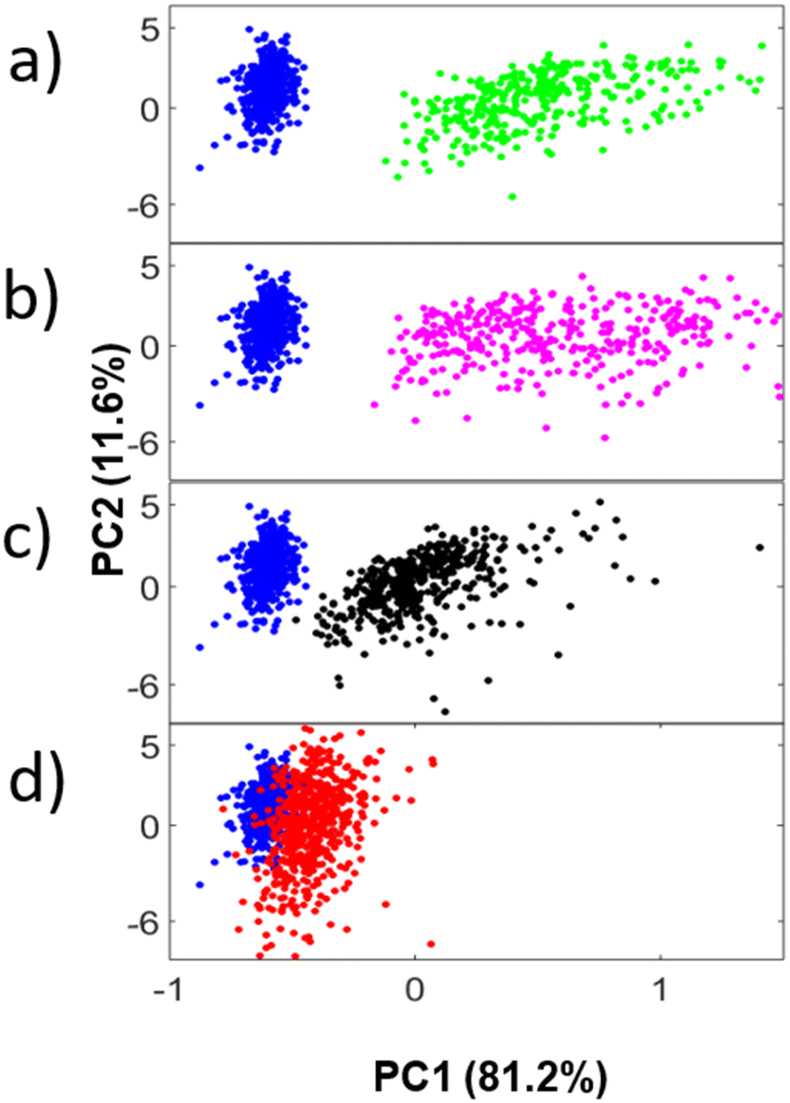 | ||
| Fig. 1 PCA scores of FTIR spectra, examining the CO stretch between 1720 cm−1 to 1750 cm−1 for the invasive cell line PC-3 treated with AA at time points (a) t = 0 min (blue) vs. t = 5 min (green) and (b) t = 0 min (blue) vs. t = 15 min (magenta) and (c) t = 0 min (blue) vs. t = 30 min (black) and (d) t = 0 min (blue) vs. t = 60 min (red). | ||
 | ||
| Fig. 2 PCA scores median cluster position plots of FTIR spectra, examining the CO stretch between 1720 cm−1 to 1750 cm−1 from invasive (a) PC-3 and (b) LNCaP C4-2B and (c) LNCaP C4-2 and (d) DU-145 and non-invasive (e) PNT2 and (f) LNCaP cell lines AA Treated (blue) and COX-2 inhibited and treated with AA (red) for 0, 5, 15, 30 and 60 min. Note that the lines are just a guide for the eye. | ||
There is a clear difference between invasive and non-invasive PCa phenotypes in the PCA median positional plots. When treated with AA, the invasive PC-3, LNCaP C4-2B, DU-145 and LNCaP C4-2 cell lines appear to shift in the median positional plots to a strong positive value compared with the untreated cells (t = 0 min) in Fig. 2a–d. There is a clear trend amongst all invasive cell lines with initial separation seen as early as t = 5 min and maximum positive positional displacement in the median plot at t = 30 min. Eventually, at t = 30 and t = 60 min the separation decreases. Thus, the chemical profile of the cell moves back towards that at t = 0 min, with the two most invasive phenotypes (PC-3 and LNCaP C4-2B cell lines), almost returning to the same chemical profile as at t = 0 min.
