
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOnline integration of capillary electrophoresis and dual detector Taylor dispersion analysis via a 3D printed instrument†
Felix S.
Atsar
a,
Hillary D.
Bourger
 b and
Christopher A.
Baker
*a
b and
Christopher A.
Baker
*a
aDepartment of Chemistry & Biochemistry, New Mexico State University, Las Cruces, NM 88003-001, USA. E-mail: cabaker@nmsu.edu
bDepartment of Chemical and Materials Engineering, New Mexico State University, Las Cruces, NM 88003-001, USA
First published on 13th January 2025
Abstract
Hydrodynamic radius (RH) is a descriptive metric of protein structure with the potential to impact drug development, disease diagnosis, and other important research areas of molecular biology. Common instrumental methods for molecular size characterization are disadvantageous due to high sample consumption, measurements made in non-physiological conditions, and/or inaccurate size determinations. Capillary Taylor dispersion analysis (TDA) is a molecular sizing method that utilizes nL sample volumes and achieves absolute size determination without calibration or comparison to standards. One key drawback of TDA is that it reports the concentration-weighted average RH, which may be limiting in the analysis of complex sample mixtures. Here, we describe the development of a 3D printed instrument to integrate capillary electrophoresis (CE) separations online with TDA size characterization. Dual laser-induced fluorescence detectors were developed to enable two-channel detection using a single PMT and fluorescence filter set, achieving detection limits for AlexaFluor 532 of 0.6 ± 0.4 nM and 1.1 ± 0.2 nM for detectors 1 and 2, respectively. Joule heating during CE separations was observed to introduce bias in subsequent TDA measurements. The effects of Joule heating were mitigated by integrating a water circulating sheath flow on the portion of the capillary used for CE. The utility of CE-TDA in bioanalysis was demonstrated by standard-free peak identification in the ficin digestion of IgG1. CE-TDA was further applied to characterizing denaturation dynamics of the Group II heat resistant protein apolipoprotein A-1 (ApoA), in which RH was observed to increase from 2.3 ± 0.2 nm at 20 °C to 5.2 ± 0.5 nm while heated at at 90 °C, then returned to a quasi-native state with RH = 2.9 ± 0.5 nm after cooling to 20 °C. CE-TDA is a powerful analysis mode with potential to impact various domains of bioanalysis. The instrument developed in this work offers a low barrier to entry for researchers interested in adopting CE-TDA.
Introduction
Accurate size determination can provide essential insights on the origins, mechanisms, interactions, and functions of biomolecules. Protein structure and function are intrinsically linked, and hydrodynamic radius (RH) is a descriptive metric of protein structure with the potential to impact drug development, disease diagnosis, and other important challenges in molecular biology.1 As such, bioanalytical methods offering straightforward, rapid, sensitive, selective, and accurate molecular size characterization are needed to accelerate the development of diagnostic methods and therapeutic molecules.2 Common instrumental methods for molecular size characterization present disadvantages such as high sample consumption, measurements made in non-physiological conditions, and/or inaccurate size determinations.1,3Size Exclusion Chromatography (SEC) is a common method for protein sizing, but limited sizing range and modest size resolution may limit its utility in some bioanlaytical applications.2,4 SEC is susceptible to inaccuracy when sizing protein aggregates due to physical filtration, non-specific binding interactions with the column matrix, or aggregate breakdown due to hydrodynamic shear forces.5 Ahrer et al. reported that accurately measuring molecular mass by comparison to protein standards can be challenging, leading to misidentification of a protein's native state or an underestimation of its molecular mass.6 Significant sample dilution occurs on the column which can alter the equilibrium distribution of non-covalent complexes, while changes in the ionic strength and pH of the mobile phase can result in inaccuracies of measured hydrodynamic radius due to electrostatic and hydrophobic effects.7–9 Various additives and modifiers have been used to suppress adsorption and hydrophobic interactions in SEC, illustrating the need for extensive method development to obtain accurate sizing.10
Dynamic light scattering (DLS) characterizes the scattering of light by particles undergoing Brownian motion.5 DLS size measurements are biased towards large aggregates because scatter intensity is proportional to the sixth power of analyte radius. As a result, DLS can overestimate Z-average and suffer from sensitivity challenges for small molecule analytes. These characteristics may present hurdles for sizing peptides or other small biomolecules, especially in the presence of larger proteins or aggregates.2,11,12
Capillary Taylor dispersion analysis (TDA) is a molecular sizing method that utilizes small sample volumes (typically low nL) and achieves absolute size determination without calibration or comparison to standards. In TDA, mean diffusion coefficient is determined by characterizing the band broadening of an analyte plug under well-controlled laminar flow conditions via the Taylor–Aris equation.13–15 Diffusion coefficients can be interpreted as RHvia the Stokes–Einstein equation. TDA offers a much wider dynamic sizing range than SEC, with accurate sizing possible from Å to μm scales. As a single-phase technique, TDA does not suffer from the same non-specific column interactions or complications of shear-induced aggregate breakdown observed in SEC.8,9 Since capillary TDA employs the same concentration-sensitive detection methods common in capillary electrophoresis (CE), it does not present the same size-dependent signal bias observed in DLS.12
One key drawback of TDA is that it reports the concentration-weighted average RH, which may be limiting in the analysis of complex sample mixtures or polydispersed analytes. Cottet and coworkers presented curve-fitting approaches to resolving multiple RH values in a sample mixture by TDA, although these were limited to simple mixtures.16–18 The problem of accurate sizing by TDA within complex sample mixtures remains an active research challenge.
TDA is often performed using commercial CE instrumentation, sharing many of the volume reduction benefits exploited by CE. This makes the integration of CE separation with TDA size analysis a logical approach for addressing the challenge of TDA sizing from complex biological samples. Our group and others have developed methods for integrating CE separation with size characterization by TDA, most commonly by adapting commercial CE instrumentation and methodology for online integration of the two techniques.19–25 For example, we developed a miniature fluorescence detector that enabled two-point TDA detection within the confines of a commercial CE instrument, and demonstrated the utility of RH determination for peak identification without comparison to standards.24
The great potential of CE-TDA as an information-rich analysis mode is held back by the high barrier to entry presented by high-cost commercial CE instrumentation and the need for custom modification of such instrumentation to achieve two-point detection. We previously reported on the development of a low cost, high sensitivity instrument for TDA analysis with dual laser-induced fluorescence (LIF) detectors, which was fabricated by consumer-grade 3D printing.10 Our goal was to make sizing analysis by TDA-LIF more accessible to a wider community of researchers.