The non-invasive cell lines LNCaP and PNT2 cells display no such behaviour as seen in Fig. 2e and f respectively. There are no shifts in PC median position within the PCA scores when the non-invasive cell lines are exposed to AA and COX-2 inhibition represented by no sizable change within the median score plots. This signifies no change in the global cellular lipid chemistry throughout the 60-minute time course for both COX-2 active and inhibited cells.
To gain a clearer insight of the interaction with AA and PCa cell lines, AA was replaced with a deuterated form, d8-AA. By replacing 8 molecular hydrogen atoms for deuterium atoms, the bonds vibrational frequency is reduced due to the difference in atomic mass between hydrogen and deuterium. The shift in vibrational frequency means the C–D stretch is in a region of the IR spectrum with no other biological spectral vibrations at 2251 cm−1. This enables the deuterium tagged AA to be uniquely identified in the cell. With this distinctive C–D vibration, it is possible to produce a spectral profile following the path of AA throughout the time course.
The seeded PCA median positional plots of the d8-AA treated PC-3 and PNT2 cells can be seen in Fig. 3a and b respectively. The PC-3 cells experience separation in the PCA median positional plot as early as t = 5 min with the greatest positive displacement shown at t = 15 min. This is then followed by a shift in the positional centroid value in a less positive direction at t = 30 and t = 60 min, indicating more chemical similarity to t = 0 min. This result is very similar to that seen with PC-3 cells treated with AA in Fig. 2a, confirming that invasive cell lines take d8-AA and AA. The PNT2 cells experience no such separation or movement in the PCA centroid positional plot, again implying no chemical change associated with d8-AA uptake consistent with Fig. 2e.
 | ||
| Fig. 3 Seeded PCA scores relative centroid plots of FTIR spectra, examining the C–D stretch between 2200 cm−1 to 2300 cm−1 from invasive (a) PC-3 and non-invasive (b) PNT2 cell lines treated with d8-AA for 0, 5, 15, 30 and 60 min. Note that the lines are just a guide for the eye. | ||
Previous studies by Brown et al.18,20 and Tawadros17 demonstrated that AA signalling and metabolism with the production of PGE2 by COX-2, is required to induce and enable metastatic behaviour and invasion of the preferred metastatic site of the bone narrow stroma., with the production of PGE2 by COX-2 inducing PC-3 PCa cell line invasion. We therefore sought to determine if we could specifically probe intracellular production of PGE2 in PC-3 and PNT2 cell lines. Seeded PCA was used to identify and characterize the spectral profile associated with PGE2 synthesis. This was carried out by seeding the PCA with a spectrum of PGE2, which has a prominent peak at 1740 cm−1. The spectral characteristics of invasive PC-3 and PNT2 cells treated with AA ± NS398 COX-2 inhibition are described in Fig. 4a and b respectively.