With a similar goal, this work describes the development of a 3D printed instrument that integrates CE separations online with TDA size characterization. We discuss the design of an optical detection system that reduces the cost and complexity of dual LIF detection, and the development of thermal management strategies to prevent bias in TDA size characterization. We demonstrate the utility of the 3D printed CE-TDA instrument by characterizing the thermal denaturation dynamics of the Group II heat-resistant protein apolipoprotein A-1 (ApoA), and with standard-free peak identification in the CE spearation of IgG1 antibody fragments.
Materials and methods
Reagents and materials
BSA, sodium phosphate dibasic anhydrous, boric Acid (99% assay), and phosphate buffered saline (PBS) used were fisher BioReagents, sodium phosphate monobasic anhydrous, sodium bicarbonate and sodium hydroxide were Fisher Chemicals, all purchased from Fisher Scientific (Suwannee, GA). Sodium tetraborate decahydrate (99.5%) was from ACROS Organics, tetramethylrhodamine-5-(and-6)-isothiocyanate mixed isomers (TRITC) and AlexaFluor 532 (AF532) were from Invitrogen, all purchased from Thermo Fisher Scientific (Waltham, MA, USA). ApoA was from Sigma Aldrich (St Louis, MO, USA), R-Phycoerythrin (RPE) was from AnaSpec, Inc. (Fremont, CA, USA), and CF532-succinimidyl ester was from Biotium (Hayward, CA, USA). IgG1 was a murine-derived monoclonal anti-lutropin antibody purchased as an AF532 conjugate from Santa Cruz Biotechnology, Inc. (Dallas, TX). All aqueous solutions were prepared using ultrapure water.Optical design
3D printing and assembly instructions are provided in part 1 of the ESI,† and 3D printing model files are available at the NIH 3D print exchange under model # 3DPX-021372. The optical design utilized 6.0 mm N-BK7 ball lenses, 10 mm diameter 4–6 wave aluminum first surface mirrors, and 10 mm right angle prisms, all purchased from Edmund Optics Inc. (Barrington, NJ). Laser excitation was generated by two low power 532 nm laser diode modules (CPS532, Thorlabs Inc., Newton, NJ) frequency modulated using a Siglent SDG 1032X waveform generator. All 3-D printed components were printed using a Form 3B SLA 3D printer with Tough 2000 resin (RS-F2-TO20-01, FormLabs Inc., Somerville, MA). Data acquisition was managed by a USB DAQ module (USB-6341, National Instruments, Austin, TX) with in-house software using LabView (National Instruments, Austin, TX).Evaluation and mitigation of Joule heating effects
Prior to each experimental session, the capillary was flushed for 3 min each with 1.0 M NaOH, 0.1 M NaOH, and DI water, followed by a 5 min flush with CE run buffer (typ. 10 mM borate, pH of 8.3). Between each injection, the capillary was rinsed with 0.1 M NaOH and CE run buffer for 1 min each. The effect of Joule heating on TDA size determinations was assessed by measuring the hydrodynamic radius of 50 nM RPE and 100 nM AF532 following applied CE potentials up to 25 kV. A 100 μm id, 50 cm total length fused silica capillary (Polymicro Technologies, Phoenix, AZ, USA) with the detection windows positioned at 30 cm and 45 cm effective lengths was used for all CE-TDA experiments. Sample was injected by pressure (12 psi applied to the eductor) for 1 second, followed by application of voltage for 2 min. CE potential was terminated prior to observing signal at the first detector, and pressure driven flow via the eductor was used to drive the separated peaks past both detectors for TDA sizing analysis. To mitigate Joule heating effects, a sheath flow of either compressed air or room temperature water was integrated to surrounded the capillary from approximately 2 cm after the inlet up to the first detector.Ficin digestion of IgG1
3 mL of immobilized ficin resin (Thermo Scientific, Waltham, MA) was added to 20 mL of equilibration buffer (5 mM EDTA, 4 mM cysteine, 100 mM citrate, pH = 6.0) then centrifuged at 1000g for 2 minutes, discarding the supernatant. A solution of 40 μg mL−1 IgG1 in 100 mM citrate buffer (pH = 6.0) was added to the resin pellet and incubated at 37 °C with gentle mixing for 12 hours. CE-TDA analysis of the resulting fragmentation products was carried out using a background electrolyte of 10 mM borate (pH = 8.0), with an applied potential of 25 kV for the first 90 seconds followed by an applied pressure at the eductor inlet of 7 psig for the remainder of the analysis.Thermal denaturation studies
A reaction mixture of 6 μM TRITC and 90 μM BSA was prepared in 100 mM bicarbonate buffer (pH = 9.0) and allowed to react in darkness overnight. Working samples of the mixture were prepared by diluting to a final concentration of 1 μM BSA with ultrapure water.A reaction mixture of 2 μM CF532-succinimidyl ester and 1 μM ApoA was prepared in 10 mM phosphate buffer (pH = 7.5) and allowed to react in darkness overnight. Working samples of the mixture were prepared to a final concentration of 100 nM ApoA in 10 mM phosphate buffer (pH = 7.5).
20 μL of either 1 μM BSA-TRITC or 100 nM ApoA-CF532 mixtures were heated to temperatures of 60 °C, 70 °C, 80 °C, or 90 °C for 40 min, cooled in a room temperature water bath for 10 minutes, then analyzed by CE-TDA.