 | ||
| Fig. 4 (a) PGE2 seeded PCA median positional plot for PC-3 cells treated with AA COX-2 active (blue) and COX-2 inhibited (red) and (b) PGE2 seeded PCA median positional plot for PNT2 cells treated with AA COX-2 active (blue) and COX-2 inhibited (red) treated for 0, 5, 15, 30 and 60 minutes. Note that the lines are just a guide for the eye. | ||
Within PC-3 cells we could spectrally detect PGE2 production post treatment with AA, with the PCA median positional plot mimics the AA median plot previously shown in Fig. 2a. Addition of the COX-2 inhibitor NS398 or the use of the cyclodextrin vehicle control (ESI: Fig. 1†) did not cause PCA separation indicating no chemical change. However we saw no such trends with the non-invasive PNT2 cells, which showed no significant change in directional values in the PCA median positional plot at any time points. This was indicative of no change in the PGE2 cellular composition within the PNT2 post AA addition.
Fluorescence microscopy and FTIR chemical lipid mapping
To confirm that FTIR spectroscopy was detecting the uptake of AA by PC-3 cells we followed AA uptake over time using the lipid stain Nile read and immunofluorescence. Dual immunofluorescence and FTIR images were taken over 60 minutes (Fig. 5). The FTIR chemical lipid map highlights the distribution of CO ester stretches in the form of a heat map. The chemical lipid map was created by taking the ratio of peak areas of the CO ester stretch between 1720 cm−1 to 1750 cm−1 and the amide I and II envelope between 1473 cm−1 to 1710 cm−1. The amide I and II peaks were used as a normalising factor, making the assumption that the total amount of cellular protein remains constant throughout the time course of the experiment.
 | ||
| Fig. 5 PC-3 cells treated with AA investigated by (a) FM images tracking the uptake and distribution of AA with COX-2 active and (b) FTIR lipid heat map of the CO stretch between 1720 cm−1 to 1750 cm−1 with COX-2 active and (c) FM images tracking the uptake and distribution of AA with COX-2 inhibited and (d) FTIR lipid heat map of the CO stretch between 1720 cm−1 to 1750 cm−1 with COX-2 inhibited for 0, 5, 15, 30 and 60 min. | ||
The fluorescence microscopy (FM) images (Fig. 5a) display a definite increase in fluorescent intensity throughout the time course, with the maximum intensity displayed at 30 minutes followed by a reduced intensity at t = 60 min. This increase and decrease in fluorescence indicates AA uptake and metabolism and supports to the chemistry previously seen in the FTIR PCA median plot analysis (Fig. 2a). The FTIR chemical lipid map intensity hot spots match the timings and distributions seen in the FM fluorescent images. When the COX-2 enzyme is inhibited, there appears to be no change in intensity throughout the 60-minute time course for the FM images and the FTIR chemical lipid heat map seen in Fig. 5c and d respectively.
The biological effect of AA stimulation of PCa cells has previously been shown to be blocked by the addition of the ω3 PUFA DHA.20 DHA has a greater Michaelis constant (Km) for binding COX-2 than AA, leading to a reduction in AA COX-2 metabolism in the presence of DHA. We would therefore expect to see a similar change in spectral chemistry post AA stimulation in the presence of the COX-2 inhibitor NS398.
PC-3 and PNT2 cells were treated with either 20 μM DHA alone or 20 μM DHA and d8-AA for 60 minutes. PCA and seeded PCA were carried out looking at the 1720 cm−1 to 1750 cm−1 range examining the CO ester stretch for DHA tracking and 2200 cm−1 to 2300 cm−1 examining the C–D stretch for d8-AA tracking respectively.