Results and discussion
Optical design
The previously reported TDA instrument,10 was further developed to reduce overall cost and complexity, improve optical performance, and integrate CE online with TDA in a single instrument. The resulting 3D printed CE-TDA instrument is illustrated in Fig. 1A.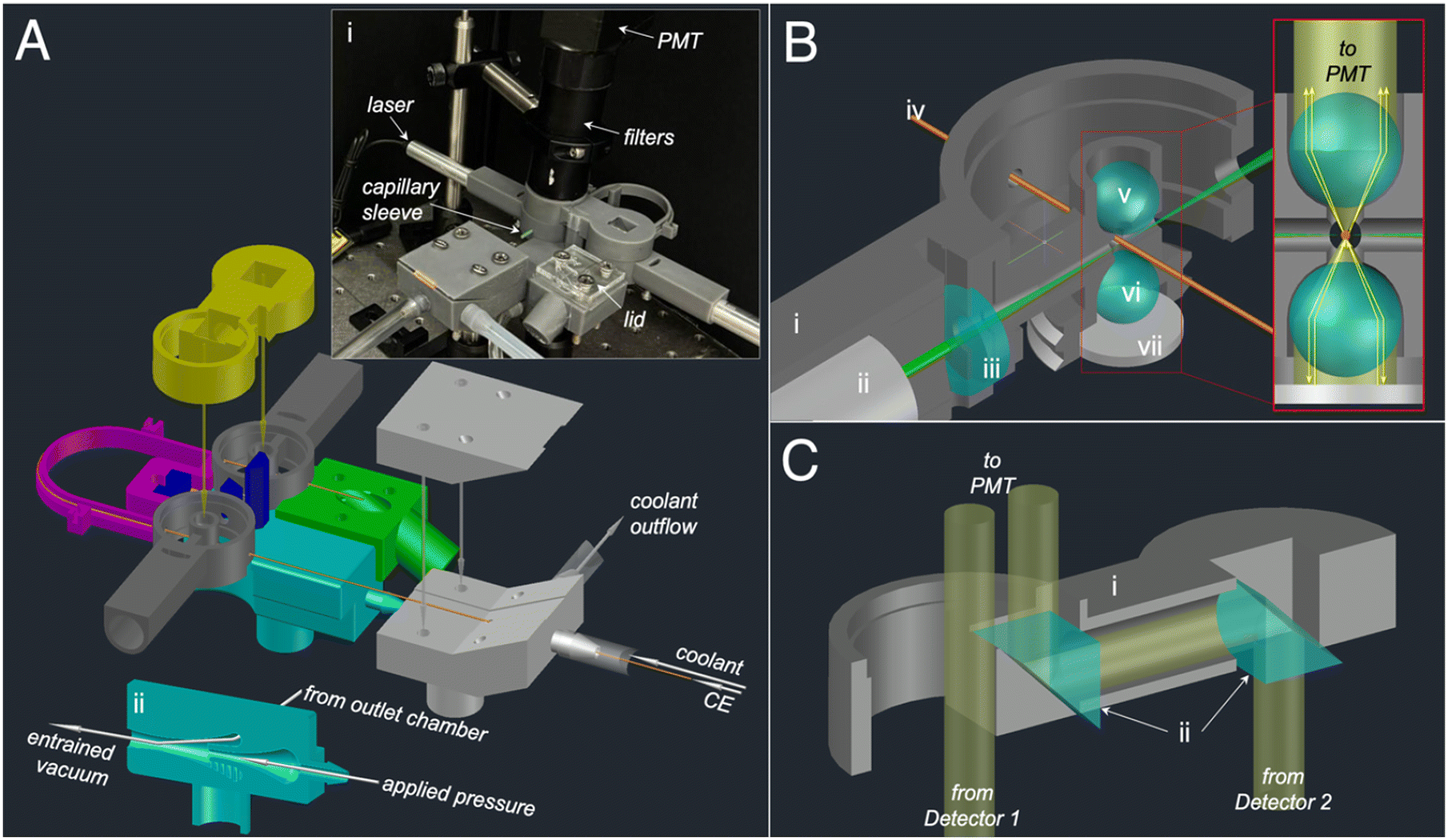 | ||
| Fig. 1 Design illustrations of the CE-TDA instrument (A) CAD illustration showing the functional modules of the instrument. The instrument is composed of 4 key subassemblies: (1) the instrument body consisting of the eductor flow system (cyan), the outlet chamber (green), two LIF detectors (dark grey), and alignment structures (blue); (2) the capillary cartridge (pink) which ensures alignment of capillary detection windows with LIF detection points; (3) the periscope (yellow) to enable side-by-side beam combination of emission from both detectors; and (4) the cooling structure (light grey), which enables a sheathflow of coolant around the capilliary. A second identical cooling structure (not shown) enables coolant inflow near the capillary inlet. Inset i shows a photograph of the assembled instrument, highlighting essential components not produced by 3D printing. Inset (ii) shows the internal geometry of the eductor, which enables pressure driven flow in the capillary by drawing vacuum pressure from the outlet chamber via a channel within the detector body. (B) A detailed illustration of the optical configuration within the LIF detector. The 3D printed detector body (i) aligns a compact laser diode module (ii) and focusing lens (iii) with the CE-TDA capillary (iv). The capillary intersects the laser excitation at a detection point positioned at the focal points of a 6 mm ball lens (v) which acts as an objective lens capturing and collimating fluorescence emission from the front side of the capillary. An identical ball lens (vi) and flat mirror (vii) form the “back side reflector”. The inset shows the action of the backside reflector to capture fluorescence emission (yellow) and return it via the detection point of the capillary to the front side objective for relay to the PMT. (C) A detailed illustration of the optical configuration within the periscope beam combiner. A 3D printed body (i) positions two 10 mm right angle prisms (ii) to combine emission from both detectors into a single 25 mm diameter lens tube (not shown) for relay to the PMT. | ||
Optical design modifications were aimed at combining fluorescence emission from both detectors into a single beam to utilize a single optical filter set and PMT transducer. Three design modifications were needed to realizing this goal. First, the beam diameter on the emission axis was reduced from ca. 25 mm to ca. 6 mm which enabled side-by-side beam combination in the 25 mm diameter optical path. The side-by-side approach was preferred over coaxial beam combination to avoid signal losses that occur when combining beams using beam splitters. Although both the original and the modified detector optics utilize a 6 mm ball lens as the primary objective for collecting fluorescence emission, the previous design employed a 25 mm parabolic mirror to reflect emission from the back side of the capillary into a nominally collimated 25 mm beam. In the revised design (Fig. 1B), the parabolic mirror was replaced by a “back-side reflector” consisting of an additional 6 mm ball lens and 10 mm flat mirror. The back-side reflector returned collected emission to be re-focused at the detection point. Back-side emission was subsequently captured by the front-side objective ball lens and transmitted to the PMT as a nominally collimated beam of 6 mm diameter.
Fig. 2 shows the signal-to-noise ratios (S/N) for 100 nM AF532 resulting from each of a series of design iterations described below. We observes S/N of 132 ± 3 and 128 ± 4 for detectors 1 and 2, respectively, when detecting fluorescence only via the front-side objective lens. S/N increased to 248 ± 13 and 288 ± 9 with the addition of the back-side reflector. The captured back-side emission was therefore shown to account for approximately half of total signal from each detector.
 | ||
| Fig. 2 Summary of detector performance vs. optical design iteration. For “front-side only”, signal was detected via a single 6 mm ball lens with dedicated filters and PMTs for each detector, and “with back-side reflector” adds to this arrangement the back-side ball lens and flat mirror. Both detectors (with back-side reflectors) were combined via the periscope structure (Fig. 1C) to produce the “combined beams” condition via a single filter set and PMT. The “frequency resolved” condition utilized the “combined beams” condition with the addition of frequency modulation/demodulation to resolve signal originating from each detector. Bar heights each represent the mean of 10 replicate measurements. Error bars represent ± 1 standard deviation. | ||
The second optical design modification involved combining the spatially separated 6 mm emission beams into a single 25 mm diameter optical path. For this we designed a 3D printed periscope which employed two right angle turning prisms to position the emission beam of detector 2 alongside that of detector 1 (Fig. 1C). This design enabled detection from both detection points via a single optical filter set and PMT, however S/N was reduced to 173 ± 3 and 130 ± 2 for detectors 1 and 2 respectively. We attributed the reduced S/N for both detectors to the combination of baseline noise from two detectors being simultaneously transduced at a single PMT.