PC-3 cells appear to uptake DHA represented by the shifts seen in blue PCA median positional curve in Fig. 6a. There is an increasing median positional value with a maximum occurring at t = 15 min followed by decrease in value throughout the remainder of the time-course. When DHA was added alongside d8-AA, there are no changes in the blue PCA median position curve associated with the C–D signature of d8-AA uptake in Fig. 6b. The PNT2 cells did not chemically respond to the addition of DHA or the co-addition of DHA and d8-AA throughout the time course represented by the red curves in Fig. 6a and b respectively. These results are both consistent with the chemistry shown in Fig. 2. It appears DHA has an inhibitory effect on d8-AA uptake on invasive cell lines as seen with COX-2 suppression through NS-398 exposure.
 | ||
| Fig. 6 PCA relative median positional plots of FTIR spectra, examining the (a) CO stretch between 1720 to 1750 cm−1 for PC-3 (blue) and PNT2 (red) cells treated with DHA and (b) C–D stretch between 2200 to 2300 cm−1 for PC-3 (blue) and PNT2 (red) cells treated with both DHA and d8-AA for 0, 5, 15, 30 and 60 min. Note that the lines are just a guide for the eye. | ||
Discussion
Recent developments and improvements in both non mass spectroscopy (MALDI/DESI) and mass spectroscopy (MS) techniques combined with bioinformatics have led to resurgence in interest in the role of lipids in cellular biology and in particular in oncology. Lipids have multiple functional roles, besides acting as energy reservoirs and structural components, acting as chemical messengers, regulating protein function and immunological bioactive molecules (reviewed in Lydic & Goo42). MS and non-MS techniques require the extraction of the lipids from the cells prior to separation by chromatography for LC-MS, lipid detection by MS and identification by bioinformatics. Whilst MS approaches are the most popular techniques, due to their sensitivity and high throughput potential, non-MS approaches are finding favour as they directly introduce lipids from the sample into MS, enabling faster analysis from smaller volumes of material and lipid imaging, albeit at a lower resolution (several hundred species as compared with 10–100000 species). However, both of these techniques are untargeted lipid profiling techniques which do not readily lend themselves to the analysis/imaging of a single lipid metabolic process within a cell.
We4,36,43–45 and others46 have previously demonstrated that FTIR spectroscopy is a powerful chemical imaging technique suitable for the detection of lipids and their metabolites in a cellular system. Here we show for the first time that using mass labelled lipids, in particular deuterated arachidonic acid, FTIR imaging can be used to chemically follow and detect differences in a targeted lipid metabolic pathway in prostate cancer cell lines with differing biopotentials.
When looking at the key chemical spectral changes and differences between invasive and non-invasive cell lines exposed to exogenous AA, a key result emerges. The PCA median positional plots in Fig. 2 imply that cells with an invasive phenotype readily uptake AA, represented by the change in median positional value in the PCA scores associated with the CO ester stretch from the exogenous AA. Seeing separation as early as 5 minutes highlights the rapidity of the AA uptake mechanism in invasive cell lines. It is apparent that there are strong chemical features associated with fatty acids in the positive direction of all the invasive cell lines. All invasive cell lines display a directional transition in the PCA median positional plots, with the cellular chemistry progressively returning back to its original pre-exposed state by t = 60 min. These timings agree with the study conducted by Brown et al.20 where they observed AA uptake within the first 30 minutes followed by a decreases at 90 minutes. This shift in direction could be a significant indication of the cellular metabolism and strong evidence that AA has a rapid effect on the invasive cell's chemistry within the first 5 minutes; by 15 minutes the cell has taken on the AA as represented by the maximum displacement in the PCA score spaces. This would suggest that exposure to AA initiates an early cellular response prior to the uptake and metabolic processing of the lipid itself. This supports earlier studies by Tawadros et al.17 and Brown et al.18 that showed that AA rapidly induces the morphological switch between mesenchymal and amoeboid morphologies required for the transendothelial migration of malignant prostate epithelial cells and essential for metastatic spread, upon exposure to AA. After this initial signalling cascade, the cell initiates the AA cascade, breaking down the AA into its downstream bioactive metabolites which are then used in different cellular processes or are ejected from the cell. The FTIR chemical analysis agrees with the complimentary FM and FTIR lipid CO heat map investigations when treating PC-3 cells with AA as shown in Fig. 5a and b respectively.
The non-invasive cell lines show no such behaviour, with no directional transition displayed in the PCA median positional plots. Applying a deuterated tag specifically helps identify AA uptake in both invasive and non-invasive cell lines. The chemistry seen through the deuterated tag in Fig. 3 for both invasive and non-invasive cell lines match the chemistry displayed initially using AA both validating the data and providing the added specificity to the lipid chemistry.
Seeded PCA was used to investigate the chemical cellular changes associated with PGE2 which were consistent with the features displayed in the AA cascade. Invasive PC-3 cells displayed separation in the PGE2 seeded PCA score whereas the non-invasive PNT2 cells showed no separation. PC-3 cells take up and metabolise AA via the COX-2 pathway to produce PGE2 which evidently increases from its PCA score space. The PNT2 cells do not display features of AA uptake or an endogenous increase in cellular PGE2 over the first 60 minutes of exposure to AA. When the COX-2 pathway was blocked, there were no signs of PGE2 production either in invasive or non-invasive cell lines: this is expected as we show a loss of ability for the invasive cell and a lack of initial ability for non-invasive cells to uptake AA.
It is known that blocking the COX-2 enzyme inhibits cellular AA metabolism.47 When the COX-2 enzyme is inhibited in invasive cell lines, both instances demonstrated no PCA separation when probing the CO ester stretch and C–D stretch in Fig. 2 and 3. This suggests that the COX-2 enzyme plays a pivotal role in the uptake mechanism; no C–D stretch was noticed when tracking the lipid chemistry, further strengthening this hypothesis. Scratch assays were performed on PC-3 cells, with clear wound closure when treated with AA after 14 hours. The wound remained open when inhibiting COX-2, demonstrating the loss of invasive potential. The data is available in ESI: Tables (1 and 2)† respectively.
The non-invasive cell line PNT2 showed no signs uptake or metabolism when probed with d8-AA. Non-invasive cell lines appear to lack the ability to take up AA.
It is evident that COX-2 activity plays a critical role in the lipid metabolism in invasive prostate cancer cell lines, through the well-established lipid cascade mechanism. To undergo metabolism, there needs to be translocation of the endogenous AA to the cellular cytoplasm. This is usually achieved by a 1 or 2 step process. AA is a fatty acid which is a pivotal component of the cellular membrane.48 AA can be used by the cell in a number of different ways. It can be incorporated in the membrane and esterified for storage, directly changing membrane fluidity or taken in directly from the exogenous surplus and utilised in PG synthasis.47
When COX-2 is blocked, by either NS938 or DHA, the results show that there is no AA uptake or metabolism. Blocking the COX-2 enzyme in invasive PCa phenotypes, seems to induce an inhibitory or blocking effect for AA to be taken up by the cell. This suggests that there is upstream signalling prior to the AA cascade which prevents exogenous AA being incorporated into the cell membrane. This implies that by blocking the COX-2 enzyme the cell has an increased selectivity towards AA incorporation into the cellular system. If the cell fails to uptake and metabolise AA, this could directly affect morphological transitions between mesenchymal and amoeboid phenotypes expressed in PC-3 cells, which is a crucial step for PCa metastasis.18
AA stimulated mesenchymal to amoeboid transitions are already well documented. The results seen in this study suggest that when the COX-2 enzyme is suppressed, invasive cells appears to lose their ability not only to metabolise AA but also their ability to uptake AA in the first place. This could affect the PCa cell and its ability to change its membrane fluidity and structure, a key step in the progression and metastasis of PCa. Prostate cancer cells require an amoeboid phenotype to penetrate endothelial tight cell junctions and metastasise into the bone marrow, the preferential PCa secondary site.