Finally, to address the overall reduction of S/N when combining beams, we implemented a frequency modulation/demodulation scheme to resolve the two detector signals by digital frequency filtering. The laser excitation sources for detectors 1 and 2 were pulsed at 29 Hz and 70 Hz, respectively, with the combined time domain signal recorded at the PMT. Digital filtering was used to reconstruct time-domain signals for the 29 Hz and 70 Hz components, resolving signal and noise from detectors 1 and 2. The narrow frequency bandwidth of the resulting signals improved S/N to 248 ± 7 and 278 ± 3 for detectors 1 and 2, respectively. Similar frequency modulation/demodulation approaches have been described for multiplexing in laser-induced fluorescence anisotropy.26 This design process reduced limit of detection (LODs) for AF532 from 6.0 ± 0.3 nM and 3.6 ± 0.2 nM to 0.6 ± 0.4 nM and 1.1 ± 0.2 nM for detectors 1 and 2, respectively.
Evaluation and mitigation of Joule heating effects on TDA sizing
Electrophoresis can induce Joule heating of the sample solution, especially at high applied potentials or in high conductivity buffers. Joule heating will have deleterious effects on the precision and accuracy of TDA by generating uncontrolled and varying solution viscosity and temperature. To evaluate this effect in the 3D printed CE-TDA instrument, CE-TDA was conducted for samples of 50 nM RPE and 100 nM AF532 at applied potentials up to 25 kV (Fig. 3). With no applied potential (i.e. by TDA only), we observed RH of 0.69 ± 0.06 nm and 5.29 ± 0.52 nm for AF532 and RPE, respectively. Both analytes showed apparent increases in RH at applied potentials of 15 kV and above when no cooling mechanism was used, indicating that sizing accuracy was affected by uncontrolled Joule heating. Interestingly, with no active cooling, fluorescent signal for RPE reduced dramatically at 25 kV such as to make TDA impossible. We hypothesize that this may have resulted from thermal denaturation of the fluorescent protein, but further work would be needed to understand the mechanisms involved.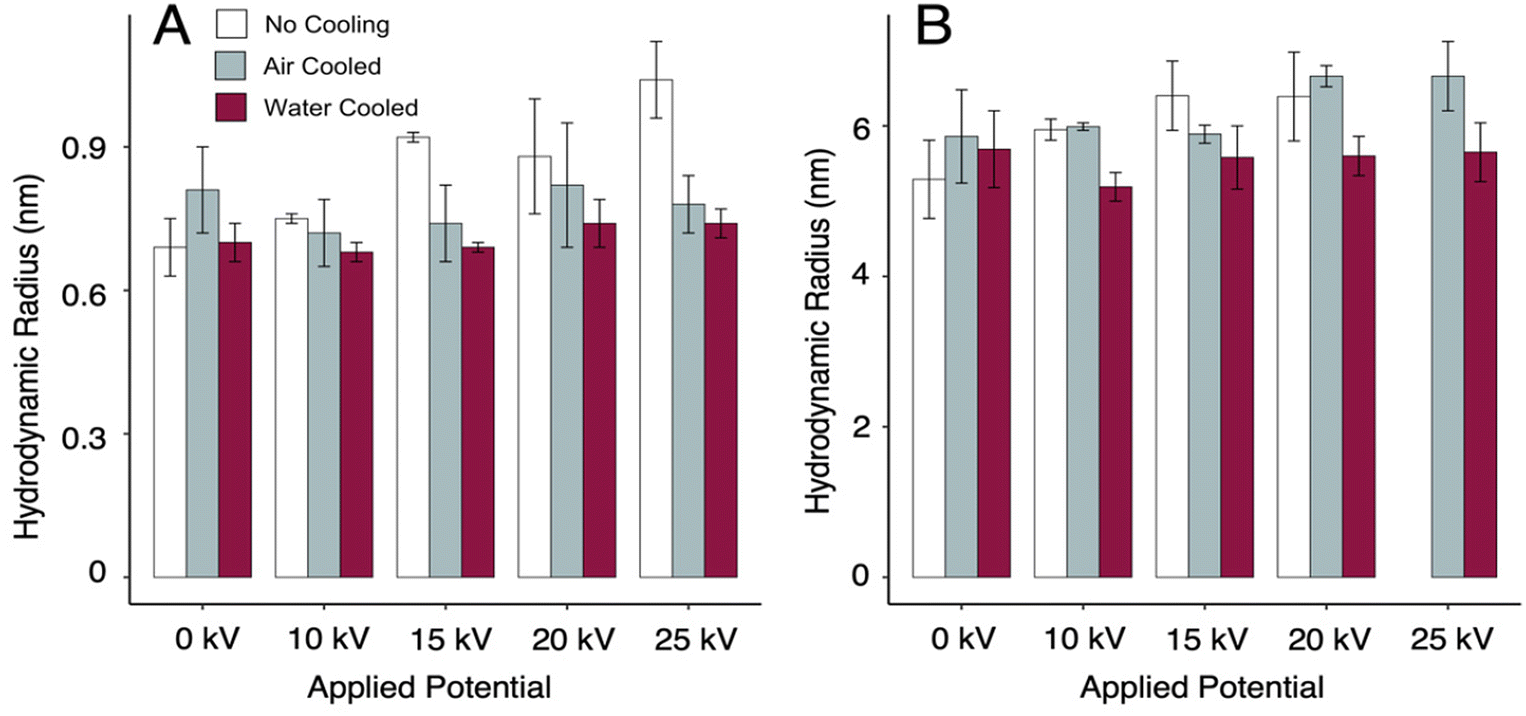 | ||
| Fig. 3 Effects of Joule heating on RH determination by CE-TDA. (A) CE-TDA sizing of the small molecule dye AF532 at various applied potentials and various capillary cooling conditions. Since denaturation cannot occur for AF532, observed RH increases (>10 kV, “no cooling”) resulted from thermal bias in the TDA size measurement. No significant RH increases were observed for either active cooling approach. (B) CE-TDA sizing of the fluorescent protein RPE. Only “water cooled” measurements were sufficient to eliminate significant RH increases above 15 kV, suggesting thermal denaturation of RPE during the CE process under the “air cooled” condition. Bar heights each represent the mean of 10 replicate measurements. Error bars represent ± 1 standard deviation. | ||
To mitigate the deleterious effects of Joule heating on TDA sizing, we integrated a sheath flow channel surrounding the capillary between the inlet and detector 1 (see Fig. 1A). Active cooling was achieved by circulating either 0 °C compressed air, generated by passing compressed air at 10 psig through a copper coil submerged in ice water, or room temperature water through the sheath flow channel. Importantly, water presents a potential electrocution hazard when used in close proximity to high voltage. Caution is advised, and alternative cooling media, such as perfluorinated solvents, may be warranted. Both cooling media were sufficient to eliminate the effects of Joule heating on TDA sizing of the small molecule AF532, with no statistical difference in RH observed for applied potentials up to 25 kV (Fig. 3A).
For RPE, active cooling with circulating water was sufficient to eliminate evidence of bias in the TDA measurement, but the same was not true for active cooling with compressed air, despite compressed air cooling restoring measurable signal for RPE at 25 kV. The difference in efficacy for compressed air cooling for AF532 vs. RPE suggests that bias in sizing RPE for CE potentials of 15 kV or more is driven by thermal denaturation during the CE step, not viscosity or temperature inconsistencies during TDA. These results highlight Joule heating as an important consideration when designing CE-TDA assays for protein and other macromolecules.
CE-TDA for peak identification
Peak identification in CE separations is typically accomplished either via information-rich detection modes, or by comparison of electrophoretic mobility with purified standards. Detection by mass spectrometry (i.e. CE-MS) provides mass-to-charge ratio as a structurally descriptive metric of the analyte. Although a powerful analysis mode, CE-MS requires costly instrumentation and extensive method development, and MS analysis occurs in the gas phase under vacuum conditions that perturb protein structure. While comparison to standards is more straightforward, lower cost, and less labor intensive than CE-MS, it is limited by the availability of pure standards. For example, when using CE to characterize binding interactions, the bound complex of interest may not be sufficiently stable to allow the preparation of purified standards. In such cases, analyses are often limited to those binding interactions which impart substantial shifts in electrophoretic mobility.Another case in which purified standards are not feasible is in the structural modification of complex natural products, such as in the enzymatic fragmentation of antibodies. Ficin is a cysteine endopeptidase utilized in the digestion of IgG antibodies, yielding antigen-binding fragments (Fab). Under various conditions, ficin digestion can yield a dimer of Fab fragments termed F(ab)2, with the crystallizable fragment (Fc) more extensively digested than the Fab fragment to give the partially digested pFC′ fragment. Digestion of antibodies into subfragments has various use cases, such as in the preparation of antibody drug formulations,27 the development of binding assays,28 and for preparation of crystal structures.29
Fig. 4 shows CE-TDA of a murine-derived IgG1 antibody, commercially labeled with AlexaFluor 532, and its ficin digestion products. Fig. 4A shows one principal peak with RH = 6.2 ± 0.3 nm, indicative of the intact immunoglobulin. After ficin digestion (Fig. 4B), three prominent peaks were observed with RH = 0.9 ± 0.05 nm (not labeled in Fig. 4B), RH = 3.0 ± 0.1 nm, and RH = 1.7 ± 0.2 nm. Various minor peaks were observed, but with insufficient signal intensity or resolution to allow confident Gaussian fits.
 | ||
| Fig. 4 Peak identification in antibody fragmentation reactions by CE-TDA. (A) Prior to ficin digestion, the murine-derived IgG1 labelled with AlexaFluor 532 was injected on the capillary at a concentration of 40 μg mL−1. CE separation at an applied potential of 25 kV was carried out for 1.5 min, followed by TDA to give RH = 6.2 ± 0.3 nm (n = 7). The trace from detector 2 is vertically offset for figure clarity. (B) CE-TDA analysis after 12 hours of ficin digestion shows 3 prominent peaks with RH = 0.9 ± 0.05 nm (not labeled, black trace tm = 1.3 min), RH = 3.0 ± 0.1 nm corresponding to Fab fragment, and RH = 1.7 ± 0.2 nm corresponding to pFC′ fragment. Peak assignments were made by comparison to crystal structures, as described in the text. | ||
Crystal structures for various IgG1 fragments were downloaded from the RCSB Protein Data Bank, and their expected hydrodynamic radii were calculated using the HullRad algorithm.30 Four crystal structures were available for murine-derived IgG1 Fab fragments,31–34 yielding RH = 3.04 ± 0.1 nm. Two crystal structures were available for murine-derived IgG1 Fc fragments,35,36 yielding RH = 2.53 ± 0.08 nm (mean ± range). One available structure of murine F(ab)2 gave RH = 3.87 nm.37 No structures were available for murine-derived pFC′ fragment, but one available structure for guinea pig-derived pFC′ resulted in RH = 1.93 nm.38
HullRad calculations of anticipated RH indicate that the peaks labelled in Fig. 4B can be assigned as Fab (RH = 3.0 ± 0.1 nm) and pFC′ (RH = 1.7 ± 0.2 nm) fragments, with minor peaks likely the byproducts of Fc digestion. This result indicates that the commercially sourced fluorescent antibody was non-selectively labelled, with labelling sites on both the Fc and Fab domains. Moreover, it illustrates the utility of CE-TDA for information rich detection sufficient for peak identification. This approach is analogous to peak identification by CE-MS in that both m/z and RH are structurally descriptive metrics.