Conclusions
Throughout this study, we have shown invasive PCa cell lines readily uptake and metabolise AA with no such activity seen for non-invasive cell lines. We hypothesise that a key factor influencing the cellular metastatic phenotype is the cell's ability to uptake and metabolise AA.When COX-2 is inhibited, it appears that AA uptake is suppressed or blocked in invasive cell lines. We suggest that there is upstream signalling preventing the incorporation and transport of AA into the cellular cytoplasm, although the specific detail of such a mechanism is unknown. The results indicate that cells are more selective than once thought with an ability to prevent uptake when the cell cannot metabolise AA. This study has also shown that FTIR spectroscopy has both the sensitivity and ability to track uptake and metabolism in cellular systems providing a new insight into the lipid cascade.
Author contributions
Thomas A. Gladwell: conceptualization, methodology, data acquisition, formal analysis, investigation, visualization, writing – original draft. Dougal Ferguson: visualization, writing – review & editing. Noel Clarke: conceptualization, writing – review & editing. Michael D. Brown: experimental design, writing – review & editing, supervision. Peter Gardner: experimental design, visualization, writing – review & editing, supervision.Ethics approval
Ethics approval was not required for these works.Data availability
The data can be made available upon reasonable request to the authors.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank Dr Alex Henderson for advice on data analysis and Mrs. Claire Hart for assistance with cell culturing protocols. In addition TAG would like to thank the University of Manchester for a Presidential Doctoral Scholarship Award. The Williamson Trust is gratefully acknowledged for funding the infrared microscope.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- D. M. Parkin, F. Bray, J. Ferlay and P. Pisani, Ca-Cancer J. Clin., 2005, 55, 74–108 CrossRef PubMed.
- L. Bubendorf, A. Schöpfer, U. Wagner, G. Sauter, H. Moch, N. Willi, T. C. Gasser and M. J. Mihatsch, Hum. Pathol., 2000, 31, 578–583 CrossRef CAS PubMed.
- M. D. Brown, C. Hart, E. Gazi, P. Gardner, N. Lockyer and N. Clarke, Br. J. Cancer, 2010, 102, 403–413 CrossRef CAS PubMed.
- M. P. Coleman, M. Quaresma, F. Berrino, J.-M. Lutz, R. De Angelis, R. Capocaccia, P. Baili, B. Rachet, G. Gatta, T. Hakulinen, A. Micheli, M. Sant, H. K. Weir, J. M. Elwood, H. Tsukuma, S. Koifman, G. A. e Silva, S. Francisci, M. Santaquilani, A. Verdecchia, H. H. Storm and J. L. Young, Lancet Oncol., 2008, 9, 730–756 CrossRef PubMed.
- D. D. Alexander, J. K. Bassett, D. L. Weed, E. C. Barrett, H. Watson and W. Harris, Nutr. Cancer, 2015, 67, 543–554 CrossRef CAS PubMed.
- N. K. Khankari, H. J. Murff, C. Zeng, W. Wen, R. A. Eeles, D. F. Easton, Z. Kote-Jarai, A. A. Al Olama, S. Benlloch, K. Muir, G. G. Giles, F. Wiklund, H. Gronberg, C. A. Haiman, J. Schleutker, B. G. Nordestgaard, R. C. Travis, J. L. Donovan, N. Pashayan, K.-T. Khaw, J. L. Stanford, W. J. Blot, S. N. Thibodeau, C. Maier, A. S. Kibel, C. Cybulski, L. Cannon-Albright, H. Brenner, J. Park, R. Kaneva, J. Batra, M. R. Teixeira, H. Pandha, W. Zheng and PRACTICAL consortium, Br. J. Cancer, 2016, 115, 624–631 CrossRef CAS PubMed.
- C. Sobolewski, C. Cerella, M. Dicato, L. Ghibelli and M. Diederich, Int. J. Cell Biol., 2010, 2010, 215158–215158 Search PubMed.
- T. Sumida, Acta Med. Nagasaki., 1965, 9, 222–241 CAS.