Denaturation dynamics characterization by CE-TDA
To illustrate the utility of CE-TDA beyond peak identification in CE, we utilized the 3D printed CE-TDA instrument to evaluate an established model of thermal denaturation in proteins. Kim and coworkers used far-UV circular dichroism spectroscopy to observe the thermal denaturation dynamics of ApoA, classifying the protein as a Group II heat-resistant protein.39 Group II proteins showed evidence of denaturation upon heating, followed by spontaneous return to a psuedo-native conformation upon cooling. Here, we demonstrate similar insights by observing the effect of heating on RH of ApoA-CF532 using CE-TDA.Fig. 5A shows the effect of heating on BSA-TRITC and ApoA-CF532 at temperatures ranging from 60 °C to 90 °C. In each case, proteins were heated at the indicated temperature for 40 minutes, then allowed to cool to 20 °C prior to measurement by CE-TDA. In the case of BSA-TRITC, RH increased from 4.2 ± 0.6 nm at 20 °C to 7.9 ± 0.1 nm at 80 °C, with no further evidence of thermal denaturation upon increasing temperature to 90 °C. This behavior agrees well with previous observations of the thermal denaturation behavior of BSA.40–45
 | ||
| Fig. 5 Thermal denaturation of BSA and ApoA characterized by CE-TDA. (A) BSA-TRITC and ApoA-CF532 samples, containing excess unreacted label, were incubated at the indicated temperature for 40 minutes, then cooled to room temperature before CE-TDA analysis of the respective mixtures. ApoA-CF532 showed delayed onset of thermal denaturation as compared to BSA-TRITC (80 °C vs. 60 °C). Sizing of unreacted dye showed no statistical differences as a function of incubation temperature, serving as internal standards to verify that observed RH changes for BSA-TRITC and ApoA-CF532 were not the result of thermal bias in the TDA measurement. (B) CE-TDA analysis of ApoA-CF532 before heating, during the incubation at 90 °C, and after cooling to room temperature revealed the Group II heat-resistant protein structural dynamics of ApoA. Increased RH at 90 °C indicates that ApoA-CF532 undergoes structural change at elevated temperatures but reverts to a more compact structure similar in RH to the native structure upon cooling. Bar heights each represent the mean of 5 replicate measurements. Error bars represent ± 1 standard deviation. | ||
The Group II heat-resistant protein ApoA showed marked differences in denaturation dynamics as compared to BSA. At 20 °C we observed a RH for ApoA-CF532 of 2.3 ± 0.3 nm, with no evidence of thermal denaturation below 80 °C. Upon heating at 80 °C, a significant increase in RH was observed to 3.1 ± 0.4 nm, which did not change significantly upon heating at 90 °C. This result demonstrates that the native conformation of ApoA is highly resistant to thermal denaturation, which agrees with previous findings showing evidence of intact alpha helical character at 100 °C.39
For both BSA-TRITC and ApoA-CF532, CE-TDA separated and sized excess unreacted dye in the labeled protein samples, allowing the excess dye to serve as internal size references. Neither TRITC (RH = 0.57 ± 0.09 nm) nor CF532 (RH = 0.59 ± 0.09 nm) showed significant differences in RH across the temperature range, indicating that observed RH changes in BSA-TRITC and ApoA-CF532 were not the result of uncontrolled temperature or viscosity in the TDA measurements.
Fig. 5B shows the dynamic behavior of ApoA-CF532 in response to thermal denaturation. When ApoA-CF532 was sized during heating but without cooling to 20 °C, a significant increase in RH was observed from 2.3 ± 0.2 nm at 20 °C to 5.2 ± 0.5 nm at 90 °C. When the same sample was cooled to 20 °C, RH reduced to 2.9 ± 0.5 nm, indicating a return to native or quasi-native structure. This result illustrates that CE-TDA can characterize transient states of protein denaturation, and agrees well with Kim and coworkers definition of Group II heat-resistant protein behavior.39
Conclusions
In this work we described the development of a 3D printed instrument for online integration of CE separations with molecular sizing by TDA. Optical design modifications were described which reduced the cost and complexity of the instrument relative to our previously reported system while achieving low nM LODs at dual LIF detection points. Joule heating was addressed as a source of potential thermal bias in TDA sizing by integrating a water-circulating sheath flow surrounding the portion of the capillary used for CE. The utility of CE-TDA was demonstrated for peak identification in CE by assigning fragment identities in the ficin digestion of IgG1. Finally, CE-TDA was demonstrated as a technology for characterizing structural dynamics in proteins, specifically by observing the denaturation dynamics of the Group II heat-resistant protein ApoA.Capillary TDA is a powerful yet underutilized molecular sizing technique that benefits from online integration with CE separations. RH is a valuable analytical metric which we have demonstrated has utility in analyte identification and characterization of structural dynamics. Our objective with the development of this instrument was to offer the research community a powerful tool and an accessible entry point into CE-TDA technology. Realizing the full potential of CE-TDA as an analysis mode requires further work, such as RH-based assay development and investigations into the scope and limitations of RH as a structural descriptor of biomolecules. We anticipate that the system developed in this work will enable such efforts.
Author contributions
Atsar, F.S.: investigation, data curation, formal analysis, methodology, writing – original draft; Bourger, H.D.: investigation, methodology, formal analysis, writing – review & editing; Baker, C.A.: conceptualization, funding acquisition, project administration, software, supervision, resources, visualization, writing – review & editing.Data availability
Data shown in this article is available via the figshare repository (https://doi.org/10.6084/m9.figshare.26997382) and STL files for the 3D printed instrument are available via the NIH 3D print exchange at https://3d.nih.gov/entries/3DPX-021372.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge Dr Laura Casto for her helpful input on data visualization, and Dr Claire Smith for her efforts maintaining the Baker Bioanalysis Lab's 3D printing infrastructure. This work was supported in part by the National Science Foundation under grant number CHE-2054748 and by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM138173. H. D. Bourger was supported in part by a training grant from National Institute of General Medical Sciences (grant # T32GM148394).References
- W. Zhang, H. Wu, R. Zhang, X. Fang and W. Xu, Structure and Effective Charge Characterization of Proteins by a Mobility Capillary Electrophoresis Based Method, Chem. Sci., 2019, 10(33), 7779–7787, 10.1039/c9sc02039j.
- H. Goetz, M. Kuschel, T. Wulff, C. Sauber, C. Miller, S. Fisher and C. Woodward, Comparison of Selected Analytical Techniques for Protein Sizing, Quantitation and Molecular Weight Determination, J. Biochem. Biophys. Methods, 2004, 60(3), 281–293, DOI:10.1016/j.jbbm.2004.01.007.
- P. M. Nowak, K. Śpiewak, M. Woźniakiewicz and P. Kościelniak, Minimizing the Impact of Joule Heating as a Prerequisite for the Reliable Analysis of Metal–protein Complexes by Capillary Electrophoresis, J. Chromatogr. A, 2017, 1495, 83–87, DOI:10.1016/j.chroma.2017.03.028.
- A. Oliva, M. Llabrés and J. B. Fariñ, Comparative Study of Protein Molecular Weights by Size-Exclusion Chromatography and Laser-Light Scattering, J. Pharm. Biomed. Anal., 2001, 25, 833–841 CrossRef.
- S. Amin, G. V. Barnett, J. A. Pathak, C. J. Roberts and P. S. Sarangapani, Protein Aggregation, Particle Formation, Characterization & Rheology, Curr. Opin. Colloid Interface Sci., 2014, 19(5), 438–449, DOI:10.1016/j.cocis.2014.10.002.
- K. Ahrer, A. Buchacher, G. Iberer, D. Josic and A. Jungbauer, Analysis of Aggregates of Human Immunoglobulin G Using Size-Exclusion Chromatography, Static and Dynamic Light Scattering, J. Chromatogr. A, 2003, 1009(1–2), 89–96, DOI:10.1016/S0021-9673(03)00433-3.
- O. Bermudez and D. Forciniti, Aggregation and Denaturation of Antibodies: A Capillary Electrophoresis, Dynamic Light Scattering, and Aqueous Two-Phase Partitioning Study, J. Chromatogr. B:Anal. Technol. Biomed. Life Sci., 2004, 807, 17–24, DOI:10.1016/j.jchromb.2004.01.029.
- J. F. Carpenter, T. W. Randolph, W. Jiskoot, D. J. A. Crommelin, C. R. Middaugh and G. Winter, Potential Inaccurate Quantitation and Sizing of Protein Aggregates by Size Exclusion Chromatography: Essential Need to Use Orthogonal Methods to Assure the Quality of Therapeutic Protein Products, J. Pharm. Sci., 2010, 99(5), 2200–2208, DOI:10.1002/jps.21989.
- J. Wen, T. Arakawa and J. S. Philo, Size-Exclusion Chromatography with On-Line Light-Scattering, Absorbance, and Refractive Index Detectors for Studying Proteins and Their Interactions, Anal. Biochem., 1996, 240(2), 155–166, DOI:10.1006/abio.1996.0345.
- M. R. Moser, C. M. Smith, G. G. Gutierrez and C. A. Baker, 3D Printed Instrument for Taylor Dispersion Analysis with Two-Point Laser-Induced Fluorescence Detection, Anal. Chem., 2022, 94(16), 6089–6096, DOI:10.1021/acs.analchem.1c04566.
- M. Kaszuba, D. McKnight, M. T. Connah, F. K. McNeil-Watson and U. Nobbmann, Measuring Sub Nanometre Sizes Using Dynamic Light Scattering, J. Nanopart. Res., 2008, 10(5), 823–829, DOI:10.1007/s11051-007-9317-4.
- A. Hawe, W. L. Hulse, W. Jiskoot and R. T. Forbes, Taylor Dispersion Analysis Compared to Dynamic Light Scattering for the Size Analysis of Therapeutic Peptides and Proteins and Their Aggregates, Pharm. Res., 2011, 28(9), 2302–2310, DOI:10.1007/s11095-011-0460-3.
- M. R. Moser and C. A. Baker, Taylor Dispersion Analysis in Fused Silica Capillaries: A Tutorial Review, Anal. Methods, 2021, 13(21), 2357–2373, 10.1039/d1ay00588j.
- W. L. Hulse and R. T. Forbes, A Taylor Dispersion Analysis Method for the Sizing of Therapeutic Proteins and Their Aggregates Using Nanolitre Sample Quantities, International Journal of Pharmaceutics, 2011, 416(1), 394–397, DOI:10.1016/j.ijpharm.2011.06.045.
- W. L. Hulse and R. T. Forbes, A Nanolitre Method to Determine the Hydrodynamic Radius of Proteins and Small Molecules by Taylor Dispersion Analysis, Int. J. Pharm., 2011, 411(1–2), 64–68, DOI:10.1016/j.ijpharm.2011.03.040.
- J. Chamieh, F. Oukacine and H. Cottet, Taylor Dispersion Analysis with Two Detection Points on a Commercial Capillary Electrophoresis Apparatus, J. Chromatogr. A, 2012, 1235, 174–177, DOI:10.1016/j.chroma.2012.02.049.
- A. Lewandrowska, A. Majcher, A. Ochab-Marcinek, M. Tabaka and R. Hołyst, Taylor Dispersion Analysis in Coiled Capillaries at High Flow Rates, Anal. Chem., 2013, 85(8), 4051–4056, DOI:10.1021/ac4007792.
- H. Cottet, J. P. Biron and M. Martin, Taylor Dispersion Analysis of Mixtures, Anal. Chem., 2007, 79(23), 9066–9073, DOI:10.1021/ac071018w.
- T. Le Saux and H. Cottet, Size-Based Characterization by the Coupling of Capillary Electrophoresis to Taylor Dispersion Analysis, Anal. Chem., 2008, 80(5), 1829–1832, DOI:10.1021/ac702257k.
- F. Oukacine, A. Morel, I. Desvignes and H. Cottet, Size-Based Characterization of Nanoparticle Mixtures by the Inline Coupling of Capillary Electrophoresis to Taylor Dispersion Analysis, J. Chromatogr. A, 2015, 1426, 220–225, DOI:10.1016/j.chroma.2015.11.024.
- J. Deschamps, S. G. Dutremez, B. Boury and H. Cottet, Size-Based Characterization of an Ionic Polydiacetylene by Taylor Dispersion Analysis and Capillary Electrophoresis, Macromolecules, 2009, 42(7), 2679–2685, DOI:10.1021/ma802109f.
- J. Østergaard and H. Jensen, Simultaneous Evaluation of Ligand Binding Properties and Protein Size by Electrophoresis and Taylor Dispersion in Capillaries, Anal. Chem., 2009, 81(20), 8644–8648, DOI:10.1021/ac901419x.
- T. Liu, F. Oukacine, H. Collet, A. Commeyras, L. Vial and H. Cottet, Monitoring Surface Functionalization of Dendrigraft Poly-l-Lysines via Click Chemistry by Capillary Electrophoresis and Taylor Dispersion Analysis, J. Chromatogr. A, 2013, 1273, 111–116, DOI:10.1016/j.chroma.2012.11.074.
- L. D. Casto, K. B. Do and C. A. Baker, A Miniature 3d Printed Led-Induced Fluorescence Detector for Capillary Electrophoresis and Dual-Detector Taylor Dispersion Analysis, Anal. Chem., 2019, 91(15), 9451–9457, DOI:10.1021/acs.analchem.8b05824.
- L. Leclercq and H. Cottet, Fast Characterization of Polyelectrolyte Complexes by Inline Coupling of Capillary Electrophoresis to Taylor Dispersion Analysis, Anal. Chem., 2012, 84(3), 1740–1743, DOI:10.1021/ac203208k.
- A. M. Schrell, N. Mukhitov and M. G. Roper, Multiplexing Fluorescence Anisotropy Using Frequency Encoding, Anal. Chem., 2016, 88(16), 7910–7915, DOI:10.1021/acs.analchem.6b02131.