- Y. Denizot, V. Desplat, C. Dulery, F. Trimoreau and V. Praloran, Mediators Inflammation, 1999, 8, 31–35 CrossRef CAS PubMed.
- Y. Denizot, C. Dulery, F. Trimoreau, V. Desplat and V. Praloran, Biochim. Biophys. Acta, Mol. Cell Res., 1998, 1402, 209–215 CrossRef CAS PubMed.
- A. P. Simopoulos, Nutrients, 2016, 8, 128–128 CrossRef PubMed.
- M. Sugano and F. Hirahara, Am. J. Clin. Nutr., 2000, 71, 189S–196S CrossRef CAS PubMed.
- P. K. Pandalai, M. J. Pilat, K. Yamazaki, H. Naik and K. J. Pienta, Anticancer Res., 1996, 16, 815–820 CAS.
- M. Hughes-Fulford, C.-F. Li, J. Boonyaratanakornkit and S. Sayyah, Cancer Res., 2006, 66, 1427–1433 CrossRef CAS PubMed.
- J. L. Masferrer, K. M. Leahy, A. T. Koki, B. S. Zweifel, S. L. Settle, B. M. Woerner, D. A. Edwards, A. G. Flickinger, R. J. Moore and K. Seibert, Cancer Res., 2000, 60, 1306–1311 CAS.
- T. Tawadros, M. D. Brown, C. A. Hart and N. W. Clarke, Br. J. Cancer, 2012, 107, 1737–1744 CrossRef CAS PubMed.
- M. Brown, J.-A. Roulson, C. A. Hart, T. Tawadros and N. W. Clarke, Br. J. Cancer, 2014, 110, 2099–2108 CrossRef CAS PubMed.
- B. Wang, L. Wu, J. Chen, L. Dong, C. Chen, Z. Wen, J. Hu, I. Fleming and D. W. Wang, Signal Transduction Targeted Ther., 2021, 6, 94 CrossRef CAS PubMed.
- M. D. Brown, C. A. Hart, E. Gazi, S. Bagley and N. W. Clarke, Br. J. Cancer, 2006, 94, 842–853 CrossRef CAS PubMed.
- V. Fradet, I. Cheng, G. Casey and J. S. Witte, Clin. Cancer Res., 2009, 15, 2559–2566 CrossRef CAS PubMed.
- X. H. Liu, A. Kirschenbaum, S. Yao, R. Lee, J. F. Holland and A. C. Levine, J. Urol., 2000, 164, 820–825 CrossRef CAS.
- X.-H. Liu, S. Yao, A. Kirschenbaum and A. C. Levine, Cancer Res., 1998, 58, 4245–4249 CAS.
- M. I. Patel, C. Kurek and Q. Dong, J. Urol., 2008, 179, 1668–1675 CrossRef CAS PubMed.
- C. Matthäus, B. Bird, M. Miljković, T. Chernenko, M. Romeo and M. Diem, Methods Cell Biol., 2008, 89, 275–308 Search PubMed.
- M. Pilling and P. Gardner, Chem. Soc. Rev., 2016, 45, 1935–1957 RSC.
- J. R. Mourant, Y. R. Yamada, S. Carpenter, L. R. Dominique and J. P. Freyer, Biophys. J., 2003, 85, 1938–1947 CrossRef CAS PubMed.
- J. Cao, E. S. Ng, D. McNaughton, E. G. Stanley, A. G. Elefanty, M. J. Tobin and P. Heraud, Int. J. Mol. Sci., 2013, 14, 17453–17476 CrossRef PubMed.
- G. Clemens, K. R. Flower, A. P. Henderson, A. Whiting, S. A. Przyborski, M. Jimenez-Hernandez, F. Ball, P. Bassan, G. Cinque and P. Gardner, Mol. Biosyst., 2013, 9, 677 RSC.
- A. L. M. Batista de Carvalho, M. Pilling, P. Gardner, J. Doherty, G. Cinque, K. Wehbe, C. Kelley, L. A. E. Batista de Carvalho and M. P. M. Marques, Faraday Discuss., 2016, 187, 273–298 RSC.
- J. Doherty, G. Cinque and P. Gardner, Appl. Spectrosc. Rev., 2016, 52, 560–587 CrossRef.
- K. R. Flower, I. Khalifa, P. Bassan, D. Démoulin, E. Jackson, N. P. Lockyer, A. T. McGown, P. Miles, L. Vaccari and P. Gardner, Analyst, 2011, 136, 498–507 RSC.
- M. Jimenez-Hernandez, M. D. Brown, C. Hughes, N. W. Clarke and P. Gardner, Analyst, 2015, 140, 4453–4464 RSC.
- N. Wald and E. Goormaghtigh, Analyst, 2015, 140, 2144–2155 RSC.
- C. A. Hart, M. Brown, S. Bagley, M. Sharrard and N. W. Clarke, Br. J. Cancer, 2005, 92, 503–512 CrossRef CAS PubMed.
- E. Gazi, T. J. Harvey, M. D. Brown, N. P. Lockyer, P. Gardner and N. W. Clarke, Vib. Spectrosc., 2009, 50, 99–105 CrossRef CAS.
- P. Bassan, A. Kohler, H. Martens, J. Lee, H. J. Byrne, P. Dumas, E. Gazi, M. Brown, N. Clarke and P. Gardner, Analyst, 2010, 135, 268–277 RSC.
- P. Bassan, A. Kohler, H. Martens, J. Lee, E. Jackson, N. Lockyer, P. Dumas, M. Brown, N. Clarke and P. Gardner, J. Biophotonics, 2010, 3, 609–620 CrossRef CAS PubMed.
- P. Bassan, A. Sachdeva, A. Kohler, C. Hughes, A. Henderson, J. Boyle, J. H. Shanks, M. Brown, N. W. Clarke and P. Gardner, Analyst, 2012, 137, 1370–1377 RSC.
- M. E. Keating, Dr. Thesis Technol. Univ. Dublin, 2019, DOI:10.21427/0tqh-r955.
- B. Gardner, J. Haskell, P. Matousek and N. Stone, Analyst, 2024, 149, 205–211 RSC.
- T. A. Lydic and Y.-H. Goo, Clin. Transl. Med., 2018, 7, 4–4 CrossRef PubMed.
- E. Gazi, N. P. Lockyer, J. C. Vickerman, P. Gardner, J. Dwyer, C. A. Hart, M. D. Brown, N. W. Clarke and J. Miyan, Appl. Surf. Sci., 2004, 231, 452–456 CrossRef.
- E. Gazi, J. Dwyer, N. Lockyer, P. Gardner, J. C. Vickerman, J. Miyan, C. A. Hart, M. Brown, J. H. Shanks and N. Clarke, Faraday Discuss., 2004, 126, 41–59 RSC.
- E. Gazi, P. Gardner, N. P. Lockyer, C. A. Hart, M. D. Brown and N. W. Clarke, J. Lipid Res., 2007, 48, 1846–1856 CrossRef CAS PubMed.
- A. Sorvina, C. A. Bader, C. Caporale, E. A. Carter, I. R. D. Johnson, E. J. Parkinson-Lawrence, P. V. Simpson, P. J. Wright, S. Stagni, P. A. Lay, M. Massi, D. A. Brooks and S. E. Plush, Oncotarget, 2018, 9, 35541–35552 CrossRef PubMed.
- K. Meirer, D. Steinhilber and E. Proschak, Basic Clin. Pharmacol. Toxicol., 2013, 114, 83–91 CrossRef PubMed.
- M. Tomita-Yamaguchi, J. F. Babich, R. C. Baker and T. J. Santoro, J. Exp. Med., 1990, 171, 787–800 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5an00126a |
| This journal is © The Royal Society of Chemistry 2025 |