- A. L. Nelson and J. M. Reichert, Development Trends for Therapeutic Antibody Fragments, Nat. Biotechnol., 2009, 27(4), 331–337, DOI:10.1038/nbt0409-331.
- J. R. Whiteaker, L. Zhao, C. Frisch, F. Ylera, S. Harth, A. Knappik and A. G. Paulovich, High-Affinity Recombinant Antibody Fragments (Fabs) Can Be Applied in Peptide Enrichment Immuno-MRM Assays, J. Proteome Res., 2014, 13(4), 2187–2196, DOI:10.1021/pr4009404.
- L. Griffin and A. Lawson, Antibody Fragments as Tools in Crystallography, Clin. Exp. Immunol., 2011, 165(3), 285–291, DOI:10.1111/j.1365-2249.2011.04427.x.
- P. J. Fleming and K. G. Fleming, HullRad: Fast Calculations of Folded and Disordered Protein and Nucleic Acid Hydrodynamic Properties, Biophys. J., 2018, 114(4), 856–869, DOI:10.1016/j.bpj.2018.01.002.
- A. V. Fokin, P. V. Afonin, I. Mikhaĭlova, I. N. Tsygannik, T. Mareeva, V. A. Nesmeianov, W. Pangborn, N. Lee, W. Duax, E. Siszak and V. Z. Pletnev, Prostranstvennaia struktura Fab-fragmenta monoklonal'nogo antitela k interleĭkinu-2 cheloveka v dvukh kristallicheskikh formakh pri razreshenii. Spatial Structure of a Fab-Fragment of a Monoclonal Antibody to Human Interleukin-2 in Two Crystalline Forms at a Resolution of 2.2 and 2.9 Angstroms, Bioorg. Khim., 2000, 26(8), 571–578 Search PubMed . Russian. PMID: 11040993.
- Z. Zhao, D. Worthylake, L. LeCour, G. A. Maresh and S. H. Pincus, Crystal Structure and Computational Modeling of the Fab Fragment from a Protective Anti-Ricin Monoclonal Antibody, PLoS One, 2012, 7(12), e52613, DOI:10.1371/journal.pone.0052613.
- A. K. Saxena, Structure of Fab Fragment of Malaria Transmission Blocking Antibody 2A8 against P. Vivax P25 Protein, Int. J. Biol. Macromol., 2012, 50(1), 153–156, DOI:10.1016/j.ijbiomac.2011.10.012.
- T. Bizebard, R. Daniels, R. Kahn, B. Golinelli-Pimpaneau, J.J. Skehel and M. Knossow, Refined Three-Dimensional Structure of the Fab Fragment of a Murine IgG1,A Antibody, Acta Crystallogr D Biol Crystallogr., 1994, 50, 768–777, DOI:10.1107/S0907444994001903.
- N. Tajima, N. Simorowski, R. A. Yovanno, M. C. Regan, K. Michalski, R. Gómez, A. Y. Lau and H. Furukawa, Development and Characterization of Functional Antibodies Targeting NMDA Receptors, Nat. Commun., 2022, 13(1), 923, DOI:10.1038/s41467-022-28559-3.
- S. H. Bryant, L. M. Amzel, R. P. Phizackerley and R. J. Poljak, Molecular-Replacement Structure of Guinea Pig IgG1 pFc’ Refined at 3.1 A Resolution, Acta Cryst. B, 1985, 41, 362–368, DOI:10.1107/S0108768185002270.
- M. J. Feige, S. Nath, S. R. Catharino, D. Weinfurtner, S. Steinbacher and J. Buchner, Structure of the Murine Unglycosylated IgG1 Fc Fragment, J. Mol. Biol., 2009, 391(3), 599–608, DOI:10.1016/j.jmb.2009.06.048.
- T. F. Lerch, P. Sharpe, S. J. Mayclin, T. E. Edwards, E. Lee, H. D. Conlon, S. Polleck, J. C. Rouse, Y. Luo and Q. Zou, Infliximab Crystal Structures Reveal Insights into Self-Association, mAbs, 2017, 9(5), 874–883, DOI:10.1080/19420862.2017.1320463.
- T. D. Kim, H. J. Ryu, H. I. Cho, C.-H. Yang and J. Kim, Thermal Behavior of Proteins: Heat-Resistant Proteins and Their Heat-Induced Secondary Structural Changes, Biochemistry, 2000, 39(48), 14839–14846, DOI:10.1021/bi001441y.
- D. K. Wilkins, S. B. Grimshaw, V. Receveur, C. M. Dobson, J. A. Jones and L. J. Smith, Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques, Biochemistry, 1999, 38(50), 16424–16431, DOI:10.1021/bi991765q.
- V. A. Borzova, K. A. Markossian, N. A. Chebotareva, S. Y. Kleymenov, N. B. Poliansky, K. O. Muranov, V. A. Stein-Margolina, V. V. Shubin, D. I. Markov and B. I. Kurganov, Kinetics of Thermal Denaturation and Aggregation of Bovine Serum Albumin, PLoS One, 2016, 11(4), e0153495, DOI:10.1371/journal.pone.0153495.
- J. De Meutter and E. Goormaghtigh, Protein Structural Denaturation Evaluated by MCR-ALS of Protein Microarray FTIR Spectra, Anal. Chem., 2021, 93(40), 13441–13449, DOI:10.1021/acs.analchem.1c01416.
- G. Yohannes, S. K. Wiedmer, M. Elomaa, M. Jussila, V. Aseyev and M.-L. Riekkola, Thermal Aggregation of Bovine Serum Albumin Studied by Asymmetrical Flow Field-Flow Fractionation, Anal. Chim. Acta, 2010, 675(2), 191–198, DOI:10.1016/j.aca.2010.07.016.
- B. Farruggia and G. A. Picó, Thermodynamic Features of the Chemical and Thermal Denaturations of Human Serum Albumin, Int. J. Biol. Macromol., 1999, 26, 317–323 CrossRef CAS PubMed https://www.elsevier.com/locate/ijbiomac .
- A. Adel, M. Nadia, O. Mohamed and G. Abdelhafidh, Study of Thermally and Chemically Unfolded Conformations of Bovine Serum Albumin by Means of Dynamic Light Scattering, Mater. Sci. Eng., C, 2008, 28(5–6), 594–600, DOI:10.1016/j.msec.2007.10.004.
Footnote |
| † Electronic supplementary information (ESI) available: PDF containing instrument assembly instructions. See DOI: https://doi.org/10.1039/d4an01208a |
| This journal is © The Royal Society of Chemistry 2025 |